
INTRODUCTION
Multiple myeloma (MM) is a kind of hematologic B cell malignancy that is featured by the accumulation of abnormal plasma cells in the bone marrow. Current treatments, including dexamethasone, chemotherapy, stem cell transplantation, proteasome inhibitors and immunomodulatory drugs, have made substantial progress toward increasing longevity in MM patients [1, 2]. However, one major limitation of the existing drugs is the eventual emergence of drug resistance for nearly all patients, regardless of the treatment regimen or the initial response to treatment [3]. It is well known that bone marrow microenvironment changes, such as cell adhesion and cytokine-related induction of the JAK/STAT and PI3K/AKT pathways, are responsible for resistance to conventional and novel therapies [4, 5]. Because MM is characterized by a high level of genomic instability and complex cytogenetic aberrations [6, 7], many of these alterations, which include chromosomal translocation, methylation, gene mutation, and microRNA (miRNA) abnormalities, can contribute to drug resistance [3, 8, 9]. Moreover the aberrant expression of oncogenes, such as c-MAF, c-Myc, and cyclin D has been found to contribute to drug resistance in MM [10–12]. Therefore, there is a need to develop new therapeutic strategies for the treatment of MM.
Myc pathway activation increases with the disease stage in MM, and a conditional MYC transgene murine model develops a plasma cell malignancy during the late stages of B cell differentiation that shares clinically relevant features of MM [13]. This result supports the central role of c-Myc activity in the pathogenesis of MM. In addition, c-Myc overexpression occurs via different mechanisms, including gene amplification, translocation and mutation. c-Myc chromosomal rearrangements were found in 15% of newly diagnosed MM patients [14], nearly 50% of advanced MM patients [15, 16], 55% of human MM cell lines [14], and such rearrangements have also been implicated in drug resistance in MM [11]. Furthermore, the inhibition of c-Myc activity by a short-hairpin RNA targeting c-Myc has been shown to be lethal to a number of human myeloma cell lines [17]. In addition, a small molecule inhibitor of BRD4 that suppresses c-Myc transcription shows therapeutic effects against MM in vitro and in vivo, and the small molecule inhibitor 10058-F4a, that specifically inhibits c-Myc-Max heterodimerization, prevents the transactivation of c-Myc, induces cell death in human myeloma cell lines and primary cells of myeloma patients [18, 19]. Thus, c-Myc is likely a promising therapeutic target for MM treatment.
Virotherapy has been shown to have anticancer effects on cancer cells but not normal cells in solid and hematological malignancies. MM represents an attractive target for virotherapy in comparison to many solid tumors because viruses can easily access the tumor cells in the bone marrow and bloodstream. Currently, four RNA viruses (measles virus, vesicular stomatitis virus, reovirus and coxsackievirus A21) and two DNA viruses (adenovirus and vaccinia virus) have shown oncolytic potential against MM [20]. These viruses kill cancer cells via selective viral replication in cancer cells but lose their oncolytic activity following inactivation [21, 22]. To date, inactive viral particles have not been included in oncolytic virotherapy for multiple myeloma.
The Sendai virus (Hemagglutinating Virus of Japan; HVJ) belongs to the paramyxovirus family and has six viral proteins encoded by a negative-sense single-strand RNA genome: nucleocapsid (N); phosphoprotein (P); large (L) matrix (M); fusion (F); and hemagglutinin-neuraminidase (HN). When the Sendai viral genome was cleaved into small pieces by UV irradiation, the viral proteins remained functional [23]. We have documented that the inactivated Sendai virus envelope (HVJ-E) has multiple anti-tumor effects, including the activation of anti-tumor immunity and induction of cancer cell-selective apoptosis [24–26]. Moreover, we demonstrated that HVJ-E RNA fragment-dependent activation of the cytoplasmic RNA receptor retinoic acid-inducible gene-I (RIG-I) and mitochondrial antiviral signaling protein (MAVS) activates the upregulation of TRAIL and Noxa, which enables HVJ-E-mediated cancer-selective apoptosis [26]. The therapeutic potential of HVJ-E has been demonstrated in solid tumors [27–30], but there are no reports concerning whether this virus can be used for the treatment of hematopoietic malignances.
In the present study, we showed that HVJ-E induced cancer-selective apoptosis in multiple myeloma cells with c-Myc addiction. In addition to decrease the phosphorylation of c-Myc at S62 and promote c-Myc protein degradation, HVJ-E induced fusion-mediated elevation of cytoplasmic Ca2+ results in c-Myc transcriptional inhibition. Furthermore, we addressed the mechanisms behind cytoplasmic Ca2+ increasing induced suppression of c-Myc transcription, activation of SMADs family involved in c-Myc downregulation. These results also provide the possibility that HVJ-E will be a therapeutic tool for MMs.
RESULTS
HVJ-E induces apoptosis in human multiple myeloma cell lines but not in PBMCs from healthy donors
To investigate whether HVJ-E exhibits an anti-cancer effect against MM, human myeloma cell lines (NCI-H929, MM1S, RPMI-8226 and U266) and normal human peripheral blood mononuclear cells (PBMCs) from healthy donors were treated with HVJ-E for 72 hours, and cell viability was measured using the MTS assay (Supplementary Figure 1A–1E). The cell viability of NCI-H929, MM1S, and RPMI-8226, but not U266 and PBMCs, was significantly decreased with HVJ-E in a dose-dependent manner. We showed that HVJ-E exhibited cytotoxicity against various MM cells but not healthy PBMCs. Human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAECs) were also treated with HVJ-E, and the proliferation of HUVECs and HAECs was not inhibited (Supplementary Figure 1F–1G), which is consistent with our previous report [31].
Next, to investigate whether apoptosis is the mechanism underlying HVJ-E-mediated cell death in MM, we assayed certain markers of apoptosis in HVJ-E-treated MMs and PBMCs. Large populations of HVJ-E-treated NCI-H929 and MM1S cells but not U266 cells and PBMCs were Annexin-V-positive (Figure 1A and 1B). Further analysis of leukocyte subsets in PBMCs from healthy donors showed that the Annexin-V-positive apoptotic cells did not change in the CD3+ T cell, CD19+ B cell and CD14+ monocyte populations after HVJ-E treatment (Supplementary Figure 2A and 2B). Although CD15+ neutrophils undergo spontaneous apoptosis after 48 hours of incubation in vitro, as reported in previous studies [32], no significant difference in CD15+Annexin-V+ cells was observed in the presence and absence of 48 hours of HVJ-E treatment in neutrophils (Supplementary Figure 2C and 2D). We also examined apoptotic cells among HUVECs and HAECs in the presence and absence of wild-type (WT) and HN-depleted HVJ-E. As shown in Supplementary Figure 2E and 2F, the ratio of Annexin-V-labeled apoptotic cells was not changed by treatment with either WT-HVJ-E or HN-depleted HVJ-E. These results suggest that HVJ-E elicits no harmful effect on healthy leukocytes and endothelial cells after systemic administration. Moreover, the cleavage of caspase-8, caspase-3 and poly(ADP-ribose) polymerase (PARP) was detected in HVJ-E-treated NCI-H929 and MM1S cells but not U266 cells by western blotting analysis (Figure 1C). A pan-caspase inhibitor (Z-VAD-FMK) prevented HVJ-E-mediated activation of caspase-3 (Figure 1D) and cell death (Figure 1E), indicating that HVJ-E induced caspase-dependent apoptosis in MM.
