
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common malignancies and is the third leading cause of cancer-related death worldwide [1–4]. HCC incidence in China is anticipated to continue to increase. HCC is the second leading cause of cancer-related deaths in China, and its mortality rate in China ranks first in the world. Currently, half of the patients with HCC are diagnosed at a late stage when conditions are unsuitable for surgical resection and liver transplantation. Meanwhile, the remaining cases of HCC are diagnosed at an early stage but most do not receive curative therapy [1–4]. Epidemiologic and clinical studies have identified many risk factors that affect HCC, including the overexpression of oncogene and low expression of tumor suppressors. However, the molecular mechanisms underlying the development of HCC, especially the correlations between genes and their functions have not yet been completely elucidated. Here, we aimed to investigate the correlation between nuclear factor-kappa B (NF-κB) and COMMD7 in hepatocellular carcinoma.
NF-κB regulates cell survival, differentiation, and inflammation, and plays important roles in cancer, inflammatory diseases and development of the immune system [5–7]. It was demonstrated that NEMO, a crucial regulator of the NF-κB pathway, interacted with COMMD7 [8]. COMMD7 is a member of the COMMD family, which is overexpressed in HCC [9] and associated with tumor growth and invasion [10]. Silencing of COMMD7 inhibited human HepG2 cell growth [11]. Endogenous depletion of COMMD7 results in stabilization of p65 (Ser468) and greater nuclear accumulation of p65 following TNF-α stimulation [8]. COMMD7 interacts with COMMD1 and together they down-regulate NF-κB activity [8]. In COMMD7 silenced HepG2 cells, the inhibition rate of NF-κB was 75%, suggesting that COMMD7 regulates the nuclear translocation of NF-κB and the consequent gene transcriptions involved in HCC growth [10]. However, it was demonstrated that mutational changes in individual proteins can cause fundamental functional changes well beyond the pathway they function in by a positive feedback loop embedded in the crosstalk [12–15]. Interestingly, many oncogenes associated with tumor development and progression are activated by NF-κB [16–18]. It is unclear whether NF-κB is also an upstream signaling factor of COMMD7, and it is unclear whether there is crosstalk between NF-κB and COMMD7 that causes an oncogenic positive feedback loop.
Here, we examined the expression of COMMD7 and NF-κB in patients with hepatocellular carcinoma, and analyzed the correlation between COMMD7 and NF-κB. Then, we detected the expressions of COMMD7 in human hepatocellular carcinoma cell lines and found that it was significantly upregulated inHepG2 and SMMC-7721 cells. Using these cell lines, the interaction of NF-κB and COMMD7 promoter was further assessed by luciferase reporter assay, chromatin immunoprecipitation (ChIP), and electrophoretic mobility shift assays (EMSA). Finally, the roles of NF-κB and COMMD7 in cell proliferation, cell apoptosis, migration and invasion of hepatocellular carcinoma cell lines, and the tumor growth were studied. We found that NF-κB correlated with the expression of COMMD7, playing important roles in the development of HCC. Therapeutic strategies for HCC should be explored based on the correlation between NF-κB and COMMD7.
RESULTS
Expression of COMMD7 is up-regulated in hepatocellular carcinoma tissues
To investigate the role of COMMD7 in hepatocellular carcinoma, we compared the difference in expression of COMMD7 between hepatocellular carcinoma and para-carcinoma tissues using immunohistochemistry (IHC) (Figure 1A), qRT-PCR (Figure 1C) and Western Blot assays (Figure 1D). As shown in Figure 1A and 1B, IHC revealed that the staining of COMMD7 and p65 were differentially distributed between hepatocellular carcinoma and para-carcinoma tissues. The expression levels of COMMD7 inhepatocellular carcinoma were much stronger than the para-carcinoma tissues, which suggests that COMMD7 is involved in the pathological changes of hepatocellular carcinoma.
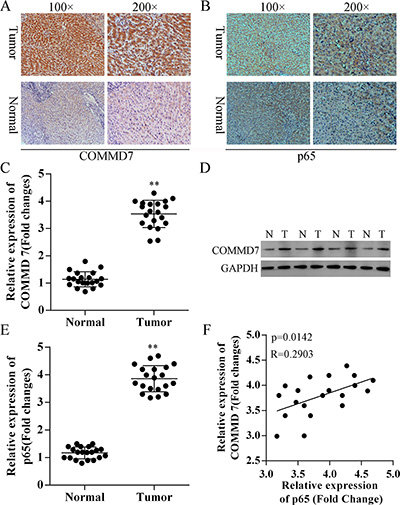
Figure 1: Correction between COMMD7 and NF-κB in hepatocellular carcinoma. (A) COMMD7 and (B) p65 expression in hepatocellular carcinoma and para-carcinoma tissues using immunohistochemistry (IHC). Representative images at different magnifications from independent experiments are shown. Tumor: hepatocellular carcinoma; Normal: para-carcinoma tissue. (C) qRT-PCR and (D) Western Blot assays revealed the up-regulation of COMMD7 in hepatocellular carcinoma (tumor, T), compared with para-carcinoma tissues (normal, N). (E) qRT-PCR also revealed the up-regulation of NF-κB in hepatocellular carcinoma. (F) Correlation analysis revealed a significant correlation between COMMD7 and the expression of p65 subunit of NF-κB. **p < 0.01 vs. normal.
