
INTRODUCTION
Chronic Lymphocytic Leukaemia (CLL) presents with a very heterogeneous clinical course from indolent to aggressive disease [1-5]. In spite of promising clinical results with recent signaling inhibitors, CLL remains incurable with standard therapy.
A current model proposes that mature CD5+/CD19+ CLL cells, which retain their sensitivity to external signals, cycle continuously between a quiescent state in the peripheral blood and a proliferative state in lymphoid organs [6]. Antigen-driven signals are involved in the progression of CLL [7-11], notably within the lymph node microenvironment [12, 13]. Binding of antigen to their receptor (BCR), initiates a set of signaling cascades leading to activation of protein kinases such as Spleen- (Syk) and Bruton -(Btk) tyrosine kinases or Phosphatidyl inositol 3-delta kinase (PI3K-δ). Targeting of these kinases [14, 15] interferes with the BCR/microenvironment crosstalk and allows peripheral cell redistribution [16]. Interestingly, inhibitors of Syk, Btk or PI3K-δ [17] also impair CLL cell migration induced by pro-survival chemokines such as CXCL12 upon binding to its cognate receptor CXCR4 [16]. Highly expressed at the surface of peripheral blood CLL cells due to its efficient recycling [18], CXCR4 mediates CLL cell chemotaxis and migration beneath and underneath CXCL12 secreting stromal cells [16]. In contrast, CXCR4 expression at the membrane of CLL cells is weak within the lymph node [13]. This is likely due to a ligand-dependent internalization of the receptor, which involves CXCR4 carboxy-terminal serine rich domain bound to various endocytic proteins [19-21]. Interestingly, in T cells, phorbol ester-induced CXCR4 endocytosis is sensitive to PKC inhibitors [19].
Initially identified as PKCμ, protein kinases D1 (PKD1) along with PKD2 and PKD3 form a new serine kinase subfamily [22-24]. The three proteins share two C1-domains, which bind diacylglycerol and phorbol esters (PMA) and an auto-inhibitory PH-domain [25]. Upon stimuli, both PKC-dependent and -independent activation of PKD members occur at serine residues [26, 27]. Phosphorylation of both Ser744 and Ser748 in the activation loop of the kinase domain is followed by trans- or auto-phosphorylation at Ser916 (a marker of PKD activation [28]) in the C-terminal region [29]. In adult mice, PKD2 is selectively expressed in murine lymphoid cells and controls their functions during adaptive immune responses [24]. Moreover, an in situ study revealed auto-phosphorylation of PKD2 in reactive lymph nodes and lymphoid tumors [30].
We have previously shown that in vitro BCR engagement induces plasma membrane CXCR4 decrease in CLL cells from progressive patients. Receptor internalization was related to decreased cellular chemotaxis towards CXCL12 gradient and correlated with shorter progression-free survival [10]. In this study, we addressed the molecular mechanisms underlying BCR-dependent CXCR4 down-regulation. We demonstrated that phosphorylation/activation of PKD in response to BCR stimulation, which involves PI3K-δ, is required for CXCR4-phosphorylation and its down-regulation. This regulatory pathway is functionally implicated in ex-vivo cell migration towards CXCL12 and correlated to the presence of lymph nodes in CLL patients.
RESULTS
PI3K and PKD2/3 activities mediate BCR-dependent CXCR4 down-regulation in CLL cells
We have previously demonstrated that the capacity for CLL B cells to down-regulate CXCR4 upon BCR engagement was correlated to shorter PFS [10]. We further strengthened this correlation on a new and larger cohort of 73 untreated CLL patients (Supplementary Figure S1 and Supplementary Table S1). Since enlarged lymph nodes, as CLL major proliferation sites, are an important clinical indicator of progression, we next investigated BCR-mediated CXCR4 downregulation capacity in patients presenting or not with lymphadenopathy (Table 1). Interestingly, all but one patients, with cells unable to downregulate CXCR4 (14/15), were stage A patients and did not harbor lymphadenopathy. In contrast, among cases with cells able to downregulate CXCR4, a majority had tumor burden and shorter time to first treatment (41/57). In lymph nodes, CXCR5 and CD62L are major players in homing, trafficking and adhesion of lymphocytes and in their tissue egress [31-35]. Strikingly, sustained antigenic stimulation of CLL cell samples promoted a similar CXCR5 downregulation and CD62L membrane release, suggesting the presence of a BCR responsive subclone (Figure 1).
Table 1: Extent of BCR-mediated CXCR4 down-regulation is correlated to lymphadenopathy from CLL patients.
BCR-mediated CXCR4 down-regulation |
|||
Low capacity (n = 15) |
High capacity (n = 57) |
||
Lymph node enlargement |
Absence |
14 |
16 |
Presence |
1 |
41 |
|
CLL cases (n = 72) were divided based on their cellular percentage of CXCR4 down-regulation in response to BCR trigering: Low capacity = CXCR4 decrease ≤ 5% and High capacity = CXCR4 decrease > 5%. Statistical analysis of the absence or presence of lymph nodes in both groups demonstrated that high BCR-mediated CXCR4 down-regulation was strongly linked to lymphadenopathy in CLL patients (‘Yates’ continuity corrected Chi2 test, p < 0.001).