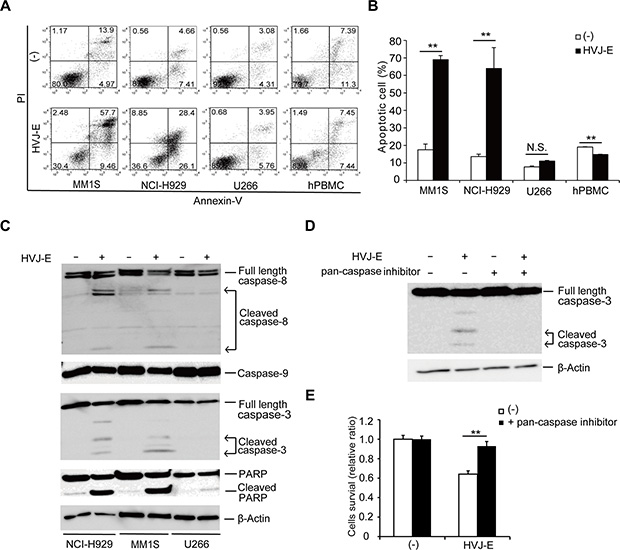
Figure 1: HVJ-E induces caspase-dependent apoptosis in MM cells. (A) Apoptotic cells were measured in HVJ-E-treated NCI-H929, MM1S, U266 and PBMCs from healthy donors at a multiplicity of infection (MOI) of 1000 for 48 hours using PI (vertical) and Annexin V (horizontal) double staining analysis by FACS. (B) Quantification plot of the ratio of the apoptotic (Annexin V+) cells shown in Figure 1A. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test). (C) Detection of caspases (caspase-8, -3, -9) and poly (ADP-ribose) polymerase (PARP) in MM cells (NCI-H929, U266, MM1S) after treatment with HVJ-E at an MOI of 1000 for 48 hours by western blotting analysis. (D) Immunoblotting results for caspase-3 cleavage after HVJ-E treatment (1000 MOI, 24 hours) in the presence or absence of pan-caspase inhibitor in NCI-H929 cells. (E) NCI-H929 cells were treated with HVJ-E at an MOI of 1000 for 48 hours in the presence or absence of pan-caspase inhibitor, and cell viability was assessed using the MTS assay. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test).
Furthermore, we investigated various proteins related to apoptosis and the expression levels of antiapoptotic Bcl-2 family members B-cell lymphoma-extra-large (Bcl-xL) and B-cell lymphoma 2 (Bcl-2) remained unchanged in responses to HVJ-E treatment in NCI-H929 and MM1S cells (Supplementary Figure 3A). However, we demonstrated that HVJ-E treatment enhanced the expression of the proapoptotic protein Bcl-2-like protein 11 (Bim), which consists of three major isoforms as a result of alternative splicing (BimEL, BimL and BimS), but not BH3 interacting-domain death agonist (Bid), Bcl-2-associated death promoter (Bad), Bcl-2-associated X protein (Bax) or p53-upregulated modulator of apoptosis (Puma) in NCI-H929 cells in a time-dependent manner (Supplementary Figure 3B). By immunoblotting analysis, we also found that HVJ-E induced Bim upregulation in RPMI-8226 and MM1S cell lines (Supplementary Figure 3C). Moreover, HVJ-E-mediated cell death was attenuated by Bim knockdown (Supplementary Figure 3D), suggesting that the Bim upregulation stimulated HVJ-E-induced apoptosis in myeloma cells.
In vivo MM tumor suppression by HVJ-E treatment
To investigate the efficacy of HVJ-E against MM in vivo, we incubated an MM1S xenograft in NOG mice. As shown in Figure 2A, HVJ-E treatment substantially lowered the tumor burden in comparison to the PBS group. Moreover, we designed an NCI-H929 xenograft model in NOG mice. NCI-H929 is a human MM cell line derived from a right pleural effusion of a 62-year-old patient who was treated for relapse after treatment with vincristine, melphalan, cytoxan and prednisone [33]. As shown in Figure 2B, HVJ-E treatment significantly suppressed tumor growth in the NCI-H929 xenograft in comparison to PBS treatment. Taken together, these results further support the potential of HVJ-E for the treatment of MM.
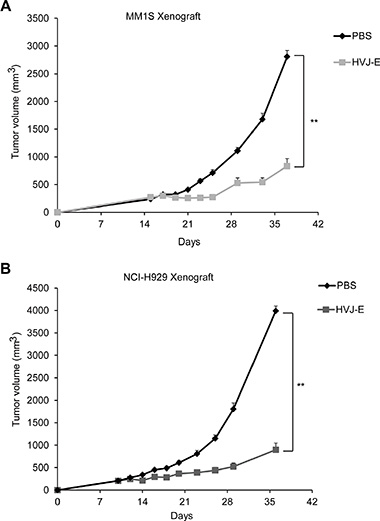
Figure 2: HVJ-E inhibits MM cell growth in vivo in MM xenograft mouse models. Tumor growth curves were measured in MM1S- (A) and NCI-H929-bearing (B) NOG mice. The tumor volume (mm3) (average and SEM) for each group of mice (n = 4–5/group) is shown. The mice were treated with PBS or 5,000 HAU of HVJ-E 3 times per week. A significant delay in tumor growth was noted in HVJ-E-treated mice in comparison to PBS-treated control mice (**P < 0.01, Student’s unpaired t-test). The bars represent the mean ± SEM. Similar experiments were repeated twice, and the same results were obtained.
Increases in cytosolic free calcium contribute to HVJ-E-mediated cytotoxicity in MM cells
Previous studies have shown that HVJ-E-mediated cell death is induced by the recognition of HVJ-E RNA genome fragments with RNA receptor retinoic-acid inducible gene-I (RIG-I) or an elevation of the cytosolic Ca2+ concentration [26, 34]. We found that HVJ-E failed to induce RIG-I expression in MM cells (NCI-H929, U266 and MM1S; Supplementary Figure 4A). Moreover, transfection of HVJ-E RNA genome fragments did not inhibit the viability of NCI-H929 cells (Supplementary Figure 4B). These results suggest that RIG-I signaling is not involved in HVJ-E-induced apoptosis in MM cells. Therefore, we next focused on the cytosolic Ca2+ concentration of HVJ-E-treated MM cells. NCI-H929 and MM1S cells were exposed to HVJ-E for 0.5, 1 and 3 hours, and cytoplasmic Ca2+ levels were measured using Fluo-4-AM. Cytoplasmic Ca2+ peaked after 0.5 hours of HVJ-E treatment (Figure 3A, Supplementary Figure 5A). In addition, NCI-H929 and MM1S cells were treated with HVJ-E and a Ca2+ chelator (BAPTA-AM) to prevent the effects of Ca2+ elevation, and BAPTA-AM almost entirely abrogated HVJ-E-induced cytotoxicity in NCI-H929 cells (Figure 3B, Supplementary Figure 5B).