Based on these results, qRT-PCR and western blot were subsequently performed to further validate these findings (Figure 1C and 1D). Moreover, as shown in Figure 1E, low expression of the p65 subunit of NF-κB in para-carcinoma tissues was also observed. Correlation analysis revealed a significant correlation between COMMD7 and the expression of p65 (Figure 1F).
Expression of COMMD7 is up-regulated in hepatocellular carcinoma cells
The expression of COMMD7 was detected by immunofluorescence, qRT-PCR, and western blot. Compared to HL7702 human liver cells, HepG2, SMMC-7721, Huh7 and Hep3B human hepatocellular carcinoma cells expressed higher level of COMMD7 (Figure 2A), and COMMD7 mRNA (Figure 2B). Moreover, the protein levels of COMMD7 in HepG2 and SMMC-7721 cells were higher than Huh7 and Hep3B cells, and all of them were higher than that in HL7702 cells (Figure 2A and 2C). HepG2 and SMMC7721 cells were identified by double staining with CD34 and p65 (Supplementary Figure S2). Then, HepG2 and SMMC-7721 cells were used as cell models to investigate the role of COMMD7 in hepatocellular carcinoma.
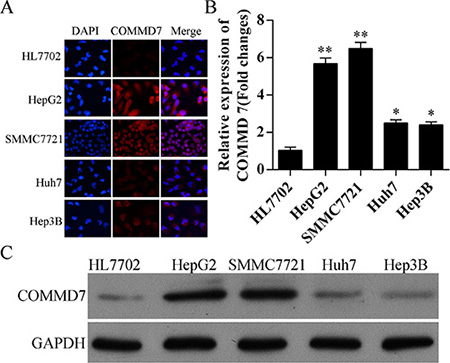
Figure 2: Expression of COMMD7 is also up-regulated in hepatocellular carcinoma cells. (A) Immunofluorescence of COMMD7 and (B) expression of COMMD7 mRNA levels in HL7702 human liver cells, and HepG2, SMMC-7721, Huh7 and Hep3B human hepatocellular carcinoma cells were detected by qRT-PCR, and (C) protein level was detected by Western Blot, respectively. *p < 0.05; **p < 0.01 vs. HL7702.
NF-κB directly binds to the COMMD7 promoter
We prepared oligonucleotide probes for the putative NF-κB binding regions (Figure 3A). Mutations (MUT1, 2, 4, 5 and 6) in the COMMD7 motif significantly diminished the luciferase activity by COMMD7 in 293T cells (p < 0.05, MUT2 vs. WT; p < 0.01, MUT1, 4, and 5 vs. WT; p < 0.001, MUT6 vs. WT) (Figure 3B).
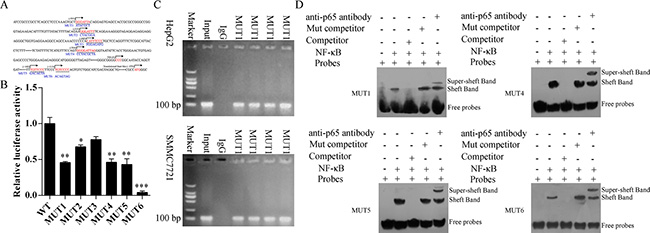
Figure 3: NF-κB binds to the endogenous COMMD7 promoters. (A) Oligonucleotide sequences corresponding to the potential COMMD7 promoters are shown, with distance from the transcription start site (TSS) indicated. Arrows denote site orientation. (B) 293T cells were transfected with the indicated reporter plasmids and co-transfected with NF-κB-expressing or empty pcDNA3 plasmids. Mean values (n = 3) of luciferase reporter activity ± standard deviation are shown. *p < 0.05; **p < 0.01 mutation (MUT) vs. wide-type (WT). (C) Binding of NF-κB to the endogenous COMMD7 promoters. Chromatin-immunoprecipitated NF-κB-binding non-enriched (rabbit IgG) DNA, enriched (NF-κB) DNA, and 0.01% whole cell extract DNA in HepG2 or SMMC-7721 were amplified by PCR. (D) Electrophoretic mobility shift assay (EMSA). 32P-labeled oligonucleotide probes corresponding to the COMMD7 promoter 1, 4, 5 and 6, complexed with NF-κB in the presence or absence of anti-NF-κB antibody or specific/mutant competitors were detected by EMSA.
To further determine whether NF-κB binds to the endogenous COMMD7 promoters, we performed ChIP assay using HepG2 and SMMC-7721 cells (Figure 3C). Consistent with the above results, we found that NF-κB was bound to this region in both HepG2 and SMMC-7721 cells. NF-κB was bound to the COMMD7 promoter containing putative NF-κB binding motifs in both HepG2 and SMMC-7721 cells.