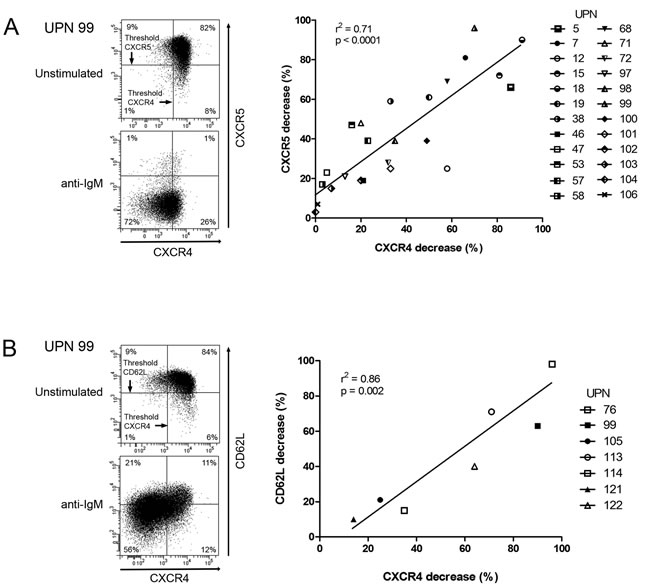
Figure 1: CXCR4, CXCR5 and CD62L are co-down-regulated in response to BCR triggering. CLL cells were stimulated for 24 hours with anti-IgM antibodies. In CD19+/CD5+ cells, CXCR4 and CXCR5 A., as well as CXCR4 and CD62L B. expressions were determined by flow cytometry (left panels) and percentages of CXCR4, CXCR5 and CD62L decreases were calculated and graphed (right panels).
In order to delineate the BCR effectors responsible for these modulations, we targeted early kinases of the pathway. As shown in Figure 2A and Supplementary Figure S2, inhibition of PI3Ks using a PI3K-δ specific inhibitor (Idelalisib) [14] or a pan-PI3K inhibitor (LY294002) [36], hindered BCR-dependent CXCR4 decrease in stimulated CLL cells. Importantly, treatment with the pan-PKC inhibitor Gö6983, which poorly inhibits PKD [37], or with the PKC inhibitor GF109203X that inhibits classical PKC isoforms, but not PKD [38], did not prevent CXCR4 decrease upon anti-IgM ligation [39]. Conversely, incubation of the cells with Gö6976, a selective inhibitor of classical PKC isoforms and purified PKD [40], blocked almost completely CXCR4 decrease (Figures 2B, 2C and Supplementary Figure S3). Then, dose response analysis with the potent and selective PKD inhibitor CID755673 [41], further assessed the functional involvement of PKDs in BCR-mediated CXCR4 decrease (Figure 2D left panel and Supplementary Figures S4 and S5A). Moreover, treatment with CID755673 blocked significantly BCR-mediated CXCR5 decrease (Figure 2D middle panel and Supplementary Figure S5B), demonstrating that PKDs also target CXCR5 clearance. In contrast, the membrane release of the CD62L selectin was not significantly altered by CID755673 treatment (Figure 2D right panel and Supplementary Figure S5C) but rather was inhibited by Gö6976 (Supplementary Figure S5D).
Altogether, these data demonstrate that membrane CXCR4/CXCR5 decrease in response to sustained BCR stimulation involves PKDs and that BCR-CD62L signaling cascade was rather controlled by PKCs.
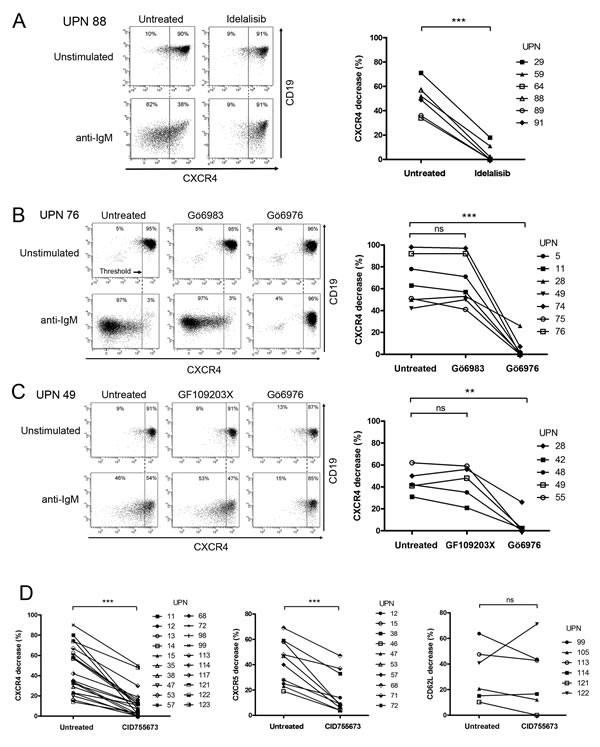
Figure 2: PI3K and PKD activities mediate BCR-dependent CXCR4/CXCR5 down-regulation but not CD62L release in CLL cells. CLL cells were incubated for 24 hours in the presence (anti-IgM) or not (Unstimulated) of immobilized anti-IgM antibodies and in the presence or not (Untreated) of A. Idelalisib (50 µM), B. C. Gö6983 (1 µM), Gö6976 (1 µM), GF109203X (1 µM) or D. CID755673 (50 µM). CXCR4 and CD19 expressions were determined by flow cytometry. A.-C. Left panels show a representative sample for each treatment. Right panels depict the percentage of CXCR4 decrease upon stimulation that was calculated (cf. material and methods) and graphed from various CLL samples. D. shows BCR-mediated CXCR4 (left panel), CXCR5 (middle panel) and CD62L (right panel) decreases with (CID755673) or without (Untreated) PKD inhibitor. ** p < 0.001; *** p < 0.0001; ns, not significant.