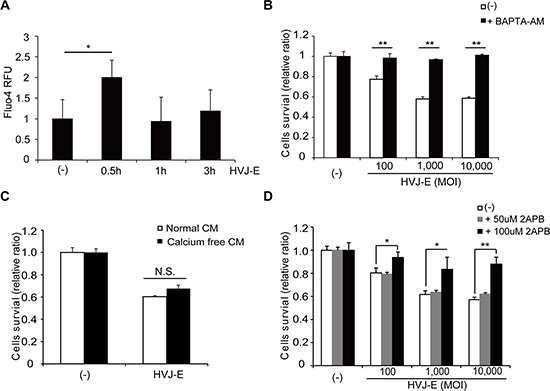
Figure 3: Effects of the increase in cytoplasmic Ca2+ on HVJ-E-induced cell death. (A) Analysis of cytoplasmic Ca2+ levels following 0.5, 1 and 3 hours of HVJ-E treatment in NCI-H929 cells using Fluo-4-AM. The data represent the mean ± SD of three independent experiments (*P < 0.05, Tukey-Kramer test). (B) NCI-H929 cells were treated with different doses of HVJ-E in the presence or absence of the calcium chelator BAPTA-AM, and cell viabilities were assessed after 48 hours using the MTS assay. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test). (C) Effect of the inhibition of Ca2+ influx from the extracellular space on HVJ-E-induced cell death. The MTS assay was performed after HVJ-E treatment for 48 hours in NCI-H929 cells cultured in normal or calcium-free medium. CM: cultured medium. The data represent the mean ± SD of three independent experiments (Tukey–Kramer test, p = N.S., no significant difference). (D) NCI-H929 cells were treated with different doses of HVJ-E in the presence or absence of the IP3R inhibitor 2-aminoethoxydiphenylborate (2-APB) for 48 hours, and the cell viabilities were examined using the MTS assay. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test).
Next, we investigated the source of the increase in free Ca2+ in the cytoplasm of HVJ-E-treated MMs. Although the NCI-H929 cells were cultured in Ca2+-free medium to block Ca2+ influx from the extracellular space, HVJ-E exhibited cell toxicity in common with the culture in normal medium (Figure 3C). However, HVJ-E-mediated cell death was inhibited in a dose-dependent manner by 2-aminoethoxydiphenyl borate (2-APB), which is an inhibitor of the inositol 1,4,5-trisphosphate receptor (IP3R)/Ca2+ channel in the endoplasmic reticulum (ER), (Figure 3D, Supplementary Figure 5C). These results suggested that HVJ-E elevated cytoplasmic Ca2+ levels by efflux from the ER but not influx from the extracellular space to induce apoptosis in MM cells.
Increase of cytosolic calcium by HVJ-E treatment induced apoptosis by suppressing c-Myc expression in MM
In this study, we demonstrated that in contrast to MM1S and NCI-H929 cells, U266 cells showed a lack of response to HVJ-E (Figure 1A, Supplementary Figure 1D). We considered that this differential susceptibility to HVJ-E might result from different expression of genes targeted by HVJ-E, which determine cell fate. Previous studies demonstrated that aberrant c-Myc expression is associated with myeloma cell survival and proliferation [2, 5]. Therefore, we focused on the regulation of c-Myc expression in HVJ-E-treated MM cells. As shown in Figure 4A, c-Myc expression was detected in NCI-H929, MM1S and RPMI-8226 cells, and c-Myc expression was significantly suppressed by HVJ-E treatment. However, c-Myc expression was barely detected in U266 cells, as reported in a previous study [8]. c-Myc expression in NCI-H929 cells was suppressed by HVJ-E in a dose-dependent manner (Figure 4B). To further understand the correlation between c-Myc expression and apoptosis, we knocked down c-Myc expression in NCI-H929 and MM1S cells. c-Myc knockdown led to a decrease in cell viability (Figure 4C) and the cleavage of the apoptosis marker caspase-3 (Supplementary Figure 6). Moreover, c-Myc overexpression in MM1S cells inhibited HVJ-E-mediated c-Myc downregulation and cell death (Figure 4D, 4E). Taken together, these results suggest that HVJ-E-induced c-Myc downregulation causes apoptosis in MM cells, and the endogenous c-Myc expression level determines the susceptibility of MM cell lines to HVJ-E.
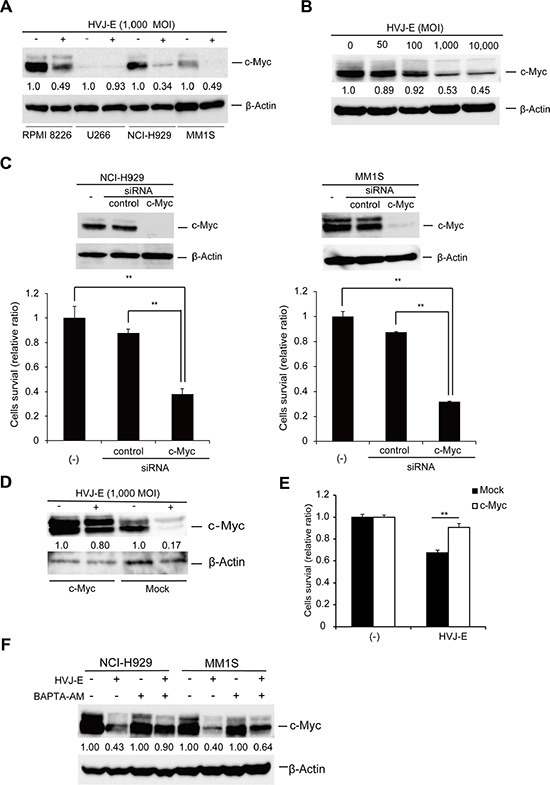
Figure 4: HVJ-E-induced apoptosis is associated with c-Myc downregulation. (A) Immunoblotting study of c-Myc expression after HVJ-E treatment (1000 MOI, 24 hours) in MM cell lines (RPMI-8226, NCI-H929, U266, and MM1S). (B) Dose-dependent effects of HVJ-E treatment on c-Myc expression in NCI-H929. (C) c-Myc and expression were evaluated by immunoblotting analysis in untreated, control and c-Myc siRNA-transduced NCI-H929 and MM1S cell lines, Cell viability was measured using the MTS assay at 72 hours after the transduction of control or c-Myc siRNA into NCI-H929 and MM1S cells. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test). (D) Immunoblotting of whole-cell lysates from empty vector- or c-Myc expression vector-transduced MM1S cells after treatment with HVJ-E (1000 MOI, 24 hours). (E) Measurement of the cell viability of either empty or c-Myc-overexpressing MM1S cells treated with HVJ-E (1000 MOI, 36 hours) using the MTS assay. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test). (F) Detection of c-Myc expression in NCI-H929 and MM1S cells after treatment with HVJ-E with or without BAPTA-AM for 20 hours by immunoblotting.
