
INTRODUCTION
HGPS is a rare but severe premature aging disease. Affected patients die in their teens from myocardial infarction or stroke as a result of rapidly progressive atherosclerosis [1-3]. Most cases are caused by a G608G(GGC > GGT) single-base mutation within exon 11 of the LMNA gene [4, 5], which codes for lamin A/C via alternative splicing. This mutation activates a cryptic splice site that prevents proper processing of prelamin A to mature lamin A and produces a permanently farnesylated and carboxymethylated toxic product called “progerin”. These post-translational progerin modifications seem to play a major role in the pathophysiology of disease [6-9]. Progerin disrupts the nuclear lamina, a compartment essential for nuclear structure and function, provoking an array of cellular aberrations, including nuclear morphological abnormalities, increased DNA damage and genomic instability, epigenetic alterations, and disrupted cell signaling, all of which ultimately cause premature senescence [10-16].
Therapies for this fatal disease are desperately needed. The combination of farnesyltransferase inhibitors (FTIs), statins and bisphosphonates, which reduce prenylation of progerin, are the best treatment strategy currently available to physicians, but only extend life by 1.6 years on average [17, 18]. Further, FTIs can induce a variety of noxious side effects. Thus, there is a tremendous need for new treatments with fewer side effects. Interestingly, progerin accumulation is also observed in old individuals, suggesting its participation in the normal aging process [19]. Identifying therapeutic strategies that reduce the toxic levels of progerin could have applicability for HGPS patients and for the general aging population.
Vitamin D is essential for proper calcium and phosphate homeostasis, via its biologically most active metabolite 1,25D. Most genomic actions of vitamin D are mediated by the vitamin D receptor (VDR), being the only high-affinity nuclear receptor for 1,25D [20]. However, expression of VDR in tissues that are not associated with calcium and bone homeostasis suggested that the vitamin D/VDR axis could exert other functions. Alterations in the vitamin D/VDR status have considerable effects at the cellular and organismal level and contribute to a wide variety of diseases [21, 22]. Interestingly, VDR knockout mice develop a premature aging phenotype similar to HGPS patients, with early alopecia, growth retardation, muscle atrophy, cardiovascular disease, and reduced lifespan [21, 23]. Additionally, vitamin D/VDR signaling is important for protecting against atherosclerosis [24, 25], the pathology ultimately underlying death in HGPS patients. Consequently, we investigated whether the vitamin D/VDR axis could have an impact on the phenotype of HGPS patient-derived cells.
Here, we demonstrate for the first time that proper lamina organization is required for appropriate VDR function and that HGPS patient cells become VDR deficient upon passage in culture. Importantly, 1,25D supplementation, which counteracts VDR loss, improves many of the phenotypes of HGPS cells, including nuclear morphology and unrepaired DNA damage while delaying the onset of senescence. Most significantly, we demonstrate that prolonged 1,25D treatment leads to a dramatic decrease of progerin levels. These results advance targeting the vitamin D/VDR axis as a potential therapeutic strategy for improving HGPS patient health.
RESULTS
Alterations in the nuclear lamina cause VDR deficiency
The nuclear lamina is a critical scaffold for many transcription factors. In lamin A/C-deficient cells, major changes occur in gene signaling and transcription factor localization [26, 27]. Because VDR is an important transcription factor and can accumulate at the nuclear envelope [28], we tested whether disruptions in the nuclear lamina impact VDR levels. We discovered that lamin A/C depletion via lentiviral transduction with specific shRNAs results in a marked decrease in VDR levels in human primary normal fibroblasts derived from parents of HGPS patients (referred to as NF) (Figure 1A). Lamin A/C loss correlates with decreases in BRCA1, and to a lesser extent in 53BP1 levels, key factors in DNA repair by homologous recombination (HR) and non-homologous end joining (NHEJ) respectively, as previously observed in mouse fibroblasts and breast cancer cells [29]. Similar decreases in VDR and DNA repair factors were observed upon depletion of lamin A/C in human primary vascular smooth muscle cells (VSMC) (Figure 1B), a cell type particularly susceptible to alterations in lamin A/C function [30, 31]. In addition, quantitative RT-PCR (qRT-PCR) revealed a profound decrease in VDR transcript levels following lamin A/C depletion (Figure 1C). The reduction in VDR was confirmed in foreskin fibroblasts (BJ) transduced with two independent shRNAs targeting lamin A/C, ruling out that off-target effects are responsible for VDR loss (Figure 1D). Interestingly, disruption of the nuclear lamina by ectopic expression of progerin in these cells also results in down-regulation of VDR (Figure 1D). Importantly, VDR levels decrease during proliferation of HGPS patient-derived fibroblasts in culture (Figure 1E), as well as during proliferation of normal fibroblasts isolated from the parent of the patient. However, while the VDR decrease in HGPS cells is evident by passage 25, the VDR decrease in NF is not observed until much later passages. In contrast, BJ fibroblasts did not show a decrease in VDR at the passages tested, suggesting differences in VDR expression during proliferation among different lines of human primary fibroblasts. Of note, the decrease in VDR levels was detected in proliferating cells, prior to senescence. Altogether, these data demonstrate that disruption of the nuclear lamina, either by lamin A/C loss or progerin overexpression, results in reduced VDR expression in primary human cells.
VDR depletion causes DNA damage in normal and HGPS cells
Signaling through the vitamin D/VDR axis regulates indirectly the expression of BRCA1 in some contexts [32, 33]. Given that NF and HGPS cells grown in culture exhibit reduced VDR levels (Figure 1E), we determined if VDR loss could contribute to DNA repair defects in these cells. Consistent with this notion, depletion of VDR in NF leads to decreased BRCA1 levels and accumulation of DNA damage, as shown by elevated γH2AX levels, mirroring the phenotype of lamin A/C-depleted cells (Figure 1F). These changes were cell cycle independent, as shown by the similar cell cycle profiles between VDR-proficient and VDR-deficient NF at the time of sample collection for protein analysis (Figure S2A). DNA damage accumulation upon VDR depletion in NF was confirmed by immunofluorescence, showing a marked increase in the percentage of γH2AX-positive cells (Figure 1G). Importantly, VDR depletion eventually led to a growth arrest with characteristics of senescence, as shown by positivity for β-galactosidase (Figure 1H), recapitulating the phenotype of lamin A/C-depleted cells (Figure 1H), BRCA1-depleted cells [34], and BJ fibroblasts depleted of VDR [33]. Importantly, the effect of VDR loss causing BRCA1 reduction and accumulation of DNA damage was confirmed in a second line of NF (Figure 1I) and in HGPS patient-derived fibroblasts of early passage (Figure 1I and1J). These data uncover a role for VDR in the maintenance of DNA repair. VDR loss leads to BRCA1 down-regulation, accumulation of unrepaired DNA damage, and entry into senescence, suggesting that the DNA repair defects observed in cells with a disrupted nuclear lamina could be exacerbated by VDR deficiency.