Oligonucleotide probes with mutations of the COMMD7 motif sites were synthesized and EMSAs were performed (Figure 3D). Upon incubating the COMMD7-promoter 1, 4, 5 and 6 oligonucleotide probes with recombinant NF-κB protein, we detected the binding of NF-κB to these sites (lane 2). Addition of specific competitor of p65 abolished the binding (lane 3), but incubation with mutant competitor increased the putative bands (lane 4). Interestingly, addition of anti-p65 antibody reduced the intensity of those bands, with an upward super-shift observed (lane 5). Taken together, these results suggest that COMMD7 is a NF-κB target gene in hepatocellular carcinoma cells, and NF-κB directly correlates with COMMD7.
Stable transfection of NF-κB shRNA or COMMD7 shRNA suppresses the proliferation of hepatocellular carcinoma cells
After stable transfection, the expression of p65 subunit of NF-κB and COMMD7 were detected by confocal microscopy and western blot. Stable transfection of NF-κB shRNA in HepG2 cells significantly down-regulated the expression of p65 subunit of NF-κB and the expression of COMMD7 (Figure 4A–4C). Transfection of COMMD7 shRNA in HepG2 cells significantly down-regulated the expression of COMMD7 (Figure 4D–4E). Knockdown of COMMD7 significantly reduced the proliferation of HepG2, and knockdown of NF-κB further attenuated the proliferation (Figure 4F). In agreement with this conclusion, in SMMC-7721 cells, NF-κB shRNA also significantly down-regulated the expression of p65 and COMMD7 (Figure 4G–4I), and COMMD7 shRNA significantly down-regulated the expression of COMMD7 (Figure 4J and 4K). The cell proliferation of SMMC-7721 was reduced by COMMD7 shRNA, and further attenuated by NF-κB shRNA (Figure 4L).
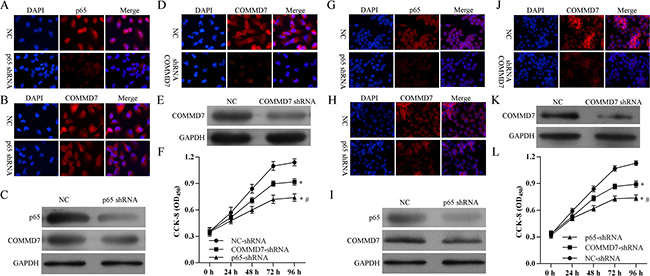
Figure 4: Stable transfection of NF-κB shRNA or COMMD7 shRNA inhibited the proliferation of hepatocellular carcinoma cells. (A–F): HepG2; (G–L): SMMC-7721 cells. The expression of p65 subunit of NF-κB and COMMD7 were detected by confocal microscopy and western blot. Expression of p65 subunit (A, C and G, I) of NF-κB and COMMD7 (B, C and H, I) after NF-κB shRNA transfection. Expression of COMMD7 (D, E and J, K) after COMMD7 shRNA transfection. (F and L) Cell proliferation was detected by MTT assay. *p < 0.05, vs. NC-shRNA; #p < 0.05, vs. COMMD7-shRNA.
Stable transfection of NF-κB shRNA or COMMD7 shRNA enhances the apoptosis of hepatocellular carcinoma cells
Following transfection of HepG2 cells with COMMD7 shRNA we found that cell apoptosis, especially the early apoptosis was enhanced, as detected by flow cytometry (Figure 5A and 5B). Knockdown of NF-κB showed a significant increase in early apoptosis in HepG2 cells, compared to cells transfected with COMMD7 shRNA (Figure 5A and 5B). Similar to HepG2 cells, in SMMC-7721 cells, the total apoptosis and early apoptosis of SMMC-7721 cells were increased by COMMD7 shRNA, and further enhanced by NF-κB shRNA (Figure 5D and 5E). The prosurvival roles of COMMD7 and NF-κB were further confirmed by Hoechest staining (Figure 5C and 5F).
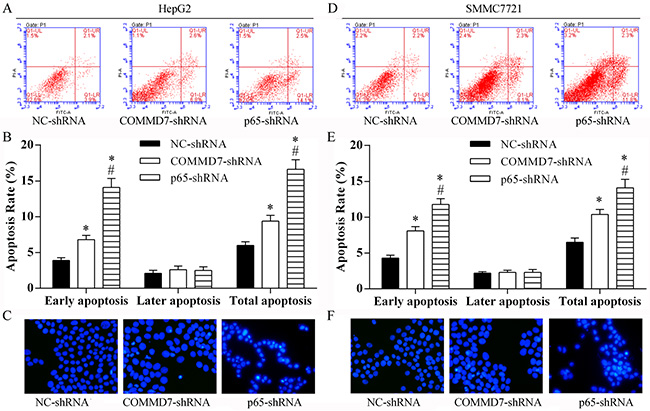
Figure 5: Stable transfection of NF-κB shRNA or COMMD7 shRNA enhanced apoptosis of hepatocellular carcinoma cells. (A–C): HepG2; (D–F): SMMC-7721 cells. (A and D) Cell apoptosis in each group was determined using the Annexin V-FITC/PI flow cytometry, and (B and E) proportion of apoptosis cells was measured. *p < 0.05, vs. NC-shRNA; #p < 0.05, vs. COMMD7-shRNA. (C and F) The roles of COMMD7 and NF-κB in cell apoptosis were further confirmed by Hoechest staining.