To determine whether the increase in cytosolic Ca2+ was required for c-Myc downregulation, c-Myc expression was examined in the presence of BAPTA-AM-treated MM cells. NCI-H929 and MM1S cells were treated with 1,000 MOI HVJ-E in the presence or absence of BAPTA-AM, and c-Myc expression was examined by immunoblotting (Figure 4F). HVJ-E alone remarkably decreased c-Myc expression, while BAPTA-AM prevented HVJ-E-induced c-Myc downregulation. These data suggest that the HVJ-E mediated increase in Ca2+ in the cytoplasm is an upstream signaling element in the downregulation of c-Myc, which determines HVJ-E-mediated cytotoxicity in MM cells.
We next examined the key factor underlying the induction of HVJ-E-mediated apoptosis via an increase in cytosolic Ca2+. We showed that a Ca2+ ionophore (A23817) significantly decreased the viability of NCI-H929 and MM1S cells compared to U266 cells and PBMCs (Figure 5A). Meanwhile, in addition, A23817 induced c-Myc downregulation in NCI-H929 and MM1S in a time-dependent manner (Figure 5B). These results confirmed that an increase in cytosolic free Ca2+ induced cell death in MMs independently of HVJ-E treatment.
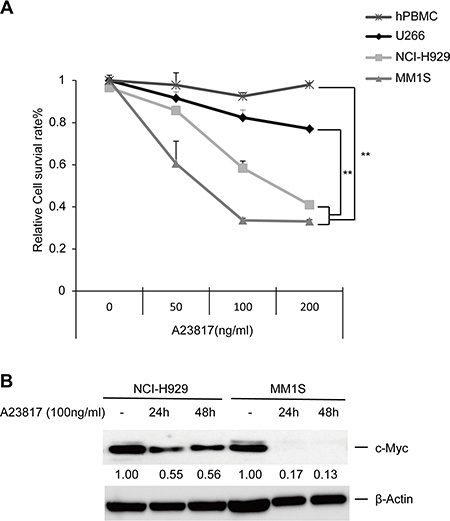
Figure 5: Increasing levels of cytoplasmic Ca2+ induce cell death and c-Myc downregulation in MM cells. (A) MM cell lines (NCI-H929, MM1S and U266) and PBMCs were exposed to different doses of A23817. The relative cell survival ratio was measured after 48 hours using the MTS assay. The data represent the mean ± SD of three independent experiments (**P < 0.01, Tukey–Kramer test). (B) Time-dependent effects of A23817 treatment (100 ng/ml) on c-Myc expression in NCI-H929 and MM1S cells, as analyzed by immunoblotting.
c-Myc downregulation by calcium signal results from both enhanced degradation of c-Myc protein and suppression of c-Myc transcription
c-Myc expression can be controlled by post-translational and transcriptional regulation [35, 36]. A post-translational pathway that affects c-Myc protein stability is dependent on the phosphorylation of two conserved amino acids: Serine 62 (S62) and Threonine 58 (T58). The elevation of S62 and loss of T58 phosphorylation of c-Myc protein increases Myc protein stability in several solid cancers [37–39]. The decrease in S62 phosphorylation could be a potential approach for reducing the levels of c-Myc protein in cancers. Here, we observed that during the HVJ-E-induced time-dependent downregulation of c-Myc protein, c-Myc S62 phosphorylation decreased at an earlier time point compared with the reduction of c-Myc whole protein and c-Myc T58 phosphorylation in NCI-H929 cells (Figure 6A).
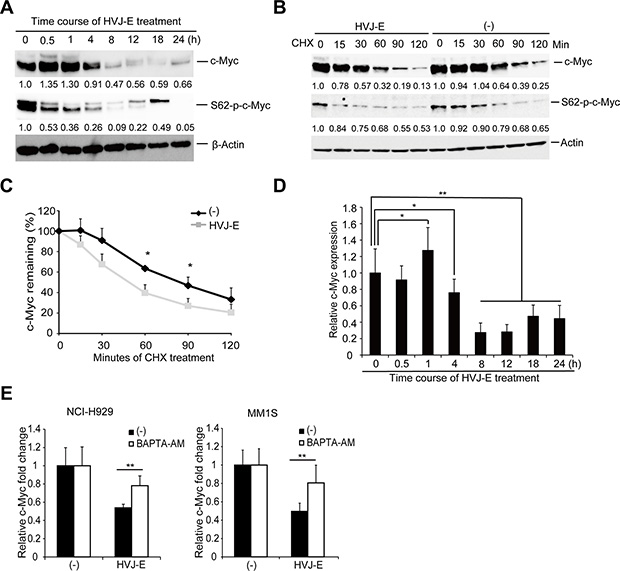
Figure 6: HVJ-E decreases S62-p-c-Myc expression and destabilizes c-Myc protein, BAPTA-AM prevented HVJ-E-induced c-Myc transcription downregulation. (A) Immunoblotting analyses of the time-dependent effects of HVJ-E treatment on c-Myc and S62-p-c-Myc expression in NCI-H929 cells. (B) NCI-H929 cells were treated with cycloheximide (CHX) with or without HVJ-E at the indicated time points, and c-Myc and S62-p-c-Myc expression levels were analyzed by immunoblotting analysis. (C) c-Myc levels were quantitated relative to the levels of β-actin in Figure 6B and graphed as the percentage of c-Myc protein remaining after CHX treatment. The data represent the mean ± SD of three independent experiments (*P < 0.05, Student’s unpaired t-test). (D) Quantitative RT-PCR analysis of c-Myc levels in HVJ-E-treated NCI-H929 cells (1000 MOI, 0–24 hours). The data are presented as a ratio of c-Myc expression at each time point compared with the baseline c-Myc expression (*P < 0.05, **P < 0.01, t = 0 h, Tukey–Kramer test). (E) Measurement of c-Myc transcriptional expression in NCI-H929 and MM1S cells after treatment with HVJ-E with or without BAPTA-AM for 20 hours by quantitative RT-PCR analysis. The data are presented as a ratio of c-Myc expression compared with baseline c-Myc expression. The data represent the mean ± SD of three independent experiments (**P < 0.01, Student’s unpaired t-test).
Next, we tested whether HVJ-E would accelerate c-Myc degradation. We cotreated NCI-H929 cells with HVJ-E and cycloheximide (CHX) to inhibit de novo protein synthesis, and c-Myc protein levels were measured by immunoblot analysis over 2 hours. As shown in Figure 6B and 6C, HVJ-E treatment significantly increased c-Myc protein degradation. These results indicated that HVJ-E downregulated c-Myc expression by decreasing the S62 phosphorylation of c-Myc protein and accelerating the rate of c-Myc protein degradation.