Involvement of a BCR/PI3K/PKD/CXCR4 phosphorylation axis
Then, we investigated expression levels of the 3 PKD isoforms in CLL cells. Quantitative PCR showed that both malignant and normal B cells expressed PKD2 and PKD3 but not PKD1 mRNA (Figure 3A). Western blotting analysis of 6 CLL cell lysates using anti-PKD1/2 (recognizing both PKD1 and PKD2), anti-PKD2 and anti-PKD3 antibodies confirmed variable expression of PKD2 and PKD3 proteins and absence of detectable PKD1 as compared to 293T cells expressing the 3 isoforms [42] (Figure 3B). Since extracellular stimuli result in rapid PKD phosphorylation [23], we evaluated their phosphorylation status using either anti-phospho-Ser744/748 antibody (phosphorylation site present in all PKD isoforms) or anti-phospho-Ser916 antibody (site present in PKD1 and PKD2 only). Upon anti-IgM stimulation, phospho-Ser744/748 and phospho-Ser916-PKD were detectable already at 30 minutes and lasted for at least 24 hours (Figure 4A and 4B). Pharmacological inhibition of PI3Ks using LY294002 or the PI3K-δ specific Idelalisib substantially reduced PKD phospho-Ser744/748 level (Supplementary Figure S6), suggesting that PI3K-δ was an upstream kinase in CLL cells. In contrast, CID755673 weakly inhibited anti-IgM-dependent phosphorylation of Ser744/748, but strongly interfered with phosphorylation on Ser916 [41] (Figure 4B) indicating the specific blocking of PKD self-phosphorylation. Furthermore, quantification of BCR-dependent phospho-Ser744/748 PKD increase in 6 CLL cell samples significantly correlated with the decrease of surface CXCR4 observed upon anti-IgM-triggering (Figure 4C). Collectively, BCR triggering allows PI3K-δ−dependent PKD phosphorylation with the subsequent down-regulation of membrane CXCR4.
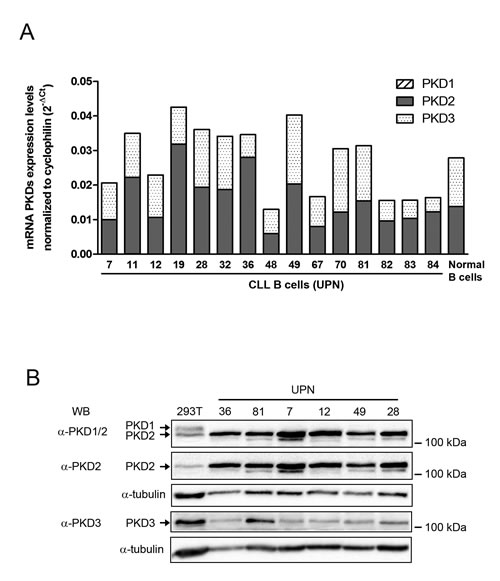
Figure 3: PKD2 and PKD3 are expressed in CLL cells. A. PKD1, 2 and 3 mRNA expression levels were quantified by RT-qPCR in CLL and normal B cells, normalized to cyclophilin and graphed. B. Protein extracts from 293T and CLL cell samples were separated on SDS-PAGE and analyzed by immuno-blotting with the indicated antibodies; α-tubulin expression was used as a loading control.
BCR-unresponsive CLL cells are still PMA-responsive
Based on the extent of CXCR4 downregulation in response to sustained BCR stimulation, our cohort of 73 CLL patients was divided into 2 groups with one characterized by a high response ( > 5%; n = 58) and another one with low response (≤ 5%, n = 15) (Supplementary Table S1). Analysis of the latters showed that phosphoSer744/748 PKD levels did not increase upon anti-IgM stimulation (Figure 5A); PKD or PKC inhibitors neither modified CXCR4 levels (Figure 5B and Supplementary Figure S7). All CLL samples, irrespective to their BCR-dependent CXCR4 down-regulation, remained responsive to PMA stimulation exhibiting increase of phospho-Ser744/748 and -Ser916 PKD levels, and strongly decreased their membrane CXCR4 (Figure 5C and Supplementary Figure S8), confirming their functional capacity.
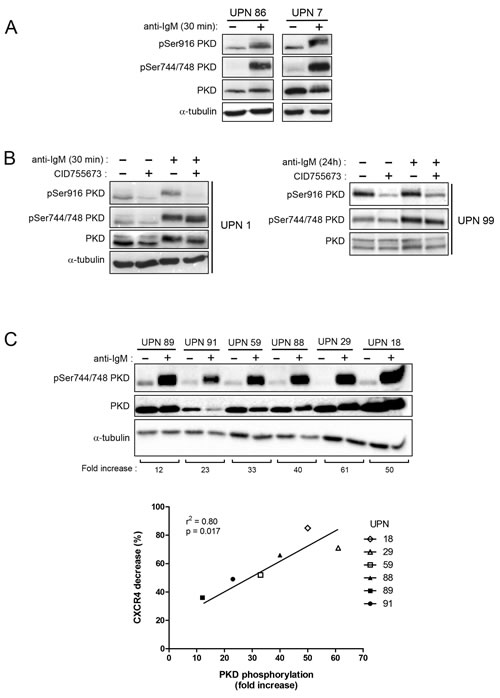
Figure 4: BCR engagement leads to specific PKD phosphorylation correlated with CXCR4 down-regulation in CLL responsive cells. A. B. CLL cell samples were stimulated (+) or not (-) with anti-IgM for 30 minutes or 24 hours in the presence (+) or not (-) of 50 µM CID755673. Phospho-Ser744/748, phospho-Ser916 and total PKD (PKD1/2) contents were analyzed by western blotting using the indicated antibodies. C. CLL cell samples were stimulated (+) or not (-) with anti-IgM for 30 minutes and fold increase of phospho-Ser744/748 PKD (normalized to total PKD) were calculated for each UPN and graphed relative to BCR-dependent CXCR4 decrease (Supplementary Table S1).