Stable transfection of NF-κB shRNA or COMMD7 shRNA abolishes the invasion of hepatocellular carcinoma cells
The invasion of HepG2 (Figure 6A and 6B) and SMMC-7721 cells (Figure 6C and 6D) was significantly abolished by COMMD7-shRNA, and further abolished by NF-κB shRNA, respectively.
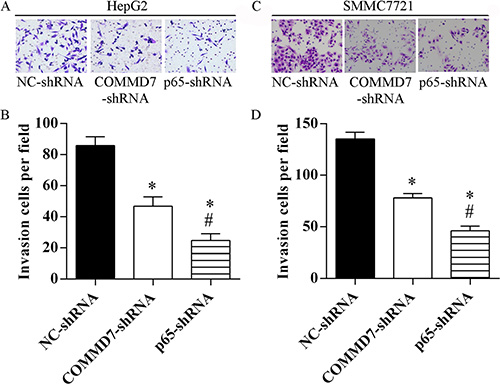
Figure 6: Stable transfection of NF-κB shRNA or COMMD7 shRNA suppressed the invasion of hepatocellular carcinoma cells. (A and B): HepG2; (C and D): SMMC-7721 cells. (A and C) Invasion assay was assessed by transwell chamber and migrated cells were counted (B and D). *p < 0.05, vs. NC-shRNA; #p < 0.05, vs. COMMD7-shRNA.
Stable transfection of NF-κB shRNA or COMMD7 shRNA suppresses tumorigenicity in vivo
To confirm the above findings, an in vivo tumor model was used. Stable transfected HepG2 (Figure 7A–7C), SMMC-7721 (Figure 7D–7F) and control cells were injected separately into nude mice. Similar results were found in HepG2 and SMMC-7721 cells. Five weeks after injection, the group with COMMD7 shRNA formed substantially smaller tumors than the control group (Figure 7A, 7B, 7D, and 7E). The expression of COMMD7 in tumors in the group with COMMD7 shRNA was lower than that in the control group (Figure 7C and 7F). The group with NF-κB shRNA formed substantially smaller tumors than the group with COMMD7 shRNA (Figure 7A, 7B, 7D, and 7E). The expression of p65 in tumors in the group with COMMD7 shRNA was lower than that in the control group (Figure 7C and 7F). Furthermore, COMMD7 is downregulated in tumors in the group with NF-κB shRNA (Figure 7C and 7F), suggesting the expression of COMMD7 is correlated with the NF-κB level.
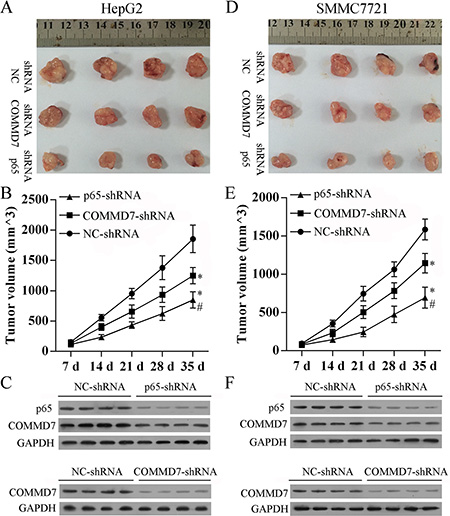
Figure 7: Stable transfection of NF-κB shRNA or COMMD7 shRNA suppressed tumorigenicity in vivo. Tumor volume (V) was measured by caliper daily and calculated using the formula V = (L × W2)/2, where L was the length and W was the width of the tumor. Growth curves were plotted using average tumor volume within each experimental group every week after respective inoculation of HepG2 cells (A–C) or SMMC-7721 cells (D–F) that were stable transfected with COMMD7 shRNA or NF-κB shRNA, and control cells. Five weeks later, the mice were euthanized, and the dissected tumors were collected (A and D), the tumor growth curves (B and E), and the expression of p65 or COMMD7 in tumors (C and F) were performed. *p < 0.05, vs. NC-shRNA; #p < 0.05, vs. COMMD7-shRNA.
DISCUSSION
NF-κB and COMMD7 transcriptional activity appear to play important roles in driving the progression of hepatocellular carcinoma (HCC), especially in tumor progression [9, 18, 19]. It was demonstrated that COMMD7 (previously named BC047440) is highly expressed in cytoplasm of HCC tissues and human HCC cell lines, while being scarcely expressed in adjacent tissues and normal liver tissue and LO2 cells [10, 20]. Interestingly, our data have shown that COMMD7 is overexpressed in HCC with a correlation to NF-κB. COMMD7 was also aberrantly overexpressed in human HCC cell lines, but was significantly less expressed in human liver cell line HL7702.