The HVJ-E-mediated acceleration of c-Myc degradation was significant but slight because the levels of remaining c-Myc protein became nearly equivalent in the presence or absence of HVJ-E treatment after 90 min of CHX treatment (Figure 6B and 6C). Therefore, we hypothesized that other factors might also participate in the HVJ-E-mediated c-Myc downregulation. Next, we focused on c-Myc transcriptional regulation and examined endogenous c-Myc mRNA levels by real-time PCR in HVJ-E-treated NCI-H929 cells. As shown in Figure 6D, HVJ-E induced a transient increase in c-Myc mRNA within one hour and a decrease to below the baseline level after four hours of treatment. Moreover, BAPTA-AM inhibited the HVJ-E-induced suppression of c-Myc transcription in NCI-H929 and MM1S cells (Figure 6E). Taken together, these results suggest that the HVJ-E-induced increase in cytoplasmic Ca2+ mediated the downregulation of c-Myc expression mainly by suppressing c-Myc transcription.
Calcium-mediated suppression of c-Myc transcription depends on SMADs activation
Finally, we investigated the mechanism by which c-Myc transcription was inhibited by the cytoplasmic Ca2+ increase in MMs. A previous study demonstrated that the cytosolic increase in Ca2+ induces Smad1 phosphorylation to promote Smad1 nuclear translocation [40]. In addition, either SMAD1 or SMAD1/5/8 regulates c-Myc transcription by binding to SMAD4 to form a Smad complex [41–43]. As shown in Figure 7A, the transient knockdown of Smad4 in NCI-H929 cells by siRNA inhibited the HVJ-E-induced suppression of c-Myc transcription. HVJ-E induced the phosphorylation of Smad1/5/8 as well as the repression of c-Myc in NCI-H929 and MM1S cells (Figure 7B). Moreover, in HVJ-E-treated NCI-H929 cells, the immunoblotting results clearly revealed a significant increase in phosphorylated SMAD1/5/8 in the nuclear fraction following HVJ-E stimulation, and BAPTA-AM abolished the HVJ-E-mediated phosphorylation and nuclear translocation of SMAD1/5/8 (Figure 7C, Supplementary Figure 7). Taken together, Figure 7 suggests that the increase in cytosolic Ca2+mediating Smad activation is involved in the downregulation of HVJ-E-induced c-Myc transcription in MM.
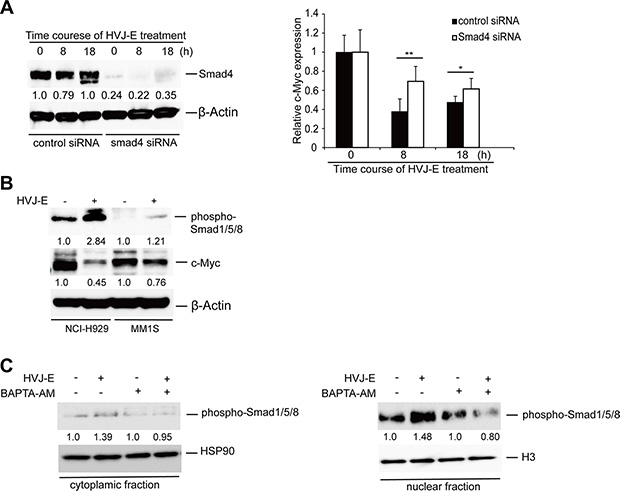
Figure 7: The involvement of SMADs in HVJ-E-induced suppression of c-Myc transcription. (A) Left panel: Time-dependent Smad4 expression was measured in control siRNA- and Smad4 siRNA-transduced NCI-HJ929 cells by immunoblotting analysis. Right panel: Time-dependent effect of HVJ-E on c-Myc mRNA expression in control siRNA- and Smad4 siRNA-transduced NCI-H929 cells by quantitative RT-PCR analysis. The data are presented as the ratio of c-Myc expression compared with baseline c-Myc expression. The data represent the mean ± SD of three independent experiments (*P < 0.05, **P < 0.01, Student’s unpaired t-test). (B) Protein levels of phospho-SMAD1/5/8 and c-Myc were analyzed in the MM cell lines NCI-H929 and MM1S with or without HVJ-E treatment (1000 MOI, 8 hours). (C) Measurement of the cytoplasmic and nuclear fraction of activated SMAD1/5/8 in NCI-H929 cells after treatment with HVJ-E with or without BAPTA-AM for 8 hours by immunoblotting.
DISCUSSION
In the present study, we demonstrated that HVJ-E effectively induced apoptosis in various MM cells by downregulating c-Myc via an elevation of cytoplasmic Ca2+ levels. Calcium-induced c-Myc downregulation resulted from both suppression of c-Myc transcription via activation of SMAD1/5/8 and acceleration of c-Myc protein degradation. MM cells (NCI-H929, MM1S, and RPMI8226) which were highly expressing c-Myc were preferentially killed by increase of calcium signal via fusion with HVJ-E, but U266 MM cell line and PBMCs were not killed. Therefore, MM cells with c-Myc addiction can be killed by HVJ-E via elevation of cytosolic calcium. c-Myc is an important transcription factor for the regulation of cancer cell proliferation and highly controls malignant state in various cancer cells [44, 45]. In previous reports, even transient inactivation of c-Myc leads to tumor collapse in mice models [46–48]. Therefore, there is a possibility that HVJ-E is able to induce cell death in malignant cancer cells with c-Myc addiction via the increase of cytoplasmic Ca2+ without affecting normal cells lacking c-Myc addiction. Moreover, as shown in this report, besides inhibiting c-Myc transcription, developing methods that accelerate the rate of c-Myc destruction by destabilizing c-Myc protein may ultimately prove effective in tumor settings [49, 50].
It is well known that Ca2+ level modification plays important roles in regulating cell death (for example, apoptosis [51, 52] and autophagy [53]). However, in this study, we demonstrated that HVJ-E induced Ca2+-dependent apoptosis, but autophagy was not involved in HVJ-E-induced cell death (unpublished data). Numerous Ca2+-dependent downstream effectors can efficiently transduce extracellular signals into apoptosis [52, 54]. However, protein kinase C (PKC) [55], calpains activation and ER stress [54] were not the downstream events involved in Ca2+ level modification-induced apoptosis by HVJ-E (unpublished data). Several papers suggested the relationship between the increase in cytosolic Ca2+ and decrease in c-Myc transcription. The Ca2+ ionophore A23817 downregulates c-Myc expression in human promyelocytic leukemia and T lymphoblastic leukemia cells (HL-60, MOLT-4) by inhibiting RNA elongation [56, 57]. However, those reports showed no evidence that the downregulation of c-Myc is responsible for A23817-induced cell death by increasing cytosolic Ca2+levels. The crosslinking of IgM in murine B-lymphoma cells induces G1 arrest and apoptosis by p27Kip1 accumulation, which results from either c-Myc downregulation or increase of cytosolic calcium [58]. This report indicates that c-Myc downregulation and cytosolic calcium increase are independently involved in apoptosis of lymphoma cells. No relationship between two pathways is clearly shown. Thus, till now, there have been no reports putting links among cytosolic Ca2+ increase, c-Myc downregulation and apoptosis.