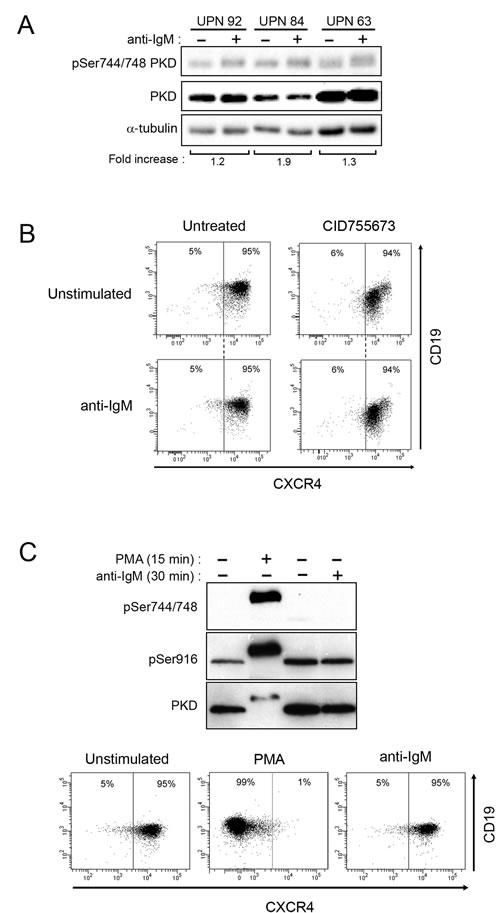
Figure 5: BCR-unresponsive cells respond to PMA treatment. A. CLL cells were stimulated and analyzed as described in Fig. 4C. B. Upon stimulation or not with anti-IgM and with or not 24 h-treatment with CID755673, CXCR4 and CD19 expressions were determined by flow cytometry (UPN 50). C. CLL cells (UPN 4) were stimulated (+) or not (-) with anti-IgM or PMA (200 nM) for the indicated time. Phosphorylated (Ser744/748 and Ser916) and total PKD (PKD1/2) were analysed by western blotting (top). After 24 hours, CXCR4 and CD19 expressions were determined by flow cytometry (bottom).
PKD is a BCR effector for CXCR4 phosphorylation and function
CXCR4 internalization upon CXCL12 binding involves phosphorylation of serine residues [20, 43]. Our in silico analysis revealed that Ser325 corresponds to a potential PKD phosphorylation consensus site (VSRGSS325); we investigated such a hypothesis.
Immunoblot analysis revealed a strong increase of phospho-Ser324/325 CXCR4 in stimulated CLL cells as compared to unstimulated ones, without any change of phospho-Ser338/339 or total CXCR4 levels in stimulated cells (Figure 6A). Moreover, treatment with CID755673 strongly impeded CXCR4 Ser324/325 phosphorylation (Figure 6B).
As expected [10], CXCR4 down-regulation in response to BCR engagement resulted in a significant reduction of the migration capacity of CLL cells towards CXCL12 in vitro (Figure 6C). Treatment with CID755673 restored the migration capacity to CXCL12 gradient (Figure 6C).
Taken together, our results argue for the phosphorylation of Ser325 upon BCR engagement via PKD leading to CXCR4 internalization and reduced mobility of the cells towards CXCL12.
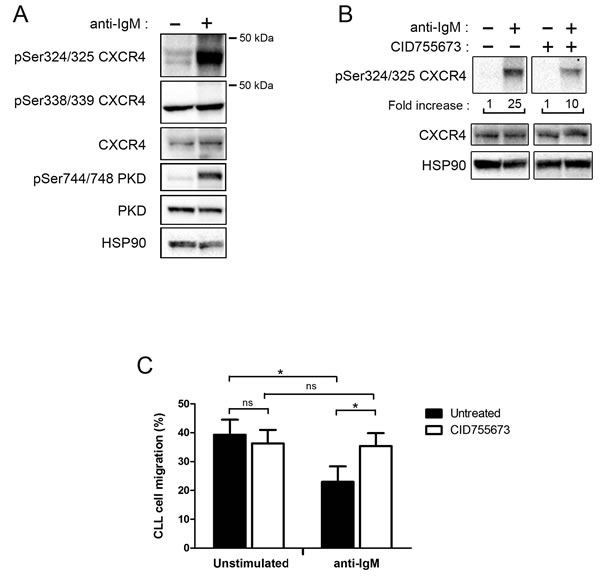
Figure 6: BCR engagement allows CXCR4 phosphorylation and reduces CLL migration toward CXCL12 via PKD. A. CLL cells (UPN 96) were stimulated (+) or not (-) with anti-IgM antibodies for 30 minutes. Phospho-Ser324/325-, phospho-Ser338/339- and total-CXCR4, as well as p-Ser744/748- and total-PKD (PKD1/2) expression levels were analysed by western blotting using the indicated antibodies; anti-HSP90 antibody showed equal amount of protein in both conditions. B. CLL cells (UPN 88) were stimulated as described in A. and incubated in the presence (+) or not (-) of 50 µM CID755673. Fold increases of phospho-Ser324/325 CXCR4 are indicated below (both panels are issued from the same western blot). C. CLL cells were stimulated or not with anti-IgM and treated in the presence or not of 50 µM CID755673. After 24 hours, cells were subjected to a migration assay, counted by flow cytometry and graphed. Values represent the mean ± SEM of 4 independent experiments. * p < 0.05
Extent of BCR-mediated CXCR4 is linked to tumor burden
Finally, we challenged the BCR-induced CXCR4 downregulation in CLL cells with respect to their IGHV mutational status (Figure 7). According to their clinical progression, almost all cases (35/36) harboring unmutated IGHV exhibited a high BCR-mediated CXCR4 downregulation cell profile (mean ± SEM of CXCR4 decrease = 44.9% ± 20.3). In contrast, among the 36 IGHV mutated cases, two groups were identified, based on their cellular ability to downregulate CXCR4 in response to BCR stimulation. The first group including 14 IGHV mutated cases with low or none CXCR4 downregulation (mean ± SEM of CXCR4 decrease = 1.4% ± 2.0) remained in stage A. None of these patients had any lymph node progression in a median follow up of 8.4 years [Q1:4.4-Q3:9.9]. The second group, including 22 IGHV mutated cases, showed cells with a strong CXCR4 down-regulation (mean ± SEM of CXCR4 decrease = 49.2% ± 19.0). Among those, 14 patients (64%) developed clinical lymphadenopathy in a median follow up of 9.2 years [Q1:6.4-Q3:10.7]. These data indicated that CXCR4 downregulation reflects BCR signaling capacity, irrespective to the IGHV mutational status.