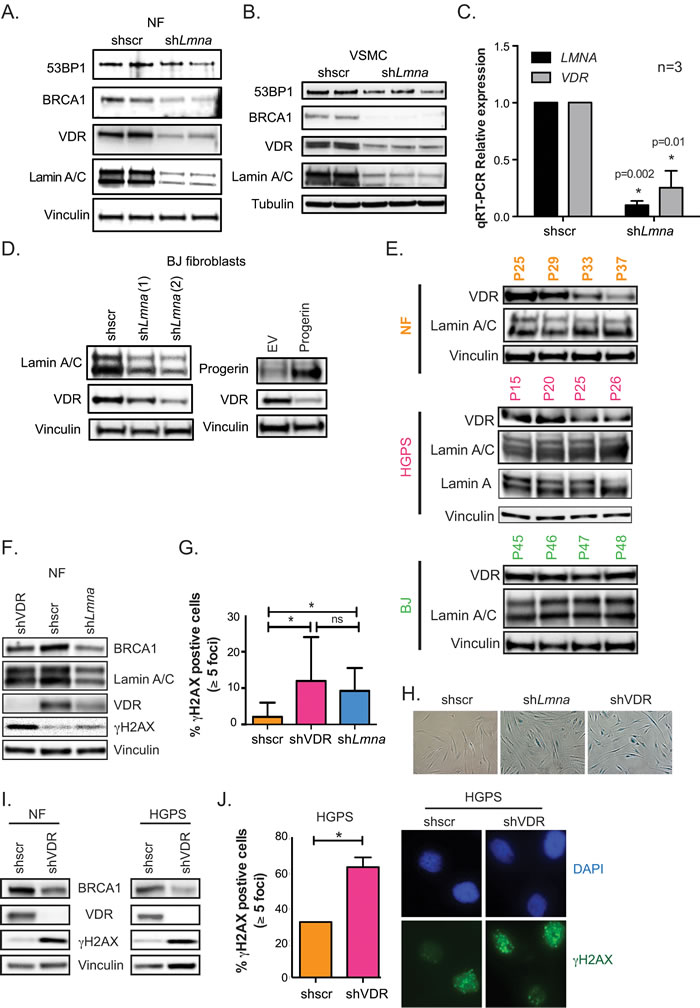
Figure 1: VDR deficiency in cells with disrupted nuclear lamina. A. Human NF lentivirally transduced with shRNA targeting lamin A/C (shLmna) or scrambled (shscr), and processed for immunoblotting (2 independent transductions). Vinculin is loading control. B. The same experiments as in (A) performed in human VSMC. Tubulin is loading control. C. Relative expression of LMNA and VDR transcripts by qRT-PCR in VSMC after depletion of lamin A/C or VDR. Results are mean±sem of 3 biological repeats. D. BJ fibroblasts were lentivirally transduced with 2 independent shRNAs targeting lamin A/C (left panels) or retrovirally transduced with progerin (right panels) and processed for immunoblotting. E. Different human fibroblasts (BJ, NF and HGPS) were collected at increasing passages in culture to monitor levels of VDR and lamin A/C by western blot. F. NF were lentivirally transduced with shscr, shLmna, or shRNA targeting VDR (shVDR) and processed for immunoblotting. G. Immunofluorescence (IF) with γH2AX antibody in NF depleted of VDR, lamin A/C, and control. Quantitated percentage of cells with more than 5 γH2AX foci. Graph represents mean±sem of 3 independent experiments. H. Images show how depletion of lamin A/C or VDR leads to accumulation of β-galactosidase positive cells, a marker of senescence (after 2 weeks). I. NF and HGPS cells were depleted of VDR and levels of BRCA1, VDR, and γH2AX monitored by immunoblotting. J. IF performed in HGPS depleted of VDR (shVDR) and control (shRNA) and percentage of γH2AX-positive cells quantitated. Images of IF showing accumulation of γH2AX in HGPS cells depleted of VDR. *p value of statistical significance (*p ≤ 0.05).
1,25D treatment ameliorates DNA repair defects in HGPS cells
HGPS cells in culture progressively accumulate progerin [10], which in turn causes nuclear deformation and fragility, accumulation of DNA damage, and altered signaling pathways [11, 16, 35]. These defects eventually cause premature entry into senescence. Given the association between VDR loss and DNA repair defects, we tested if activation of VDR signaling by 1,25D treatment ameliorates the phenotypes of HGPS patient-derived fibroblasts. We performed prolonged treatment with 1,25D or vehicle as control, because the abnormalities of HGPS cells are exacerbated with time in culture. In addition, the prolonged treatment allowed us to determine if 1,25D impacts premature entry into senescence. HGPS cells media was supplemented with 1,25D (10-7M) every 3 days for 90 days, since 1,25D is known to increase VDR transcript levels and protein stability [36]. The levels of VDR and DNA repair factors in HGPS cells were monitored by western blot prior to entry into senescence, and compared to those of NF of similar passage. We discovered that HGPS cells grown in normal media exhibit lower levels of 53BP1 and BRCA1 than NF, in addition to the decrease in VDR levels (Figure 2A and 2B). Prolonged treatment of HGPS cells with 1,25D increased VDR expression, as well as 53BP1 and BRCA1 levels (Figure 2A and 2C). Importantly, prolonged 1,25D treatment markedly reduces progerin protein levels (Figure 2A and 2C). Immunofluorescence analysis confirmed the substantially reduced progerin expression in HGPS cells treated with 1,25D (Figure 2D), when compared to vehicle-treated controls. Moreover, analysis of transcript levels from the LMNA gene by qRT-PCR revealed that prolonged 1,25D treatment reduces both total LMNA and progerin transcript levels (Figure 2E). These results indicate that 1,25D treatment has multiple beneficial effects in HGPS cells; stabilizing VDR and DNA repair factors, while reducing progerin expression. Of note, we did not detect any differences in the subcellular localization of VDR between NF and HGPS fibroblasts treated with 1,25D or vehicle control (Figure S1).