In the present study, we found that HVJ-E-induced cell death was dependent on the downregulation of c-Myc (Figure 4E) and that the intracellular Ca2+ chelator BAPTA-AM prevented the HVJ-E-induced c-Myc downregulation (Figure 4F and Figure 6E) and cell death of MM cells (Figure 3B). Taken together, our results confirm that the accumulation of cytosolic Ca2+ upstream of c-Myc downregulation leads to apoptosis in HVJ-E-treated MMs. In this study, we also found that HVJ-E treatment increased the expression of the pro-apoptotic protein Bim in MM cells (Supplementary Figure 3B, 3C). The relationship between c-Myc and Bim regulation was well documented in MM cells in a previous study [59]. Namely, shRNA-mediated c-Myc suppression subsequently increases Bim expression in myeloma cells, which indicates that the silencing of c-Myc induces myeloma cell apoptosis [59]. In our study, HVJ-E mediated both c-Myc downregulation and Bim upregulation, and the siRNA-mediated downregulation of Bim inhibited the cytotoxicity of HVJ-E in MM cells (Supplementary Figure 3D). These results suggest that HVJ-E-induced c-Myc downregulation likely results in Bim upregulation, which leads to apoptosis in MM cells.
HN glycoprotein enables HVJ-E particles to adhere to the host cell surface [60]. A previous study indicated that HVJ-E-mediated cytotoxicity is dependent on the expression of GD1a and SPG, which are HN receptors [61], on target cells. In the present study, we showed that HVJ-E exhibited cytotoxic activity against MM cells, although, MM cells had no conventional receptors for acidic gangliosides recognized by HN glycoprotein of HVJ (Supplementary Figure 8A); however, HVJ-E could still bind to MM cells (Supplementary Figure 8B). Moreover, HN-depleted HVJ-E could adhere to the cell surface of NCI-H929 cells (Supplementary Figure 9A) and induce apoptosis via membrane fusion (Supplementary Figure 9B and 9C). For these reasons, we suggest that MMs may express an unknown receptor for F binding to induce membrane fusion. MM is a systemic cancer, and systemic treatment is required for MM therapy. Because HN is responsible for not only HVJ-E particle-adhesion but also hemagglutination, systemic administration of HVJ-E is not practical. Although HN depletion of HVJ-E decreases both hemagglutinating activity and cell adhesion ability [62], HN-depleted HVJ-E displays added cancer cell affinity and utility for cancer therapy [63]. Here, we showed that MMs had an affinity for HN-depleted HVJ-E. Therefore, the systemic administration of HN-depleted HVJ-E will be a promising tool for MM therapy.
MATERIALS AND METHODS
Cell culture
Human MM cell lines (NCI-H929, MM1S, U266, and RPMI-8226) were obtained from the American Type Culture Collection (ATCC; VA, USA). MM1S, U266, and RPMI-8226 cells were cultured in RPMI 1640 medium (Nacalai Tesque, Inc., Kyoto, Japan) containing 10% fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Nacalai Tesque). NCI-929 cells were cultured in streptomycin and 2mercaptoethanol (Nacalai Tesque) at a final concentration of 0.05 mM. PBMCs were obtained from healthy donors after obtaining informed consent. Five milliliters of whole blood were collected from three healthy donors, and PBMCs were isolated from the blood by cell density centrifugation over a Ficoll-Paque density gradient. PBMCs were washed three times with sterile PBS (Nacalai Tesque). PBMCs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and RPMI 1640 mixed 1:1 and supplemented with 100 U/ml of penicillin, 100 μg/ml of streptomycin, 5% heat-inactivated AB human serum (Sigma-Aldrich, Inc., Tokyo, Japan), 10 mM HEPES, and 60 μg/ml of arginine-HCl.
Preparation of HVJ-E
HVJ (VR-105 parainfluenza 1 Sendai/52, Z strain from the ATCC) was amplified in the chorioallantoic fluid of 14-day-old chick eggs, purified by centrifugation and inactivated by UV irradiation (99 millijoules/cm²). The inactivated virus lost the ability to replicate and synthesize viral proteins but retained its membrane fusion activity.
Chemical treatments
To inhibit caspase activity, 100 μmol/L of the pan-caspase inhibitor Z-VAD-FMK (Medical and Biological Laboratories, Inc., Nagoya, Japan) was added to the cells one hour prior to HVJ-E treatment. Calcium-free conditions were prepared using calcium-free RPMI 1640 medium (US Biological, Inc., MO, USA), and in the cytoplasmic Ca2+ inhibition experiments, 5 μmol/L BAPTA-AM or 50 μmol/L 2-aminoethoxydiphenyl borate (2-APB) (Sigma-Aldrich) was added to the cells 1 hour before exposure to HVJ-E. To increase cytoplasmic Ca2+, A23817 was used which was purchased from Sigma-Aldrich (Tokyo, Japan).
Cytotoxicity assays
Cell viability was determined using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay kit (Promega, Inc., WI, USA). Briefly, after treatment with the compounds at the indicated concentrations for the indicated durations, 100 μl of Cell Titer 96 Aqueous One Solution reagent was added to each well, and the plates were incubated at 37°C in 5% carbon dioxide for 2 hours. After transferring 100 μl of incubation medium from each well into a new 96-well plate, the absorbance at 490 nm was measured.
Assessment of apoptosis
Apoptosis was evaluated using a PE Annexin V Apoptosis Detection Kit (BD Biosciences, Inc., CA, USA). Briefly, 1 × 105 cells were resuspended in 500 μl of 1× binding buffer, to which was added 5 μl of Annexin V and 5 μl of propidium iodide. The cells were incubated in the dark for 15 minutes. For the flow cytometry analysis, the percentages of the positive cells in the upper right (Annexin V+/PI+: late apoptotic cells) and lower right quadrants (Annexin V+/PI−: early apoptotic cells) were summed to obtain the total number of apoptotic cells.
Measurement of cytoplasmic Ca²+ in HVJ-E-treated MM cells
The level of intracellular cytosolic calcium was measured using the Fluorescent Indicator Fluo-4 direct calcium assay kit (Invitrogen). NCI-H929 cells were stimulated in the presence of HVJ-E at an MOI of 1,000 for 0, 0.5, 1 and 3 hours. After the incubation, the cells were stained with Fluo-4 according to the manufacturer’s instructions. The fluorescence was measured using Infinite M200 at an excitation of 494 nm and emission of 516 nm.
siRNA and plasmid transfection
Transfection was performed by electroporation using three 10-second pulses at a voltage of 1300 with the Neon® transfection system (Life Technologies) according to the manufacturer’s instructions. For c-Myc and Smad4 transient silencing, NCI-H929 and MM1S cells were transfected with SMARTpool Accell human c-Myc siRNA (E-003282-00-0005; Thermo Fisher Scientific, Inc., UT, USA) or Smad4 siRNA (E-003902-00-0005; Thermo Fisher Scientific), and Accell Green Non-targeting siRNA (D-001950-01-20; Thermo Fisher Scientific) was used as a control. We used 100 pmol of siRNA/2.5 × 105 cells. For c-Myc overexpression, plasmid vectors containing a cytomegalovirus (CMV) promoter with or without (control) human c-Myc cDNA were purchased from Thermo Fisher Scientific. One microgram of plasmid per 2.5 × 105 cells was used.