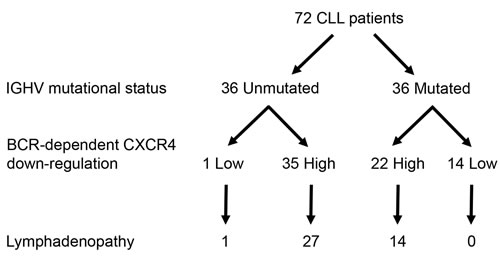
Figure 7: Extent of BCR-mediated CXCR4 down-regulation is related to lymph node enlargement from IGHV mutated CLL patients. According to their IGHV mutational status (Unmutated and Mutated) and then on their BCR-mediated CXCR4 down-regulation (Low (≤5%) and High ( > 5%)), the 72 CLL samples were divided into 4 sub-groups. Indicated numerals represent the number of patients in each sub-group. (‘Yates’ continuity corrected χ2 test: p < 0.001).
DISCUSSION
We have previously shown that ex-vivo BCR engagement leads to a decrease of membrane CXCR4 expression in CLL cells from patients with unfavourable prognostic factors and at risk of disease progression [10]. In the present study, we further emphasized the BCR-dependent down-regulation of CXCR4 that was linked to tumor burden in both IGHV mutated and unmutated patients. We also deciphered the phosphorylation cascade leading to CXCR4 phosphorylation and to its subsequent endocytosis. This cascade consisted on a novel BCR-dependent pathway in which PI3K-δ phosphorylates PKD, which in turn phosphorylates CXCR4 at Ser324/325. We provided evidence that interrupting this pathway through inhibition of PKD rescued chemokine-driven cell migration that might contribute to CLL cell egress from lymph nodes in vivo.
PKD are important effectors in response to various stimuli in normal and pathological models [22-24]. Our data demonstrate that CLL cells co-express PKD2 and PKD3, but not PKD1. This pattern of expression is compatible to those described by immunohistochemistry in reactive lymphoid tissues and in neoplastic cells of lymphoid origin [30].
Both PKC-dependent and -independent pathways have been described in PKD activation [27, 28, 44-46]. In our study, we evidenced the upstream functional implication of PI3K-δ, rather than PKCs, in PKD-dependent CXCR4 and CXCR5 endocytoses upon antigen stimulation. Targeting PI3K-δ, which is abundantly expressed and constitutively activated in CLL cells [47], with Idelalisib has shown extensive efficacy for relapsed and refractory CLL with the drastic reduction of lymphadenopathy [14-16]. Besides uncovering a new regulation downstream of PI3K addressing CXCR4, we also showed that PKDs are involved in the endocytosis of an additional chemokine receptor, CXCR5. In contrast, shedding of CD62L rather implicates PKC in agreement with the described PKC/secretase/CD62L axis [48]. These data indicate that PKDs act as common signalling intermediates in BCR-dependent modulation of both CXCR4 and CXCR5 in CLL cells. It will be interesting to address whether these BCR signalling effectors are also involved in the regulation of several seven-transmembrane G protein-coupled receptors, including S1PR1 that controls lymphocytes egress from lymph nodes [49]. Interestingly, PKD involvement in BCR-mediated CXCR4/CXCR5 downregulation observed in our CLL samples appears to be different from the CXCL12/CXCR4 signaling pathway, which leads to CXCR4, but not CXCR5 and CD62L downregulation in the mouse model of TCL-1 CLL-like disease [50]. Based on the identification of CXCR4 as a partner of the BCR signalosome [51], a concomitant internalization between cell surface BCR, CXCR4 and CXCR5 upon antigen stimulation would not be surprising according to antigen targeted-regulation of these chemokine receptors.
In CLL cells, anti-IgM stimulation resulted in phosphorylation of Ser744 and/or Ser748 residues positioned in the activation loop of the PKD2 and PKD3 kinase domains and of Ser916 residue, a site of auto-phosphorylation present in the PKD2 isoform only [22-24, 28]. In absence of PKD1, CXCR4 down-regulation blocked by the CID755673, which interferes with PKD autophosphorylation at Ser916, pointed out phospho-PKD2 as the effector responsible for CXCR4 internalization.
Our experiments also indicate that in BCR-unresponsive cells, PKD is nevertheless functional. Whereas BCR engagement leads to only a weak phosphorylation of PKD and low or absence of CXCR4 internalization, treatment with the DAG analogue PMA overpassed this initial threshold response. This suggests that BCR-unresponsive CLL cells display a functional deficiency at the level of the BCR signalosome [11] rather than a defect of downstream effectors such as PKD.
Our in silico analysis identified CXCR4 Ser325 and CXCR5 Thr338 as consensus sites for PKD phosphorylation (Figure 8). In line with this analysis, we demonstrated that CXCR4 Ser324/325 is a phosphorylated target of PKD upon BCR triggering in CLL cells. To our knowledge, no genetic variant of this residue has been described in CLL cells [52, 53]. A previous study described high levels of phospho-Ser339 CXCR4 in CLL cells compared to normal cells [54]. Our results not only confirmed this basal phosphorylation but also showed the absence of further activation upon BCR triggering. This suggests that phospho-Ser338/339 CXCR4 is not a target of BCR/PKD pathway in CLL cells but more probably a target of Btk/PKC upon CXCL12- or EGF-stimulations [50, 55]. Thus, our results in line with others propose that depending on the stimulus, different kinases are required for an appropriate CXCR4 regulation leading to alternative phosphorylation patterns [19-21, 43, 50, 54, 56, 57].