Next, we investigated potential mechanisms behind the rescue of DNA repair factors (53BP1 and BRCA1) upon 1,25D treatment of HGPS cells. Our previous studies in lamin A/C-depleted cells demonstrated activation of cathepsin L (CTSL)-mediated degradation of 53BP1. We also showed that 1,25D and the broad cathepsin inhibitor E-64 inhibit CTSL activity, leading to stabilization of 53BP1 in lamin A/C-depleted cells [37]. Here, we determined if CTSL could be involved in the decrease of 53BP1 in HGPS cells. A second line of HGPS cells under prolonged treatment with 1,25D or vehicle (Control) was subjected to acute treatment with E-64 for 24 h (Figure 2F). As expected, control HGPS cells exhibit low levels of 53BP1, BRCA1, and VDR, which are stabilized by 1,25D treatment. Interestingly, E-64 treatment restores the levels of 53BP1 in HGPS control cells, suggesting that the same mechanism of 53BP1 degradation discovered in lamin A/C-depleted cells is active in progerin-expressing HGPS cells. In contrast, E-64 was unable to rescue the levels of BRCA1 or VDR, indicating that the loss of these factors in HGPS cells does not involve CTSL. These studies reveal a mechanism behind the loss of 53BP1 in HGPS cells. Importantly, 1,25D treatment reduces significantly the accumulation of DNA damage in HGPS cells, as shown by a marked decrease in the percentage of γH2AX-positive cells (Figure 2G and 2H), and a reduction in average intensity of labeling with γH2AX in 1,25D-treated HGPS cells (Figure 2I), when compared to vehicle-treated cells. Our data demonstrate that VDR deficiency contributes to DNA repair defects in HGPS patient cells and that 1,25D treatment rescues expression of VDR and key DNA repair factors. Vitamin D-based regimens could represent a strategy to restore DNA repair capabilities in HGPS cells, a major cause of the pathology associated with this disease.
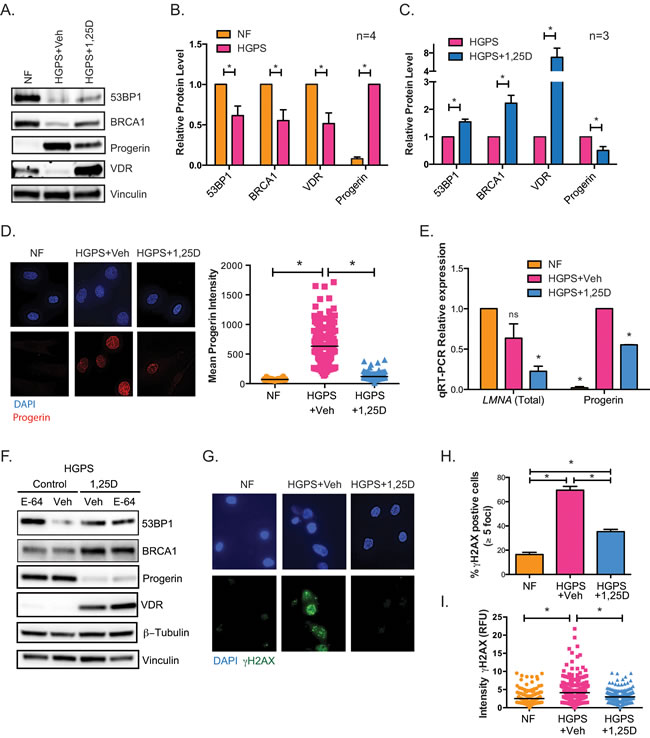
Figure 2: Phenotypes of HGPS cells are rescued by vitamin D. A. HGPS cells were grown in culture with 1,25D (10-7M) or vehicle and collected for western blot prior to entering senescence (passage 25). Levels of DNA repair factors 53BP1 and BRCA1, VDR, and progerin were compared to NFs. B. Densitometry of immunoblots comparing levels of 53BP1, BRCA1, VDR, and progerin between NF and HGPS cells (mean±sem of 4 biological repeats). C. Densitometry as in (B) comparing HGPS cells under prolonged treatment with 1,25D or vehicle as control (mean±sem of 3 biological repeats). D. DAPI and IF staining of progerin shows that accumulation of progerin in HGPS cells is reduced by prolonged 1,25D treatment (passage 27). Graph shows quantitation of progerin labeling intensity (relative fluorescence units) in NFs and in HGPS cells subjected to prolonged treatment with vehicle or 1,25D. DAPI staining was used to demarcate nuclei and intensity of progerin labeling measured using ImageJ program. A total of 200 cells were quantitated in each condition. E. HGPS fibroblasts were grown in culture for at least 90 days with 1,25D or vehicle, and qRT-PCR performed to monitor levels of total LMNA and progerin transcripts (passage 29). NF of similar passage were used as control. Results are the mean±sem of 3 independent experiments. F. Fibroblasts derived from a second HGPS patient were subjected to prolonged 1,25D treatment or vehicle control (passage 27). These two lines were treated with the cathepsin inhibitor E64 or vehicle for 24 h, and samples processed for immunoblotting. G. DAPI and IF staining of γH2AX shows that accumulation of DNA damage in HGPS cells is reduced by prolonged 1,25D treatment. H. Quantitation of percentage of cells positive for γH2AX in 3 biological repeats. I. Quantitation of γH2AX labeling intensity (relative fluorescence units) in NF and in HGPS cells under prolonged 1,25D treatment. DAPI staining was used to demarcate nuclei and intensity of γH2AX labeling measured using ImageJ program. A total of 200 cells were quantitated. *p value of statistical significance (*p ≤ 0.05).
1,25D reduces nuclear defects and delays senescence
In addition to genomic instability, HGPS cells exhibit gross nuclear architectural disruptions and nuclear blebbing as a result of progerin accumulation at the nuclear envelope [6, 38]. The severity of these nuclear aberrations often correlates with the degree of cellular decline. Given the robust effect of 1,25D reducing progerin and DNA damage in HGPS cells, we investigated whether 1,25D improves nuclear morphological abnormalities in these cells. HGPS cells under prolonged treatment with 1,25D or vehicle were processed for immunofluorescence with lamin A antibody and counterstained with DAPI, to visualize the distribution of lamin A in the nucleus (Figure 3A). In normal fibroblasts, a thin layer of lamin A was distributed evenly at the nuclear periphery, highlighting the edge of the nucleus that was ovular in shape. In contrast, HGPS cells had grossly disrupted nuclear morphology and extensive nuclear blebbing. Lamin A/progerin was localized at the nuclear periphery, but was unevenly distributed. Importantly, cells treated with 1,25D showed a significant improvement in nuclear morphology. Analysis of over 500 cells per condition revealed a marked decrease in the percentage of cells with abnormally shaped nuclei (Figure 3B). Moreover, HGPS cells grown in culture exhibit a marked increase in nuclear volume when compared to NF (Figure 3C), and 1,25D treatment reduces the nuclear volume. These data demonstrate that 1,25D treatment improves profoundly nuclear morphology in HGPS patient cells, which in turn has the potential to ameliorate cellular decline.