In previous studies, it was demonstrated that COMMD7 has an inhibitory role on NF-κB signaling [8, 21]. COMMD7 binds to IKK complex through NEMO in the nucleus and induced degradation of p65 leading to the termination of NF-κB signaling [8]. Silenced COMMD7 expression in HEK293T cells induced a nuclear accumulation of p65 and a persistent activation of NF-kB upon a continuous TNFα activation [8]. This inconsistent may due to different cell types or cytokine stimulation. Knockdown of COMMD7 significantly and specifically decreases the in vitro proliferation of viable HepG2 cells [10, 11]. In COMMD7-silenced HepG2 cells, we observed the impairment of the nuclear translocation of NF-κB by EMSA assay, and the inhibition (75%) of NF-κB using luciferase reporter assay [10]. These findings suggest that COMMD7 is correlated with the nuclear translocation of NF-κB and the consequent gene transcriptions involved in HCC growth.
We demonstrated that NF-κB directly binds with COMMD7 promoter and serves as an activator for COMMD7 transcription, providing a novel molecular mechanism by which NF-κB stimulates transcription of the COMMD7 gene. This is also supported by the inhibition of NF-κB by knocking down of COMMD7 [10]. Thus, NF-κB correlated or is correlated with COMMD7.
The silencing of COMMD7 inhibited human HepG2 cell growth showing an anti-proliferative effect [11]. We also showed that cell proliferation was inhibited by COMMD7 silencing, and NF-κB silencing inhibited the expression of COMMD7 and further attenuated cell proliferation in both HepG2 and SMMC-7721 cells. Cell apoptosis was promoted by COMMD7 silencing, and NF-κB silencing further promoted apoptosis. Cell migration and invasion were also inhibited by COMMD7 silencing, and NF-κB silencing further inhibited the cell migration and invasion. Although the detailed molecular mechanism underlying the anti-proliferative and anti-invasion effect by COMMD7 silencing is unknown, our findings suggested that it was positively correlated with NF-κB. It has partly revealed the specific target genes and mechanisms of transcriptional activation in these processes. Further studies are required to determine co-activation that mediate this effect.
Taken together with our previous study [22], we have now identified an important mechanism of crosstalk between NF-κB and COMMD7 that serves to increase the activity of both pathways and drive HCC progression. COMMD7 activated NF-κB signaling through TRAF6 [22] and silencing of COMMD7 impaired the TNF-α-activated NF-κB signaling [11], showing an anti-proliferative effect. On the other hand, silencing of NF-κB reduced the expression of COMMD7, also showing anti-proliferative, pro-apoptotic, and anti-invasion effects. In addition, the overexprsssion of NF-κB induced an increase of COMMD7 in both HepG2 and SMMC-7721 cells (Supplementary Figure S1). This suggests that COMMD7 is correlated with a novel NF-κB positive feedback loop in hepatocellular carcinoma. These results suggest that the therapeutic strategies for HCC should be explored keeping in mind the correlation between NF-κB and COMMD7, so as to improve the specificity and sensitivity of therapy and to reduce toxicity.
MATERIALS AND METHODS
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the Xinqiao Hospital of Third Military Medical University review board. Written informed consent was obtained for all patient samples. Animal experiments were approved by the Institutional Committee for Animal Research and were performed in conformity with national guidelines for the care and use of laboratory animals.
Clinical samples
Frozen and paraffin-embedded hepatocellular carcinoma, and patient-matched para-carcinoma tissue samples were obtained from Chinese patients who underwent curative resection in the Xinqiao Hospital of Third Military Medical University. No cancer cells were detected at the resection margin. None of the patients had received chemotherapy or radiotherapy before surgery. All specimens were confirmed by pathological examinations. Clinical staging was performed according to the International Union for Cancer Control (UICC). The clinicopathological data is summarized as follows. Patients (10 Males and 10 Females) ages ranged from 41.6 years to 63.8 years (median age: 54.5 yrs). Time from diagnosis to surgery ranged from 9 to 22 days (median: 14 days). The histological grades of primary hepatocellular carcinoma are stage II and III. The tissue samples were frozen in liquid nitrogen and stored at −80°C until the measurement of expression of COMMD7.
Immunohistochemistry (IHC) and immunofluorescence (IFC)
Paraformaldehyde (4%)-fixed and paraffin-embedded tissues were cut into 3-μm-thick sections, de-waxed in xylene and then progressively rehydrated by gradient concentrations of ethanol. Antigens were retrieved by heating the tissue sections at 100°C for 30 min in citrate solution (10 mmol/L, pH 6.0). The sections were cooled and immersed in methanol in the presence of 0.3% hydrogen peroxide for 15 min to block the endogenous peroxidase activity. The sections were subsequently rinsed in PBS for 5 min and then incubated with primary antibody against COMMD7 (1:150, Abcam) or p65 (1:100, Abcam) at 4°C overnight. Sections incubated without the primary antibody were used as negative controls. The sections were then incubated with horseradish peroxidase-labeled goat against mouse/rabbit secondary antibody (Maixin Biotechnology, Fuzhou, China). Diaminobenzene or DAPI was used as the chromogen, and hematoxylin was used as the nuclear counterstain.