Based on the crucial roles of CXCR4 and CXCR5 in CLL cells trafficking within the lymph nodes [16, 32, 50] and given that CXCR4 is associated with poor clinical outcome in CLL patients [10, 58, 59], we explored the clinical relevance of BCR-PKD-CXCR4/CXCR5 signaling pathway. Nearly all unmutated IGHV cases (35/36) exhibited responsiveness to BCR engagement in terms of CXCR4 downregulation. This correlation is not surprising regarding the cellular capacity to respond to BCR triggering and is consistent with disease progression as well as ultimately constant need of treatment in patients with unmutated IGHV [60, 61]. In contrast, in IGHV mutated cases, ability to down-regulate CXCR4 in response to BCR ligation identified two subgroups. In the first subgroup, 14 patients showed no or low CXCR4 decrease and none of them exhibited lymph node enlargement with a median follow up of 8.4 years; only 4 patients needed treatment and, for other reason than nodal progression. In the other subgroup of 22 IGHV mutated cases, with a stronger BCR/CXCR4 downregulation profile, a majority (64%) had tumor progression with lymphadenopathy, and 7 of them required treatment for nodal progression with a median follow up of 9.2 years. Altogether, our proposed model of BCR-mediated CXCR4 downregulation is in agreement with in vivo deuterium glucose labeling experiments showing that CXCR4low peripheral cell fraction is representative of the proliferative pool of cells [62].
Our data support that the intensity of CXCR4 downregulation reflects the proportion of the BCR responsive subclone in a given patient. Indeed, we have shown previously that CXCR4 and CD62L downregulation occurred concomittanly upon BCR triggering [10] and we currently demonstrated that intensity of CXCR4 and CXCR5 downregulation took place in a similar subset of cells. PI3K-δ inhibitors allow CLL cells to exit from lymph nodes in vivo and represent an effective therapy. Interestingly, PKD inhibitors were shown to block pancreatic cancer growth in a xenograft mouse model [63]. Given the PKD implication in BCR-dependent CXCR4/CXCR5 internalization and CLL cell migration, targeting PKD could be an interesting alternative therapeutic option that would circumvent the PI3K-δ inhibitors side effects that target ΑΚΤ/mTor signaling pathways [64].
In conclusion, we identified a new PI3K-δ/PKD signaling pathway that leads to CXCR4/CXCR5 downregulation and is activated upon BCR engagement in CLL progressive cases (Figure 8). This work highlights the importance of BCR responsiveness towards CXCR4 downregulation, irrespective of the IGHV mutational status, on progression of tumor burden.
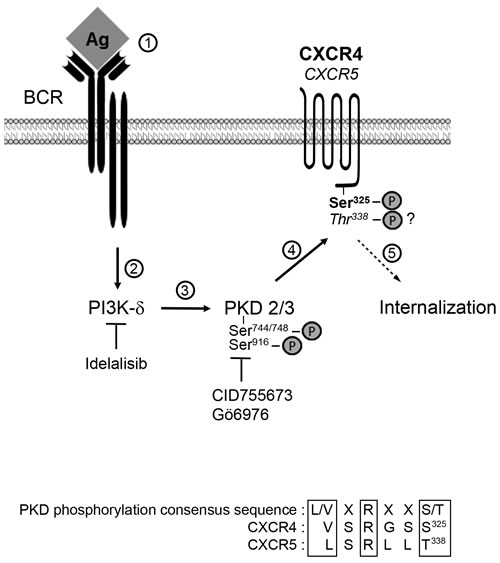
Figure 8: Cascade of events upon BCR trigerring in cases of lymphadenopathy. Antigen binding to BCR (step 1) allows activation of PI3K-δ (step 2) and the subsequent increased phosphorylation of PKD2/3 on Ser744/748 and Ser916 (step 3). Activated PKD2/3 in turn phosphorylates CXCR4 Ser324/325 (and possibly CXCR5 Thr338) leading to GCPR internalization (step 5). Inhibitors of PI3K-δ and PKD2/3 are indicated. Consensus PKD phosphorylation sites altogether with the C-terminal sequence of CXCR4 and CXCR5 are aligned.
MATERIALS AND METHODS
CLL cell isolation and culture
CLL blood samples were obtained from untreated patients, after informed consent and validation by the local research ethics committee from the Avicenne Hospital (Bobigny, France), in accordance with the Declaration of Helsinki. CLL B cells were purified by negative selection using RosetteSep Human B Cell Enrichment Cocktail (StemCell Technologies, Grenoble, France). Purity (98,71% ± 1,41) was assessed as previously described [7]. CLL B cells were cultured fresh or after viable thawing in RPMI 1640 supplemented with 10 % FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine (PAA, Les Mureaux, France). For BCR stimulation, CLL B cells (4x106 cells/well/12-well plate) were incubated with immobilized rabbit anti-IgM antibody (10 μg/mL; Jackson Immunoresearch, Montlucon, France) and incubated or not with Gö6976, Gö6983 (Calbiochem, Saint-Quentin-en-Yvellines, France), GF109203X, LY294002 (Sigma-Aldrich, Saint-Quentin-Fallavier, France), CAL101 (Selleckchem, Souffelweyersheim, France), CID755673 (Tocris Bioscience, Lille, France) or phorbol 12-myristate 13-acetate (PMA) (Cell Signaling Technology, Saint-Quentin-Fallavier, France). Human 293T and HepG2 cell lines were maintained in DMEM supplemented with 10 % FBS.
Flow cytometry analysis
CD5, CD19 and CXCR4 membrane expression levels were analyzed by flow cytometry (FACS-CANTO II; Becton Dickinson) after labeling with the indicated conjugated monoclonal antibodies (mAbs): PE-Cy7-anti-CD5, APC-Cy7-anti-CD19, APC-anti-CXCR4, PE-CXCR5 and FITC-CD62L or the respective control IgG isotype mAbs (BD Biosciences, Pont-de-Claix, France). Cell viability and plasma membrane CXCR4, CXCR5 and CD62L expression levels were determined in CD19+/CD5+ CLL cells. Data acquisition and analysis were performed using BD FACSDiva software. A threshold was arbitrarily set up case-by-case on untreated cells to include at least 90 % of CXCR4high or CXCR5high or CD62Lhigh cells. BCR- and PMA-dependent CXCR4 or CXCR5 or CD62L membrane decreases were calculated as follows:
100 - ((% CXCR4high/CXCR5high/CD62Lhigh after IgM stimulation ×100) / (% CXCR4high/CXCR5high/CD62Lhigh before IgM stimulation)).