To test the hypothesis that 1,25D improves the proliferation of HGPS cells in culture, we compared proliferation rates between cells subjected to prolonged treatment with 1,25D (10-7M) or vehicle. Initially, HGPS cells treated with 1,25D grew at a slightly slower rate than those treated with vehicle. This was not entirely surprising, since 1,25D has anti-proliferative effects, particularly in cancer cells [39] (Figure S2B). However, with passage in culture, 1,25D-treated HGPS cells began to grow at a higher rate than those treated with vehicle (Figure 3D). Eventually, HGPS cells treated with vehicle became growth arrested, and presented characteristics of senescence such as positivity for β-galactosidase (Figure 3D and 3E), while those treated with 1,25D continued proliferating. These results were confirmed a second time in the same HGPS line (Figure 3F), as well as in a different HGPS line (Figure 3G). 1,25D-treated HGPS cells eventually entered senescence, but at much later passages. Thus, prolonged treatment with 1,25D allows HGPS patient cells to delay premature entry into senescence.
In summary, our studies demonstrate that 1,25D treatment of HGPS cells reduces accumulation of DNA damage, improves nuclear morphology, and allows bypass of premature senescence. Thus, the 1,25D/VDR axis represents a novel target for treatment of laminopathies such as HGPS, in which expression of a mutant form of lamin A causes cellular decline and organismal degeneration.
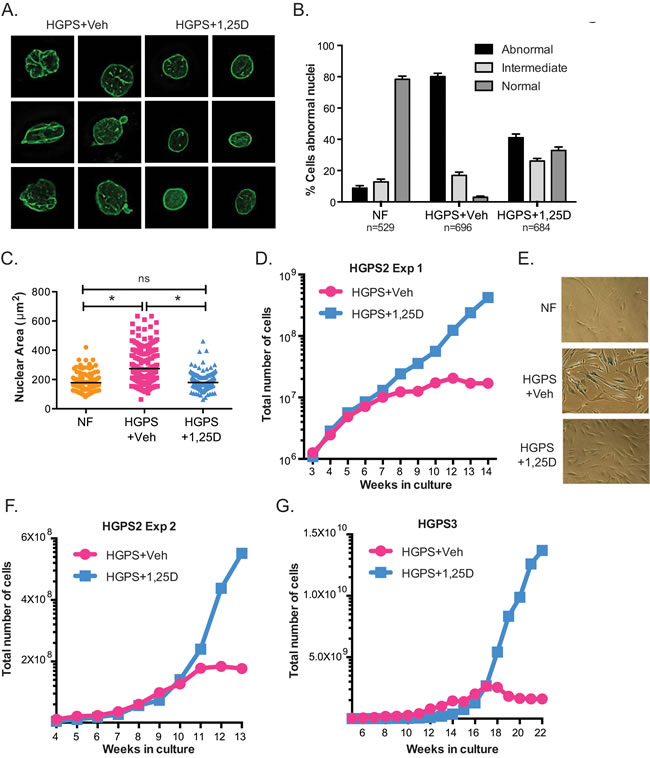
Figure 3: Vitamin D rescues nuclear abnormalities and delays senescence in HGPS cells. A. DAPI staining and IF with lamin A antibody performed in NF and HGPS fibroblasts under prolonged treatment with vehicle or 1,25D (passage 25). Representative images are shown. B. Quantitation of percentage of cells showing aberrant nuclear morphology (extensive protrusions, lobulations, herniations, etc), an intermediate phenotype (slight change in nuclear morphology), and normal nuclear morphology. More than 500 cells were counted per condition in 3 independent blinded experiments. C. The nuclear volume of cells processed for IF as in (A) was calculated using Leica’s microscope software. A total of 200 cells were quantitated. D. Proliferation rate monitored during culture of HGPS cells in media containing 1,25D or vehicle. Note how vehicle-treated HGPS cells growth arrested after approximately 8-10 weeks in culture while 1,25D-treated cells continued proliferating. E. β-galactosidase assay performed in NF and HGPS cells treated with 1,25D or vehicle, once vehicle-treated cells growth arrested (passage 33). F. Proliferation rate of a second long-term treatment of the same line of HGPS fibroblasts with 1,25D or vehicle control. G. Proliferation rate of fibroblasts from a different HGPS patient.
1,25D/VDR axis modulates LMNA gene expression
Given that prolonged 1,25D treatment decreased LMNA and progerin expression, we investigated if the LMNA gene is directly regulated by the 1,25D/VDR axis, especially because VDR is a nuclear receptor with transcriptional activity. To this end, we tested the effect of short 1,25D treatments on LMNA gene expression, and monitored the binding of VDR to the proximity of the LMNA gene by chromatin immunoprecipitation followed by sequencing (ChIP-seq). First, genome-wide RNAseq analysis of NF treated with 1,25D (10-7M) or vehicle for 24 hours, revealed a decrease in total LMNA transcript levels after 1,25D treatment (Figure 4A). In addition, qRT-PCR performed in NF after a 4-hour and a 24-hour treatment with 1,25D confirmed a modest but consistent decrease in LMNA transcripts at 24, but not at 4 hours (Figure 4B). The same effect of 1,25D on LMNA gene expression was observed in BJ fibroblasts (Figure S2C) and in mouse cells. Mouse adult fibroblasts (MAFs) isolated from the ears, and immortalized with SV40 large T antigen were treated with 1,25D (10-7M) for 24 hours. As shown in Figure 4C, 1,25D treatment reduces total LMNA transcripts. Similarly, mouse embryonic fibroblasts (MEFs) treated with 1,25D for two weeks exhibit reduced LMNA transcripts (Figure S2D). These results demonstrate that 1,25D treatment can regulate total LMNA transcript levels in human and mouse cells.