For immunofluorescence, cell were fixed in 4% paraformaldehyde, followed by a wash in PBS for 5 min. Then, cells were incubated with primary antibody against COMMD7 or p65 at 4°C overnight. Cells incubated without the primary antibody were used as negative controls. Cells were then incubated with AF594-labeled goat against mouse/rabbit secondary antibody (Maixin Biotechnology, Fuzhou, China). DAPI was used to label the nucleus. Images were acquired by an FV10i confocal microscope (OLYMPUS, Japan).
Cell culture
Human hepatocellular carcinoma cell lines including HepG2, SMMC-7721, Huh7 and Hep3B, and human liver cells HL7702 were purchased from Cell Bank of Shanghai Institute of Biochemistry & Cell Biology, Chinese Academy of Sciences. Cells were cultured in RPMI-1640 medium (Invitrogen, USA) and supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin/streptomycin at 37°C in 5% CO2.
qRT-PCR
Total RNA was isolated using TRIZOL (Invitrogen Inc., USA). RNA concentration was measured by GeneQuant II (Pharmacia, Uppsala, Sweden) at 260 nm. Reverse transcription reaction and cDNA synthesis was performed according to the manufacturer’s instructions (Invitrogen). PCR analysis was performed on Applied Biosystems 7500 Sequence Detection system (ABI, USA) using SYBR Premix Ex Taq GC kit (Takara, Japan). The primers for COMMD7: 5′-AGTGGCTTTCTCCTCACTAAGACC-3′(forward) and 5′-GGAAAGATTTCTGGCTCAGCTC-3′ (reverse); for p65: 5′-CGAACTGTTCCCCCTCATCTT-3′ (forward) and 5′-CTTGGGCTGCTCAATGATCTC-3′ (reverse); for GAPDH: 5′-ACACCCACTCCTCCACCTTT-3′ (forward) and 5′- TTACTCCTTGGAGGCCATGT-3′ (reverse). Gene expression of COMMD7 and p65 was normalized to the level of GAPDH within each sample using the relative ΔΔCT method, respectively. Gene expression is shown as relative expression to control. The correlation between COMMD7 and the expression of p65 was tested by correlation analysis. The data shown is representative of three independent experiments.
Western blotting
Samples were lysed on ice for 10 min in 50 mM Tris-HCl (pH 7.5), 10% glycerol, 2% SDS, 0.1 M dithiothreitol (DTT) and 10 mM phenylmethylsulfonyl fluoride (PMSF). Proteins were separated on 10% SDS-polyacrylamide gels and electroblotted onto a nitrocellulose membrane in 25 mM Tris base and 190 mM glycine at 50 V for 3 h at 4°C. To detect the expression of COMMD7 and p65, blots were incubated with monoclonal antibodies against COMMD7 and p65 at a concentration of 1:1000, respectively. All antibodies were purchased from Santa Cruz Biotechnology, USA. Proteins were detected by enhanced chemiluminescence (ECL) as described by the manufacturer (Beyotime, China).
Luciferase reporter assay and transfections
The transcription factor binding sites (TFBS) were identified by Genomatix. pGL4.12-Luc vector (Promega) including the indicated cloned genomic regions and thymidine kinase (TK) promoter was transfected into 293T cells. A Renilla luciferase reporter pRL-TK (Promega) was co-transfected as a control for evaluating transfection efficiency. Transfections were performed with Lipofectamine 2000 or LTX (Invitrogen) according to the manufacturer’s instructions. After 24 h, cells were washed before measuring firefly and Renilla luciferase activities using the Dual-Glo™ Luciferase Assay System (Promega). In each experiment, firefly luciferase activity was normalized to Renilla luciferase activity.
Chromatin immunoprecipitation (ChIP)
Cells were cross-linked with 1% formaldehyde for 10 min at RT, followed by addition of glycine to a final concentration of 0.125 M. Next, cells were washed with PBS, collected, washed with cell lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, and 10 μg of PMSF/ml) containing protease inhibitors, and resuspended in nuclear lysis buffer (50 mM Tris–HCl pH 8.1, 10 mM EDTA, 1% SDS, and 10 μg of PMSF/ml) containing protease inhibitors. Chromatin was sheared to approximately 200–1000 bp by sonication, prior to a 1/5 dilution in ChIP dilution buffer (16.7 mM Tris–HCl pH 8.1, 167 mM NaCl,1.2 mMEDTA, 0.01% SDS, 1.1% Triton ×−100, and 10 μg of PMSF/ml) containing protease inhibitors. Then, the chromatin solution was incubated with the indicated antibody and Dynabeads® M-280 sheep anti-rabbit IgG (Dynal Biotech) overnight at 4°C. Beads were washed three times with ChIP wash buffer (50mMHEPES–KOH (pH 7.0), 0.5 MLiCl, 1 mMEDTA, 0.7% sodium deoxycholate, and 1% NP-40). Immunocomplexes were eluted from beads using ChIP elution buffer (50 mM Tris–HCl (pH 8.0), 10 mM EDTA, and 1% SDS) for 1 h at 65°C. Elutes were then incubated overnight at 65°C to reverse cross-linking, prior to the addition of 0.5 mg of protease K/ml for 2 h at 55°C. DNA was purified using the MinElute PCR purification kit (QIAGEN) and analyzed by PCR. The primer sequences used for ChIP PCR analysis were shown in Supplementary Table S1.