Immunoblot analysis
After BCR stimulation or PMA treatment, total B cell contents were extracted using 1% Nonidet P-40 lysis buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5 mM EDTA, supplemented with proteases inhibitors (Sigma-Aldrich). Proteins were separated on SDS-PAGE and analysed by western blotting with the appropriate antibodies: anti-phospho-Erk1/2, anti-PKD1/2, anti-phospho-Ser744/748 PKD, anti-phospho- Ser916 PKD, anti-HSP90 (Cell Signaling Technology), anti-PKD2 (Millipore, Molsheim, France), anti-PKD3 (Bethyl Laboratories Inc, Souffelweyersheim, France), anti-CXCR4 (Abcam, Paris, France), anti-phospho-Ser339 CXCR4 (Abcam), anti-phospho-Ser324/325 CXCR4 (ECM Biosciences, Souffelweyersheim, France) and anti-α-Tubulin (Sigma-Aldrich). Detection was achieved using chemi-luminescence (ECL, GE Heathcare, Velizy-Villacoublay, France) and visualized using the ChemiDoc MP Imaging System (BioRad, Marnes-la-Coquette, France).
RNA isolation, reverse transcription and Q-PCR
Total RNAs were isolated from cells using Trizol reagent (Invitrogen, Saint Aubin, France), purified (QIAGEN, Courtaboeuf, France) and quantified by spectrophotometry. Total RNAs (1 µg) were retro-transcribed using MMLV reverse transcriptase. Quantitative real-time PCR (Q-PCR) was performed using a 7500 SDS Thermal Cycler (Applied Biosystems, Saint Aubin, France). Complementary DNAs, 3.2 μM gene-specific sense and anti-sense primers, as well as specific FAM-MGB probes (Applied Biosystems) (Supplementary Table S2) and PCR Master Mix were mixed in a 25 μL volume. The reactions were performed as follow: 40 cycles at 98°C for 15 seconds and 58°C for 60 seconds. Each experiment was repeated in duplicates. Cyclophilin was used as internal gene control. Relative quantification of each PKD gene expression was determined using the ∆Ct method and values are expressed as 2-∆Ct.
Chemotaxis assay
Chemotaxis assays were performed as previously described [10]. Briefly, CLL cells were stimulated or not with coated anti-IgM for 24 hours and treated or not with CID755673. For each condition, cells were transferred into an upper chamber of a Transwell culture insert (Corning Costar, Illkirch, France), which was moved into a well containing RPMI supplemented with CXCL12 (100 ng/mL). After 8 hours at 37°C, non-migrated cells in the upper chambers and transmigrated cells in lower chambers were collected and counted. All assays were performed in duplicate.
Data analysis and statistics
Data and statistical analyses were performed using the GraphPad program (Prism Version 6, France).
ACKNOWLEDGMENTS
This study was supported by INSERM, University Paris 13, Labex Inflamex n°ANR11 IDEX00502 and grant of Fondation de France. SSG and MQ were the recipients of Société Française d’Hématologie and Ligue Nationale Contre le Cancer fellowships, respectively. We acknowledge Pr P.O. Schischmanoff for designing specific probes for Q-PCR analyses of PKD isoforms and Dr L. Izzi (Montréal, QC) for critical reading of the manuscript.
CONFLICTs OF INTEREST
SSG, MQ, MB, SLC, VLa, LG, NVB, CLR and DL declare no conflict of interest concerning this article. VLé received honoraria from Roche, Jannsen and Gilead. FAC received honoraria from Roche, Jannsen, Gilead and Karyopharm.
GRANT SUPPORT
INSERM, Université Paris 13, Fondation de France, Société Française d’Hématologie, Ligue Nationale Contre le Cancer.
REFERENCES
1. Caligaris-Cappio F and Ferrarini M. B cells and their fate in health and disease. Immunol Today 1996; 17:206-208.
2. Chiorazzi N, Rai KR and Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005; 352:804-815.
3. Dighiero G, Travade P, Chevret S, Fenaux P, Chastang C and Binet JL. B-cell chronic lymphocytic leukemia: present status and future directions. French Cooperative Group on CLL. Blood 1991; 78:1901-1914.
4. Hamblin TJ and Oscier DG. Chronic lymphocytic leukaemia: the nature of the leukaemic cell. Blood Rev 1997; 11:119-128.
5. Letestu R, Levy V, Eclache V, Baran-Marszak F, Vaur D, Naguib D, Schischmanoff O, Katsahian S, Nguyen-Khac F, Davi F, Merle-Beral H, Troussard X and Ajchenbaum-Cymbalista F. Prognosis of Binet stage A chronic lymphocytic leukemia patients: the strength of routine parameters. Blood 2010; 116:4588-4590.
6. Hillmen P. Using the biology of chronic lymphocytic leukemia to choose treatment. Hematology Am Soc Hematol Educ Program 2011; 2011:104-109.
7. Deglesne PA, Chevallier N, Letestu R, Baran-Marszak F, Beitar T, Salanoubat C, Sanhes L, Nataf J, Roger C, Varin-Blank N and Ajchenbaum-Cymbalista F. Survival response to B-cell receptor ligation is restricted to progressive chronic lymphocytic leukemia cells irrespective of Zap70 expression. Cancer Res 2006; 66:7158-7166.
8. Efremov DG, Gobessi S and Longo PG. Signaling pathways activated by antigen-receptor engagement in chronic lymphocytic leukemia B-cells. Autoimmun Rev 2007; 7:102-108.
9. Pleyer L, Egle A, Hartmann TN and Greil R. Molecular and cellular mechanisms of CLL: novel therapeutic approaches. Nat Rev Clin Oncol 2009; 6:405-418.