Importantly, ChIP-seq analysis on hematopoietic cell lines and BJ fibroblasts treated with 1,25D or vehicle for up to 24 hours did not show VDR binding to any genomic region near the LMNA gene transcription start site (TSS) under any condition (Figure S2). In summary, ChIP-seq, RNA-seq and qRT-PCR analyses suggest that the effect of 1,25D/VDR regulating LMNA gene expression is likely to be indirect, putatively by facilitating the function of unknown transcription factors being the main regulators of the LMNA gene. Upon ligand activation, VDR decreases LMNA gene transcripts, possibly by counteracting the function of the factor/s regulating LMNA gene expression [40]. Although further studies are required to identify the VDR-regulated factors mediating LMNA gene expression, our data provide strong evidence for the use of 1,25D to reduce expression of mutant lamins.
Lastly, we monitored the effect of short treatments with 1,25D on LMNA gene expression in HGPS cells. As shown in Figure 4D, total LMNA transcripts are decreased after a 24-hour treatment. Importantly, 1,25D treatment of HGPS cells for 4 and 7 days ameliorates some of the phenotypic abnormalities of HGPS cells. In particular, the short treatment progressively ameliorates the accumulation of DNA damage (Figure 4E), and reduces nuclear volume (Figure 4F). The effect of 1,25D on nuclear size is specific of HGPS cells, as NF did not exhibit any changes in nuclear size upon 1,25D treatment (data not shown). These results indicate that while the mechanism whereby 1,25D down-regulates progerin expression is likely indirect, 1,25D/VDR signaling has a significant beneficial effect on HGPS cellular phenotypes, which is noticeable a few days after treatment.
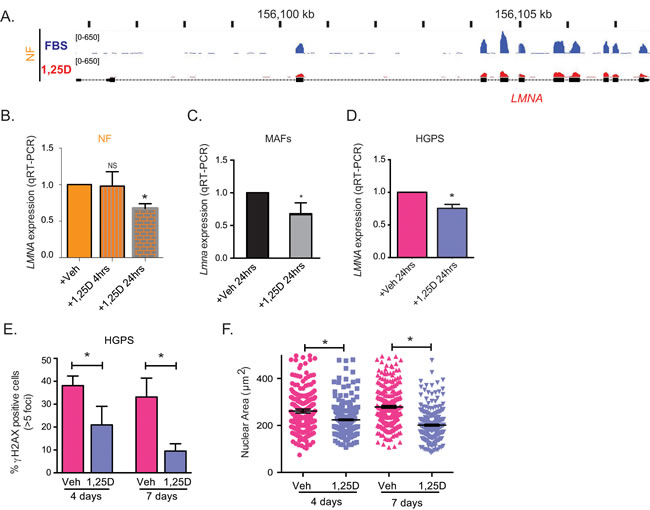
Figure 4: Regulation of LMNA gene expression by vitamin D/VDR. A. RNAseq analysis performed in NF after a 24-h treatment with 1,25D (10-7M) or vehicle (FBS) as control. Map shows the levels of transcripts containing exon sequences of the LMNA gene in control cells (blue) and 1,25D-treated cells (red). B. Relative expression of total transcripts from LMNA gene by qRT-PCR in NF treated with 1,25D for 4 h or 24 h. Results are the mean±sem of 3 biological repeats. C. Relative expression of LMNA transcripts by qRT-PCR in MAFs treated with 1,25D for 24 h. D. Relative expression of total transcripts from LMNA gene as determined by qRT-PCR in HGPS patient-derived treated with 1,25D for 24 h or vehicle control. E. IF for γH2AX in HGPS cells treated with 1,25D or vehicle for 4 and 7 days. Graph shows percentage of γH2AX-positive cells. A total of 400 cells were quantitated per condition. F. DAPI staining and IF with lamin A antibody in HGPS cells treated with 1,25D or vehicle for 4 and 7 days. The nuclear volume was calculated using Leica’s microscope software. A total of 300 cells were quantitated. All graphs represent mean±sem. *p value of statistical significance (*p ≤ 0.05).
DISCUSSION
In laminopathies such as HGPS, the nuclear lamina is severely disrupted, causing alterations in the activities of various transcription factors [35, 41, 42]. Here, we show that depletion of lamin A/C or accumulation of progerin results in decreased expression of VDR, a nuclear receptor that regulates approximately 3% of the transcriptome and mediates most genomic actions of vitamin D [20, 40]. Alterations in lamin A/C expression result in mislocalization of genes in the 3D nuclear space, which in turn impact on their transcriptional regulation. Thus, nuclear localization of the VDR gene could be altered in HGPS cells. Alternatively, progerin could impact the epigenetic status of the VDR gene, leading to repression. Moreover, since VDR regulates its own expression, a direct association of lamin A/C with VDR protein could potentially regulate VDR stability and/or transcriptional activity. Future studies need to identify the mechanisms behind VDR loss in cells with an altered nuclear lamina.
We also demonstrate that 1,25D/VDR regulates LMNA gene expression. 1,25D causes down-regulation of LMNA transcripts after 24 hours, suggesting that the effect is indirect. This is supported by the lack of VDR binding in the vicinity of the LMNA gene TSS, as shown by ChIP-seq analysis. Importantly, we observed a dramatic decrease in progerin levels in cells under prolonged 1,25D treatment, suggesting that 1,25D treatment could have dual beneficial effects for HGPS cells, ameliorating the toxicity associated with progerin expression, while rescuing VDR function (Figure 5).
Deficiency in VDR function has many detrimental effects. For instance, vitamin D/VDR deficiencies are correlated with cardiovascular disease, bone defects, and a whole variety of inflammatory diseases, and vitamin D-deficient individuals have higher risk of developing cardiac hypertrophy, hypertension, and myocardial infarction [22, 43, 44]. In addition, VDR knockout mice exhibit phenotypes observed in HGPS patients, such as premature aging, atherosclerosis, and cardiovascular disease (CVD) [24, 45], suggesting that impaired VDR activity could contribute to the pathophysiology of HGPS. In fact, we demonstrate that prolonged treatment of HGPS patient-derived fibroblasts with 1,25D rescues DNA repair defects, ameliorates nuclear morphological abnormalities, and allows senescence bypass. Based on these data, and the reported roles of vitamin D in cardiovascular protection [46], we propose that sufficient vitamin D supplementation could benefit HGPS patients.