Real-time quantitative RT-PCR
RNA was extracted from cultured cells using the RNeasy Mini Kit (Qiagen, Tokyo, Japan), and 500 ng of total RNA were converted into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Tokyo, Japan). Human c-Myc and β-actin were amplified using SYBR Premix Ex Taq (Takara Bio, Shiga, Japan). All procedures were performed according to the manufacturer’s instructions. The following primers were used:
Human β-actin-forward, 5′-GAGCTACGAGCTG CCTGACG-3′, and
Human β-actin-reverse, 5′-GTAGTTTCGTGGAT GCCACAG-3′; and
Human c-Myc forward, 5′-GACCAG- CTGGAGAT GGTGAC-3′, and
Human c-Myc reverse, 5′-GGTCGCAGATGAAA CTCTGG-3′.
Antibodies and western blotting analysis
Antibodies were obtained from the following commercial sources: anti-β-actin antibody (AC-15, A5441) was purchased from Sigma-Aldrich; the anti-caspase-3 (#9662), anti-caspase-9 (#9502), anti-caspase-8 (#9746), anti-PARP (#9542), anti-c-Myc (#5605), anti-HSP90 (#4874), anti-Smad4 (#9515) and anti-H3 (#9715) antibodies were purchased from Cell Signaling Technology (Tokyo, Japan); the anti-phospho-Smad1/Smad5/Smad8 antibodies recognizing phosphorylated Ser463/465 were purchased from Millipore (MA, USA); and the anti-phospho-Ser62-c-Myc (ab51156) and anti-phospho-Thr58-c-Myc (ab135558) antibodies were purchased from Abcam (Cambridge, MA). After treatment, the cells were washed once with cold PBS, the cell pellets were treated with RIPA buffer, and the total protein was isolated. Protein (10 μg) was resolved in polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were probed with the primary antibody described above followed by a horseradish peroxidase-linked secondary antibody (GE Healthcare Japan). The signals were detected using Chemi-Lumi One (Nacalai Tesque) with Image Quant LAS 4000 mini software (GE Healthcare, Buckinghamshire, UK). The band density was analyzed using the NIH ImageJ software.
Measurement of c-Myc protein stability
NCI-H929 cells were treated with 100 μg/ml of CHX in the presence or absence of HVJ-E. Cell lysates were prepared after the indicated time periods from 0–120 minutes. The samples were then immunoblotted to visualize total endogenous c-Myc, S62-c-Myc and β-actin. Protein levels of c-Myc, S62-c-Myc and β-actin were then quantified using Image Quant TL image analysis software (GE Healthcare). The levels of total c-Myc protein and S62-c-Myc (normalized to β-actin) in CHX-treated cells with or without HVJ-E was determined using the value for CHX-untreated cells at time 0 hours as equal to 1. Relative c-Myc levels were then visualized on an X-Y scatter graph plotted using the best-fit exponential curve for the indicated time points.
Human MM xenograft models
Six- to eight-week old NOG (NOD/Shi-scid, IL- 2Rγ KO) mice were purchased from Clea Japan (Tokyo, Japan) and maintained in a temperature-controlled pathogen-free room. All animal experiments were carried out according to the guidelines of the Institute for Animal Studies at Osaka University (Osaka, Japan). MM1S or NCI-H929 cells (5.0 × 106) in 100 μl of PBS were inoculated subcutaneously into the right flanks of NOG mice (female, 8 weeks old). When the tumors were measurable (approximately 150–200 mm3), approximately 10– 14 weeks after MM cell injection, the mice were treated intratumorally with vehicle alone (PBS) or 5,000 HAU of HVJ-E 6 times every two days.
Statistical analysis
The results are reported as the mean ± standard deviation (SD). The statistical significance of differences was evaluated using the Tukey–Kramer test or Student’s unpaired t-test, and a P value less than 0.05 was considered to be significant.
CONFLICTS OF INTEREST
The authors have declared that no conflicts of interest exists.
GRANT SUPPORT
This study was supported by the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (Project ID: 10–03) and Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (Project ID: 25290056).
REFERENCES
1. Palumbo A, Anderson K. Multiple myeloma. The New England journal of medicine. 2011; 364:1046–1060.
2. Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008; 111:2962–2972.
3. Abdi J, Chen G, Chang H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget. 2013; 4:2186–2207. doi: 10.18632/oncotarget.1497.
4. Shi L, Wang S, Zangari M, Xu H, Cao TM, Xu C, Wu Y, Xiao F, Liu Y, Yang Y, Salama M, Li G, Tricot G, et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget. 2010; 1:22–33. doi: 10.18632/oncotarget.105.
5. Yang WC, Lin SF. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. Biomed Res Int. 2015; 2015:341430.
6. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12:335–348.
7. Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, Johnson DC, Gonzalez D, Dagrada GP, Protheroe RK, Konn ZJ, Stockley DM, Gregory WM, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010; 116:e56–65.
8. Nojima M, Maruyama R, Yasui H, Suzuki H, Maruyama Y, Tarasawa I, Sasaki Y, Asaoku H, Sakai H, Hayashi T, Mori M, Imai K, Tokino T, et al. Genomic screening for genes silenced by DNA methylation revealed an association between RASD1 inactivation and dexamethasone resistance in multiple myeloma. Clin Cancer Res. 2009; 15:4356–4364.
9. Jagannathan S, Vad N, Vallabhapurapu S, Vallabhapurapu S, Anderson KC, Driscoll JJ. MiR-29b replacement inhibits proteasomes and disrupts aggresome+autophagosome formation to enhance the antimyeloma benefit of bortezomib. Leukemia. 2015; 29:727–738.
10. Annunziata CM, Hernandez L, Davis RE, Zingone A, Lamy L, Lam LT, Hurt EM, Shaffer AL, Kuehl WM, Staudt LM. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood. 2011; 117:2396–2404.
11. Greco C, D’Agnano I, Vitelli G, Vona R, Marino M, Mottolese M, Zuppi C, Capoluongo E, Ameglio F. c-MYC deregulation is involved in melphalan resistance of multiple myeloma: role of PDGF-BB. Int J Immunopathol Pharmacol. 2006; 19:67–79.
12. Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B and Shaughnessy J Jr, Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005; 106:296–303.
13. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, Fonseca R, Kremer R, Cattoretti G, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008; 13:167–180.
14. Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R, Intergroupe Francophone du M. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001; 98:3082–3086.
15. Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, Garbitt VM, Riggs DL, Brents LA, Roschke AV, Van Wier S, Fonseca R, Bergsagel PL, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014; 28:1725–1735.
16. Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000; 97:228–233.
17. Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, Zeng Y, Chen B, Epstein J, et al. IRF4 addiction in multiple myeloma. Nature. 2008; 454:226–231.
18. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146:904–917.
19. Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to c-MYC in multiple myeloma. Blood. 2012; 120:2450–2453.
20. Thirukkumaran CM, Morris DG. Oncolytic virotherapy for multiple myeloma: past, present, and future. Bone Marrow Res. 2011; 2011:632948.
21. Deng H, Tang N, Stief AE, Mehta N, Baig E, Head R, Sleep G, Yang XZ, McKerlie C, Trudel S, Stewart AK, McCart JA. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia. 2008; 22:2261–2264.
22. Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Gao H, Bahlis N, Neri P, Pho M, Stewart D, Mansoor A, Morris DG. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin Cancer Res. 2012; 18:4962–4972.
23. Kaneda Y, Nakajima T, Nishikawa T, Yamamoto S, Ikegami H, Suzuki N, Nakamura H, Morishita R, Kotani H. Hemagglutinating virus of Japan (HVJ) envelope vector as a versatile gene delivery system. Mol Ther. 2002; 6:219–226.
24. Fujihara A, Kurooka M, Miki T, Kaneda Y. Intratumoral injection of inactivated Sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol Immunother. 2008; 57:73–84.
25. Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007; 67:227–236.
26. Matsushima-Miyagi T, Hatano K, Nomura M, Li-Wen L, Nishikawa T, Saga K, Shimbo T, Kaneda Y. TRAIL and Noxa are selectively upregulated in prostate cancer cells downstream of the RIG-I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin Cancer Res. 2012; 18:6271–6283.
27. Gao H, Gong XC, Chen ZD, Xu XS, Zhang Q, Xu XM. Induction of apoptosis in hormone-resistant human prostate cancer PC3 cells by inactivated Sendai virus. Biomed Environ Sci. 2014; 27:506–514.
28. Matsuda M, Nimura K, Shimbo T, Hamasaki T, Yamamoto T, Matsumura A, Kaneda Y. Immunogene therapy using immunomodulating HVJ-E vector augments anti-tumor effects in murine malignant glioma. J Neurooncol. 2011; 103:19–31.
29. Nomura M, Shimbo T, Miyamoto Y, Fukuzawa M, Kaneda Y. 13-Cis retinoic acid can enhance the antitumor activity of non-replicating Sendai virus particle against neuroblastoma. Cancer Sci. 2013; 104:238–244.
30. Tanemura A, Kiyohara E, Katayama I, Kaneda Y. Recent advances and developments in the antitumor effect of the HVJ envelope vector on malignant melanoma: from the bench to clinical application. Cancer Gene Ther. 2013; 20:599–605.
31. Nishikawa T, Tung LY, Kaneda Y. Systemic administration of platelets incorporating inactivated Sendai virus eradicates melanoma in mice. Mol Ther. 2014; 22:2046–2055.
32. Payne CM, Glasser L, Tischler ME, Wyckoff D, Cromey D, Fiederlein R, Bohnert O. Programmed cell death of the normal human neutrophil: an in vitro model of senescence. Microsc Res Tech. 1994; 28:327–344.
33. Gazdar AF, Oie HK, Kirsch IR, Hollis GF. Establishment and characterization of a human plasma cell myeloma culture having a rearranged cellular myc proto-oncogene. Blood. 1986; 67:1542–1549.
34. Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014; 74:1056–1066.
35. Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem. 2006; 281:34725–34729.
36. Levens D. You Don’t Muck with MYC. Genes Cancer. 2010; 1:547–554. doi: 10.1177/1947601910377492.
37. Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000; 95:2104–2110.
38. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004; 6:308–318.
39. Zhang X, Farrell AS, Daniel CJ, Arnold H, Scanlan C, Laraway BJ, Janghorban M, Lum L, Chen D, Troxell M, Sears R. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proc Natl Acad Sci U S A. 2012; 109:2790–2795.
40. Eapen A, Kulkarni R, Ravindran S, Ramachandran A, Sundivakkam P, Tiruppathi C, George A. Dentin phosphophoryn activates Smad protein signaling through Ca2+-calmodulin-dependent protein kinase II in undifferentiated mesenchymal cells. J Biol Chem. 2013; 288:8585–8595.
41. Holien T, Vatsveen TK, Hella H, Rampa C, Brede G, Groseth LA, Rekvig M, Borset M, Standal T, Waage A, Sundan A. Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by Smad-dependent repression of MYC. Leukemia. 2012; 26:1073–1080.
42. Reddi AH. Initiation of fracture repair by bone morphogenetic proteins. Clin Orthop Relat Res. 1998; S66–72.
43. Tsukamoto S, Mizuta T, Fujimoto M, Ohte S, Osawa K, Miyamoto A, Yoneyama K, Murata E, Machiya A, Jimi E, Kokabu S, Katagiri T. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep. 2014; 4:7596.
44. Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002; 297:63–64.
45. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999; 18:3004–3016.
46. Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002; 297:102–104.
47. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004; 431:1112–1117.
48. Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007; 104:13028–13033.
49. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol. 2007; 9:765–774.
50. Popov N, Herold S, Llamazares M, Schulein C, Eilers M. Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle. 2007; 6:2327–2331.
51. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003; 4:552–565.
52. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000; 1:11–21.
53. Harr MW, Distelhorst CW. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol. 2010; 2:a005579.
54. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008; 27:6407–6418.
55. Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007; 7:281–294.
56. Salehi Z, Niedel JE. Multiple calcium-mediated mechanisms regulate c-myc expression in HL-60 cells. J Immunol. 1990; 145:276–282.
57. Bading H, Moelling K. Transcriptional down-regulation of c-myc expression by protein synthesis-dependent and -independent pathways in a human T lymphoblastic tumor cell line. Cell Growth Differ. 1990; 1:113–117.
58. Donjerkovic D, Zhang L, Scott DW. Regulation of p27Kip1 accumulation in murine B-lymphoma cells: role of c-Myc and calcium. Cell Growth Differ. 1999; 10:695–704.
59. Chen L, Li C, Zhang R, Gao X, Qu X, Zhao M, Qiao C, Xu J, Li J. miR-17–92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett. 2011; 309:62–70.
60. Okada Y. Sendai virus-induced cell fusion. Methods Enzymol. 1993; 221:18–41.
61. Hatano K, Miyamoto Y, Nonomura N, Kaneda Y. Expression of gangliosides, GD1a, and sialyl paragloboside is regulated by NF-kappaB-dependent transcriptional control of alpha2,3-sialyltransferase I, II, and VI in human castration-resistant prostate cancer cells. Int J Cancer. 2011; 129:1838–1847.
62. Saga K, Tamai K, Kawachi M, Shimbo T, Fujita H, Yamazaki T, Kaneda Y. Functional modification of Sendai virus by siRNA. J Biotechnol. 2008; 133:386–394.
63. Shimbo T, Kawachi M, Saga K, Fujita H, Yamazaki T, Tamai K, Kaneda Y. Development of a transferrin receptor-targeting HVJ-E vector. Biochem Biophys Res Commun. 2007; 364:423–428.