Electrophoretic mobility shift assay (EMSA)
DNA–protein binding was assayed with DNA probes that had been 32P-labeled by end-filling with Klenow fragment and NF-κB in the presence or absence of anti-NF-κB antibody or specific/mutant competitors. Anti-NF-κB antibody or specific mutant competitors were pre-incubated with NF-κB for 30 min at 25°C before addition of radiolabeled probes. Reactions were performed in binding buffer [20 mM HEPES (pH 7.9), 10% glycerol, 50 mM KCl, 0.05% NP-40, 0.5 mM EDTA, 0.5 mM DTT, and 1 mM PMSF] for 30 min at 25°C. Products of the binding reactions were separated by polyacrylamide gel electrophoresis (PAGE) on a 4% gel for 3 h at 100 V. The probes used for EMSA assay were shown in Supplementary Table S2.
Cell transfection
For knockdown of NF-κB or COMMD7, the p65 shRNA, COMMD7 shRNA and negative shRNA (50 nM) were synthesized and purified by OriGene (OriGene, Rockville, USA). All the sequences for shRNA were shown in Supplementary Table S3. HepG2 or SMMC-7721 cells were seeded in a 6-well plate (2 × 105/well) for 24 h before transfection, and then were transfected with p65 shRNA, COMMD7 shRNA or negative shRNA using FuGENE® HD transfection reagent (Promega, USA) in accordance with the manufacture’s instruction. Following transfection, cells were selected in G418, and resistant colonies were propagated and examined for the downregulation of NF-κB or COMMD7 by qRT-PCR, immunofluorescence and western blotting, and cells were used for further experiments.
Cell proliferation assay
The cell survival of HepG2 and SMMC-7721 cells was evaluated using the MTT [3-(4,5-dimethyl-thiazol-2-y1) 2,5-diphenyl tetrazolium bromide] (Sigma, USA) colorimetric assay, respectively. Cells were seeded in 96-well tissue culture plates at 2 × 104 cells per well for 0, 24, 48, 72, and 96 h. Then, HepG2 or SMMC-7721 cells were washed with PBS and incubated in 100 μL of 5 mg/mL MTT solution (Invitrogen Inc., USA) for 3 h. MTT is converted into purple colored formazen in living cells which was then solubilized with dimethylsulfoxide (DMSO) (Invitrogen Inc., USA) and absorbance of solution was read at 450 nm using a microplate reader Thermo Plate (Rayto Life and Analytical Science C. Ltd, Germany).
Cell apoptosis assay
Cell apoptosis was determined by Annexin V assay. Following transfection for 48 h, cells were harvested by trypsinization and washed with PBS, and suspended in Annexin V binding buffer. FITC-conjugated Annexin V and propidium iodide (PI; KeyGen, China) were added to the cells successively. After incubation, Annexin V binding buffer was added, and cells were analyzed by a FACScan (Becton-Dickinson, USA) flow cytometer. Annexin V(+)/P(−) and Annexin V(+)/P(+) represent cells in early apoptosis and late apoptosis/necrosis, respectively. Cell apoptosis was further confirmed by Hoechest staining.
Cell invasion assay
Cell invasion of HepG2 and SMMC-7721 cells were assayed using a transwell chamber (Millipore, USA). Transwell chamber was coated with 30 μl Matrigel and was placed into 24-well plate and incubated for 30 minutes at 37°C. After 48 h of transfection, HepG2 or SMMC-7721 cells were trypsinized and seeded in chambers at the density of 8 × 104 cells per well and cultured in RPMI 1640 medium with 2% serum, while 600 μl of 10% FBS–RPMI 1640 was added to the lower chamber. Twenty-four hours later, migrated cells were fixed with 100% methanol for 30 min and stained using crystal violet for 20 min. Non-migrated cells were removed using cotton swabs. Cell images were obtained under a phase-contrast microscope (Olympus, Tokyo, Japan).
Tumorigenicity assay in nude mice
Six-week-old female Balb/c athymic nude mice (Vitalriver Laboratory Animals, Beijing, China) were subcutaneously injected in the right flank with 2 × 106 cells in 0.1 mL PBS. Once tumors were formed, tumor volume (V) was measured by caliper daily and calculated using the formula V = (L × W2)/2, where L was the length and W was the width of the tumor. The mice were randomly divided into four groups (n = 6) for respective inoculation of SMMC-7721 cells or HEPG2 cells that were stable transfected with COMMD7 shRNA or NF-κB shRNA, and control cells for 42 days after tumors reached an average 32–65 mm3. Growth curves were plotted using average tumor volume within each experimental group every week. Five weeks later, the mice were euthanized, and the dissected tumors were collected and prepared for subsequent analyses. All animal experiments were approved by the institutional animal center.