The finding that vitamin D reduces progerin levels is of high significance (Figure 5). Studies have shown a decrease in progerin protein levels by treatment with rapamycin or sulforaphane, by activating its clearance via autophagy [47, 48]. In addition, treatment with retinoids down-regulate total LMNA gene expression and decrease progerin expression in HGPS cells [49-51]. This effect is mediated by the retinoic acid receptor (RAR), which binds to regulatory elements in the LMNA gene promoter. Future studies need to test if the combination of vitamin D with rapamycin or sulforaphane, or with retinoids has the most benefit as a treatment for HGPS, by reducing progerin transcripts and increasing progerin protein clearance by autophagy. In addition, since progerin expression is observed in cells from old individuals [19], and VDR deficiency and genomic instability increase with age [52, 53], maintaining vitamin D/VDR signaling could ameliorate pathologies associated with normal aging. However, when deciding on a treatment for HGPS it is important to consider that treatment with 1,25D and retinoids not only reduces progerin expression, but also total transcripts from the LMNA gene. There is clear evidence that the amount of progerin relative to prelamin A/lamin A dictates the severity of the disease [54, 55]. In fact, recent studies have shown a dosage-dependent effect of progerin expression in normal fibroblasts inducing phenotypes characteristic of HGPS fibroblasts [56]. Thus, further studies are needed in vitro and in vivo to determine the 1,25D or retinoid regimens that reduce progerin levels under a threshold while maintaining sufficient levels of prelamin A/lamin A as to reduce phenotype severity.
In addition to the role of vitamin D/VDR regulating LMNA gene expression, we present evidence for a role of VDR in the maintenance of DNA repair factors such as BRCA1 and 53BP1, which maintain genome integrity by facilitating DNA double-strand break repair (Figure 5). As such, VDR loss in human primary cells hinders DNA repair capabilities and induces senescence. We also show that HGPS cells exhibit decreased levels of BRCA1 upon passage in culture, which are rescued by prolonged 1,25D treatment, thus suggesting that VDR deficiency contributes to DNA repair defects in HGPS cells. We envision that vitamin D/VDR deficiencies could contribute to the genomic instability that drives aging and aging-related diseases. Reduced BRCA1 function in VDR-deficient cells could contribute to cellular decline during aging, while allowing secondary hits in the genome that promote tumorigenesis. Studies that investigate in depth the functional relationship between BRCA1 and the vitamin D/VDR axis could lead to important discoveries about the role of these tumor suppressor pathways in a variety of aging-related diseases.
Numerous studies suggest cardiovascular protection by vitamin D, including anti-atherosclerotic and anti-inflammatory activities [46, 57, 58]. Although our study is the first to demonstrate VDR deficiency in progeria, physicians recommend the intake of 400 IU vitamin D for HGPS children to forestall bone disease (http://www.progeriaresearch.org). We envision that the same VDR reduction that we find in fibroblasts from HGPS patients could extend to other cells, especially vascular cells, and that 400 IU of vitamin D could be insufficient to correct VDR content and biological actions. All these data stress the need for preclinical studies in mouse models of HGPS that explore the benefits of vitamin D-based regimens to ameliorate the progeria phenotypes at the organismal level. Also, the combination of vitamin D regimens with current therapeutic strategies such as FTIs and prenylation inhibitors, could result in increased efficacy of the treatments, and allow lowering the doses of single drugs, reducing toxicity. Importantly, our data suggest that levels of VDR and progerin could be used as biomarkers to screen patients with laminopathies and identify those that could benefit from vitamin D treatment. In addition, monitoring the levels of these proteins before and after vitamin D treatment could serve to determine the effectiveness of the treatment. Thus, vitamin D/VDR signaling holds promise as a target for treatment of HGPS patients, which could ward off the disastrous consequences of this ultimately fatal disease.
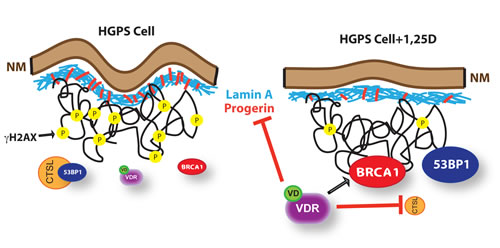
Figure 5: Model of functional relationship between nuclear lamina integrity, vitamin D/VDR axis, and expression of LMNA gene. We propose a model whereby in cells with an integral nuclear lamina, expression of DNA repair factors such as BRCA1 and 53BP1, and DNA repair capabilities are maintained by the vitamin D/VDR signaling axis. As such, depletion of VDR in normal fibroblasts results in BRCA1 loss, accumulation of DNA damage, and premature senescence. Cells with a disrupted nuclear lamina due to lamin A/C depletion or progerin accumulation (HGPS) experience a marked reduction in VDR levels, which in turn contributes to the down-regulation of BRCA1 and the activation of CTSL-mediated degradation of 53BP1. These cells accumulate DNA damage markers (γH2AX), nuclear morphological abnormalities and other cellular alterations that ultimately cause premature senescence. Importantly, treatment of HGPS cells with 1,25D to stabilize and activate VDR results in reduced progerin production, stabilization of BRCA1, inhibition of CTSL-mediated degradation of 53BP1, and amelioration of a variety of nuclear phenotypes. This suggests multiple benefits of a vitamin D based therapy for HGPS and other laminopathies.
MATERIALS AND METHODS
Cell culture
Skin fibroblasts from 4 HGPS patients, 2 with classic mutations and 2 with non-classic mutations, and from parents of HGPS patients were obtained from the Progeria Research Foundation (see SI). Cells were maintained in DMEM, 10% FBS, antibiotics/antimycotics. VSMC were obtained from Cell Applications and cultured in Lonza SmBM media with 5% FBS and LonzaSmGMTM-2 SingleQuots.
Vitamin D treatment
For long-term treatment, cells were supplemented with 10-7 M 1,25D (1α,25-dihydroxyvitamin D3) every 3 days, as described [34]. Short-term treatments were performed with 10-7 M 1,25D for 24 h, 4 days, and 7 days.
E-64 treatment
HGPS cells were incubated with the broad-spectrum cathepsin inhibitor E-64 10 µM for 24 h.
Immunoblotting
Cells were lysed in RIPA buffer (150mM NaCl, 50mM Tris-HCl pH 7.4, 1% NP-40, 0.2% SDS, 0.25% sodium deoxycholate, and 1mM EDTA), containing HALT protease and phosphatase inhibitor cocktail. Lysates were sheared using 10 passes through a 26-gauge needle followed by 10 passes through a 30-gauge needle. 60-120µg of total protein was loaded on 4-15% Criterion TGX Gel. List of antibodies used in SI.
Quantitative reverse-transcription PCR
cDNA was generated from 1μg total RNA using the GeneAmp® RNA PCR kit. qRT-PCR was performed using the 7500HT Fast Real-Time PCR system with the Taqman® Universal PCR Master Mix or Universal SYBR Green Supermix. Reactions were carried out in triplicate and target gene and endogenous controls were amplified in the same plate. Relative quantitative measurements of target genes were determined by comparing the cycle thresholds. List of probes used in SI.
Immunofluorescence
IF, microscopy and photo capture were performed exactly as described [34]. Cells were considered positive for γH2AX when exhibiting more than 5 nuclear foci. Staining intensity for γH2AX was quantitated using ImageJ. DAPI staining was used to select nuclear area, and mean signal intensity was then determined on the γH2AX staining. Background signal was accounted for and subtracted from mean signal intensity. Lamin A pictures were taken using 3D deconvolution in the Leica Application Suite. Cell morphology was then scored as normal or abnormal based on blebbing or rupture of the nucleus. Progerin staining intensity and nuclear area were quantitated from the progerin and DAPI pictures using the Leica Application Suite.
Viral transduction
Lentiviral transductions were performed as described [59]. Different shRNAs were purchased from Sigma-Aldrich, except for the progerin plasmid, which was a gift from Brian Kennedy (Buck Institute for Research on Aging, Novato, CA).
Proliferation assays
Cells were plated in triplicate at 600,000 cells/10-cm plate, and counted when confluency was near 80% utilizing Trypan Blue Viability Assay on the Nexcelom Cellometer Vision CBL. To extrapolate proliferation to the respective time periods, we used the equation Nf = N0ekt, as in [34].
Senescence-associated β-galactosidase staining
Cells were fixed in 0.2% glutaraldehyde for 5 min at 37°C, washed in PBS, and incubated for 24 h in fresh senescence β-galactosidase staining solution (1 mg 5-bromo-4-chloro3-indolyl β-D-galactoside per mL of buffer containing: 40 mM sodium phosphate/150 mM NaCl/5 mM potassium ferrocyanide/5 mM potassium ferricyanide/2 mM MgCl2 at pH 6). After 4-6 h incubation at 37°C, cells were washed in PBS and pictures taken.
Analysis of cell cycle profile
Cells were fixed in ice-cold 70% ethanol while vortexing and stored at 4°C until analysis. Fixed cells were washed with PBS, suspended in 1 ml propidium iodide staining (400 μg /ml), and incubated for 15 min at 37°C. Stained cells were analyzed for DNA content using the Nexcelom Cellometer Vision CBL, and cell cycle profiles were created by the program FCS Express 4 Flow Cytometry.
Statistical analysis
For all qRT-PCR experiments, a standard “two-tailed” student’s t-test was used to calculate statistical significance of the observed differences. Microsoft Excel v.2010 was used for the calculations. For γH2AX and progerin labeling experiments a one-way ANOVA test was performed using GraphPad Prism software. In all cases, differences were considered statistically significant when p < 0.05.
RNAseq and ChIPseq described in supplementary methods
ACKNOWLEDGMENTS
Research in the laboratory of S.G. is supported by NIGMS Grant RO1 GM094513-01, DOD BCRP Idea Award BC110089, PRF award from SLU, and Siteman Investment Program (SIP) from WUSTL. R.K. is supported by a Predoctoral Fellowship from AHA (16PRE27510016), and is recipient of the William S. Sly Fellowship in Biomedical Sciences. R.K. generated most of the data and wrote the first draft of the manuscript. M.A.N., M.C., G.B.D., and S.G. contributed with some experiments. A.D. provided expertise guidance on vitamin D treatments. D.D. performed RNAseq and ChIPseq analyses on human fibroblasts. C.C. performed genome-wide ChIP-seq and expression analyses in hematopoietic cells. A.D., D.D., and C. C. participated in discussions of the research. S.G. supervised the research and prepared the manuscript. We acknowledge the help of Dr. Jacqui Hawkins-Salsbury at early stages of the project, and James Williams and Lauren Bernier, medical and graduate students from SLU. We also thank Ziva Misulovin for her help with ChIP-seq experiments.
conflicts of interest
All other authors have no conflict of interest.
References
1. Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008; 358:592-604.
2. Pereira S, Bourgeois P, Navarro C, Esteves-Vieira V, Cau P, De Sandre-Giovannoli A and Levy N. HGPS and related premature aging disorders: from genomic identification to the first therapeutic approaches. Mech Ageing Dev. 2008; 129:449-459.
3. Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010; 30:2301-2309.
4. De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M and Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003; 300:2055.
5. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003; 423:293-298.
6. Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG and Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005; 102:10291-10296.
7. Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM and Lopez-Otin C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008; 14:767-772.
8. Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C and Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006; 311:1621-1623.
9. Ibrahim MX, Sayin VI, Akula MK, Liu M, Fong LG, Young SG and Bergo MO. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science. 2013; 340:1330-1333.
10. Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R and Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004; 101:8963-8968.
11. Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T and Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006; 103:8703-8708.
12. Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005; 11:780-785.
13. Burtner CR and Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol. 2010; 11:567-578.
14. Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, Lee HS, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010; 19:413-425.
15. Osorio FG, Barcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM and Lopez-Otin C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012; 26:2311-2324.
16. Gonzalo S and Kreienkamp R. DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Curr Opin Cell Biol. 2015; 34:75-83.
17. Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012; 109:16666-16671.
18. Gordon LB, Massaro J, D’Agostino RB, Sr., Campbell SE, Brazier J, Brown WT, Kleinman ME and Kieran MW. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014; 130:27-34.
19. Scaffidi P and Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006; 312:1059-1063.
20. Carlberg C. Genome-wide (over)view on the actions of vitamin D. Front Physiol. 2014; 5:167.
21. Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C and Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008; 29:726-776.
22. Plum LA and DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010; 9:941-955.
23. Keisala T, Minasyan A, Lou YR, Zou J, Kalueff AV, Pyykko I and Tuohimaa P. Premature aging in vitamin D receptor mutant mice. The Journal of steroid biochemistry and molecular biology. 2009; 115:91-97.
24. Szeto FL, Reardon CA, Yoon D, Wang Y, Wong KE, Chen Y, Kong J, Liu SQ, Thadhani R, Getz GS and Li YC. Vitamin D receptor signaling inhibits atherosclerosis in mice. Mol Endocrinol. 2012; 26:1091-1101.