Statistical analysis
For quantitative data, all results are expressed as the mean ± SD. Statistical significance between groups was determined using one-way analysis of variance (ANOVA) or an unpaired Student’s t-test using SPSS 18.0 (SPSS, USA). Each experiment was repeated at least three times. p < 0.05 was considered statistically significant.
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the Xinqiao Hospital of Third Military Medical University review board. Written informed consent was obtained for all patient samples. Animal experiments were approved by the Institutional Committee for Animal Research and were performed in conformity with national guidelines for the care and use of laboratory animals.
ACKNOWLEDGMENTS
This work was supported by a grant from National Natural Science Foundation of China (81372561). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
FUNDING
This work was supported by a grant from National Natural Science Foundation of China (81372561).
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
1. Singal AG, El-Serag HB. Hepatocellular Carcinoma from Epidemiology to Prevention: Translating Knowledge into Practice. Clin Gastroenterol Hepatol. 2015.
2. Clark T, Maximin S, Meier J, Pokharel S, Bhargava P. Hepatocellular Carcinoma: Review of Epidemiology, Screening, Imaging Diagnosis, Response Assessment, and Treatment. Curr Probl Diagn Radiol. 2015.
3. McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015; 19:223–238.
4. Lafaro KJ, Demirjian AN, Pawlik TM. Epidemiology of hepatocellular carcinoma. Surg Oncol Clin N Am. 2015; 24:1–17.
5. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004; 431:461–466.
6. He X, Wei Z, Zhou E, Chen L, Kou J, Wang J, Yang Z. Baicalein attenuates inflammatory responses by suppressing TLR4 mediated NF-kappaB and MAPK signaling pathways in LPS-induced mastitis in mice. Int Immunopharmacol. 2015; 28:470–476.
7. Schuliga M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules. 2015; 5:1266–1283.
8. Esposito E, Napolitano G, Pescatore A, Calculli G, Incoronato MR, Leonardi A, Ursini MV. COMMD7 as a novel NEMO interacting protein involved in the termination of NF-kappaB signaling. J Cell Physiol. 2016; 231:152–161.
9. Zheng L, Huang X, Liang P, Wen Y, Yang T, Li Y, Zhao H, Han K, Li J. BC047440 overexpression is a risk factor for tumor invasion and poor prognosis in hepatocellular carcinoma. Hepatogastroenterology. 2010; 57:919–925.
10. Zheng L, Liang P, Li J, Huang XB, Liu SC, Zhao HZ, Han KQ, Wang Z. ShRNA-targeted COMMD7 suppresses hepatocellular carcinoma growth. PLoS One. 2012; 7:e45412.
11. Zhou J, Li J, Liang P, Yang T, Huang X. Primary study of the function of a novel gene BC047440 highly expressed in human hepatocellular carcinoma. Chin J Exp Surg. 2006; 23:288–290.
12. Kim D, Rath O, Kolch W, Cho KH. A hidden oncogenic positive feedback loop caused by crosstalk between Wnt and ERK pathways. Oncogene. 2007; 26:4571–4579.
13. Lian J, Tang J, Shi H, Li H, Zhen T, Xie W, Zhang F, Yang Y, Han A. Positive feedback loop of hepatoma-derived growth factor and beta-catenin promotes carcinogenesis of colorectal cancer. Oncotarget. 2015. doi: 10.18632/oncotarget.4982.
14. Yang G, Murashige DS, Humphrey SJ, James DE. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015; 12:937–943.
15. Yang X, Friedl A. A positive feedback loop between prolactin and STAT5 promotes angiogenesis. Adv Exp Med Biol. 2015; 846:265–280.
16. Martin J, Dufour JF. Tumor suppressor and hepatocellular carcinoma. World J Gastroenterol. 2008; 14:1720–1733.
17. Moreira D, Zhang Q, Hossain DM, Nechaev S, Li H, Kowolik CM, D’Apuzzo M, Forman S, Jones J, Pal SK, Kortylewski M. TLR9 signaling through NF-kappaB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. 2015; 6:17302–17313. doi: 10.18632/oncotarget.4029.
18. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009; 1:a000141.
19. Zhao L, Zhang Y. miR-342-3p affects hepatocellular carcinoma cell proliferation via regulating NF-kappaB pathway. Biochem Biophys Res Commun. 2015; 457:370–377.
20. Zheng L, Liang P, Li J, Huang XB, Wang WW, Wang L, Feng H. Expression of BC047440 protein in hepatocellular carcinoma and its relationship to prognosis. Chin J Cancer. 2010; 29:931–936.
21. Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, Maine GN, Wilkinson JC, Mayo MW, Duckett CS. COMMD proteins, a novel family of structural and functional homologs of MURR1. J Biol Chem. 2005; 280:22222–22232.
22. Ling H, Recklies AD. The chitinase 3-like protein human cartilage glycoprotein 39 inhibits cellular responses to the inflammatory cytokines interleukin-1 and tumour necrosis factor-alpha. Biochem J. 2004; 380:651–659.