25. Weng S, Sprague JE, Oh J, Riek AE, Chin K, Garcia M and Bernal-Mizrachi C. Vitamin D deficiency induces high blood pressure and accelerates atherosclerosis in mice. PLoS One. 2013; 8:e54625.
26. Heessen S and Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007; 8:914-919.
27. Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L and Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008; 22:832-853.
28. Barsony J, Renyi I and McKoy W. Subcellular distribution of normal and mutant vitamin D receptors in living cells. Studies with a novel fluorescent ligand. J Biol Chem. 1997; 272:5774-5782.
29. Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, Stewart CL, Zhang J and Gonzalo S. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle. 2011; 10:2549-2560.
30. Zhang H, Xiong ZM and Cao K. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc Natl Acad Sci U S A. 2014; 111:E2261-2270.
31. Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acin-Perez R, Enriquez JA, Lopez-Otin C and Andres V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013; 127:2442-2451.
32. Campbell MJ, Gombart AF, Kwok SH, Park S and Koeffler HP. The anti-proliferative effects of 1alpha,25(OH)2D3 on breast and prostate cancer cells are associated with induction of BRCA1 gene expression. Oncogene. 2000; 19:5091-5097.
33. Graziano S, Johnston R, Deng O, Zhang J and Gonzalo S. Vitamin D/vitamin D receptor axis regulates DNA repair during oncogene-induced senescence. Oncogene. 2016; doi:10.1038/onc.2016.77.
34. Grotsky DA, Gonzalez-Suarez I, Novell A, Neumann MA, Yaddanapudi SC, Croke M, Martinez-Alonso M, Redwood AB, Ortega-Martinez S, Feng Z, Lerma E, Ramon y Cajal T, Zhang J, Matias-Guiu X, Dusso A and Gonzalo S. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. J Cell Biol. 2013; 200:187-202.
35. Prokocimer M, Barkan R and Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013; 12:533-43.
36. Haussler MR, Whitfield GK, Kaneko I, Haussler CA, Hsieh D, Hsieh JC and Jurutka PW. Molecular mechanisms of vitamin D action. Calcif Tissue Int. 2013; 92:77-98.
37. Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A and Gonzalo S. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. The EMBO journal. 2011; 30:3383-3396.
38. Constantinescu D, Csoka AB, Navara CS and Schatten GP. Defective DSB repair correlates with abnormal nuclear morphology and is improved with FTI treatment in Hutchinson-Gilford progeria syndrome fibroblasts. Exp Cell Res. 2010; 316:2747-2759.
39. Chung I, Han G, Seshadri M, Gillard BM, Yu WD, Foster BA, Trump DL and Johnson CS. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009; 69:967-975.
40. Carlberg C and Campbell MJ. Vitamin D receptor signaling mechanisms: integrated actions of a well-defined transcription factor. Steroids. 2013; 78:127-136.
41. Mekhail K and Moazed D. The nuclear envelope in genome organization, expression and stability. Nat Rev Mol Cell Biol. 2010; 11:317-328.
42. Andres V and Gonzalez JM. Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol. 2009; 187:945-957.
43. Haussler MR, Haussler CA, Whitfield GK, Hsieh JC, Thompson PD, Barthel TK, Bartik L, Egan JB, Wu Y, Kubicek JL, Lowmiller CL, Moffet EW, Forster RE and Jurutka PW. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the “Fountain of Youth” to mediate healthful aging. The Journal of steroid biochemistry and molecular biology. 2010; 121:88-97.
44. Kunadian V, Ford GA, Bawamia B, Qiu W and Manson JE. Vitamin D deficiency and coronary artery disease: a review of the evidence. Am Heart J. 2014; 167:283-291.
45. Simpson RU, Hershey SH and Nibbelink KA. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. The Journal of steroid biochemistry and molecular biology. 2007; 103:521-524.
46. Pilz S, Tomaschitz A, Marz W, Drechsler C, Ritz E, Zittermann A, Cavalier E, Pieber TR, Lappe JM, Grant WB, Holick MF and Dekker JM. Vitamin D, cardiovascular disease and mortality. Clin Endocrinol (Oxf). 2011; 75:575-584.
47. Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D and Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011; 3:89ra58.
48. Gabriel D, Roedl D, Gordon LB and Djabali K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell. 2015; 14:78-91.
49. Kubben N, Brimacombe KR, Donegan M, Li Z and Misteli T. A high-content imaging-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. 2016; 96:46-58.
50. Pellegrini C, Columbaro M, Capanni C, D’Apice MR, Cavallo C, Murdocca M, Lattanzi G and Squarzoni S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015; 6:29914-29928. doi:10.18632/oncotarget.4939.
51. Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW and Discher DE. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013; 341:1240104.
52. Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194-1217.
53. Lanske B and Razzaque MS. Vitamin D and aging: old concepts and new insights. J Nutr Biochem. 2007; 18:771-777.
54. Moulson CL, Fong LG, Gardner JM, Farber EA, Go G, Passariello A, Grange DK, Young SG and Miner JH. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum Mutat. 2007; 28:882-889.
55. Reunert J, Wentzell R, Walter M, Jakubiczka S, Zenker M, Brune T, Rust S and Marquardt T. Neonatal progeria: increased ratio of progerin to lamin A leads to progeria of the newborn. Eur J Hum Genet. 2012; 20:933-937.
56. Chojnowski A, Ong PF, Wong ES, Lim JS, Mutalif RA, Navasankari R, Dutta B, Yang H, Liow YY, Sze SK, Boudier T, Wright GD, Colman A, Burke B, Stewart CL and Dreesen O. Progerin reduces LAP2alpha-telomere association in Hutchinson-Gilford progeria. Elife. 2015; 4.
57. Franczyk A, Stolarz-Skrzypek K, Wesolowska A and Czarnecka D. Vitamin d and vitamin d receptor activators in treatment of hypertension and cardiovascular disease. Cardiovasc Hematol Disord Drug Targets. 2014; 14:34-44.
58. Gotsman I, Shauer A, Zwas DR, Hellman Y, Keren A, Lotan C and Admon D. Vitamin D deficiency is a predictor of reduced survival in patients with heart failure; vitamin D supplementation improves outcome. Eur J Heart Fail. 2012; 14:357-366.
59. Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, Morgado-Palacin L, Gapud EJ, Sleckman BP, Sullivan T, Sage J, Stewart CL, Mai S and Gonzalo S. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. The EMBO journal. 2009; 28:2414-2427.