10. Vlad A, Deglesne PA, Letestu R, Saint-Georges S, Chevallier N, Baran-Marszak F, Varin-Blank N, Ajchenbaum-Cymbalista F and Ledoux D. Down-regulation of CXCR4 and CD62L in chronic lymphocytic leukemia cells is triggered by B-cell receptor ligation and associated with progressive disease. Cancer Res 2009; 69:6387-6395.
11. Le Roy C, Deglesne PA, Chevallier N, Beitar T, Eclache V, Quettier M, Boubaya M, Letestu R, Levy V, Ajchenbaum-Cymbalista F and Varin-Blank N. The degree of BCR and NFAT activation predicts clinical outcomes in chronic lymphocytic leukemia. Blood 2012; 120:356-365.
12. Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, Murphy EJ, Koduru P, Ferrarini M, Zupo S, Cutrona G, Damle RN, Wasil T et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest 2005; 115:755-764.
13. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, Gibellini F, Njuguna N, Lee E, Stennett L, Raghavachari N, Liu P, McCoy JP et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011; 117:563-574.
14. Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011; 117:591-594.
15. Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, Spurgeon SE, Kahl BS, Bello C, Webb HK, Johnson DM, Peterman S, Li D et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014; 123:3390-3397.
16. Burger JA and Gribben JG. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: Insight into disease biology and new targeted therapies. Semin Cancer Biol 2014; 24:71-81.
17. Niedermeier M, Hennessy BT, Knight ZA, Henneberg M, Hu J, Kurtova AV, Wierda WG, Keating MJ, Shokat KM and Burger JA. Isoform-selective phosphoinositide 3’-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood 2009; 113:5549-5557.
18. Patrussi L, Capitani N, Martini V, Pizzi M, Trimarco V, Frezzato F, Marino F, Semenzato G, Trentin L and Baldari CT. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res 2015; 75:4153-4163.
19. Signoret N, Rosenkilde MM, Klasse PJ, Schwartz TW, Malim MH, Hoxie JA and Marsh M. Differential regulation of CXCR4 and CCR5 endocytosis. J Cell Sci 1998; 111 ( Pt 18):2819-2830.
20. Orsini MJ, Parent JL, Mundell SJ, Marchese A and Benovic JL. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J Biol Chem 1999; 274:31076-31086.
21. Marchese A. Endocytic trafficking of chemokine receptors. Curr Opin Cell Biol 2014; 27:72-77.
22. Rozengurt E, Rey O and Waldron RT. Protein kinase D signaling. J Biol Chem 2005; 280:13205-13208.
23. Fu Y and Rubin CS. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. EMBO Rep 2011; 12:785-796.
24. Matthews SA, Navarro MN, Sinclair LV, Emslie E, Feijoo-Carnero C and Cantrell DA. Unique functions for protein kinase D1 and protein kinase D2 in mammalian cells. Biochem J 2010; 432:153-163.
25. Nishikawa K, Toker A, Johannes FJ, Songyang Z and Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem 1997; 272:952-960.
26. Jacamo R, Sinnett-Smith J, Rey O, Waldron RT and Rozengurt E. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J Biol Chem 2008; 283:12877-12887.
27. Guo J, Gertsberg Z, Ozgen N, Sabri A and Steinberg SF. Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J Biol Chem 2011; 286:6500-6509.
28. Matthews SA, Rozengurt E and Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem 1999; 274:26543-26549.
29. Steinberg SF. Regulation of protein kinase D1 activity. Mol Pharmacol 2012; 81:284-291.
30. Kovalevska LM, Yurchenko OV, Shlapatska LM, Berdova GG, Mikhalap SV, Van Lint J and Sidorenko SP. Immunohistochemical studies of protein kinase D (PKD) 2 expression in malignant human lymphomas. Exp Oncol 2006; 28:225-230.
31. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N and Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol 2004; 5:943-952.
32. Heinig K, Gatjen M, Grau M, Stache V, Anagnostopoulos I, Gerlach K, Niesner RA, Cseresnyes Z, Hauser AE, Lenz P, Hehlgans T, Brink R, Westermann J et al. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia B-cell activation and proliferation. Cancer Discov 2014; 4:1448-1465.
33. Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R and Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 2012; 120:2240-2248.
34. Lafouresse F, Bellard E, Laurent C, Moussion C, Fournie JJ, Ysebaert L and Girard JP. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood 2015; 126:1336-1345.
35. Irjala H, Johansson EL, Grenman R, Alanen K, Salmi M and Jalkanen S. Mannose receptor is a novel ligand for L-selectin and mediates lymphocyte binding to lymphatic endothelium. J Exp Med 2001; 194:1033-1042.
36. Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF and Waterfield MD. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J 2007; 404:15-21.
37. Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D and Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem 1993; 268:9194-9197.
38. Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F and et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 1991; 266:15771-15781.
39. Faubert Kaplan BL and Kaminski NE. Cannabinoids inhibit the activation of ERK MAPK in PMA/Io-stimulated mouse splenocytes. Int Immunopharmacol 2003; 3:1503-1510.
40. Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ and Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett 1996; 392:77-80.
41. Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS and Wang QJ. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem 2008; 283:33516-33526.
42. Azoitei N, Pusapati GV, Kleger A, Moller P, Kufer R, Genze F, Wagner M, van Lint J, Carmeliet P, Adler G and Seufferlein T. Protein kinase D2 is a crucial regulator of tumour cell-endothelial cell communication in gastrointestinal tumours. Gut 2010; 59:1316-1330.
43. Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, Ali H and Snyderman R. Regulation of human chemokine receptors CXCR4. Role of phosphorylation in desensitization and internalization. J Biol Chem 1997; 272:28726-28731.
44. Pracht C, Minguet S, Leitges M, Reth M and Huber M. Association of protein kinase C-delta with the B cell antigen receptor complex. Cell Signal 2007; 19:715-722.
45. Matthews SA, Dayalu R, Thompson LJ and Scharenberg AM. Regulation of protein kinase Cnu by the B-cell antigen receptor. J Biol Chem 2003; 278:9086-9091.
46. Sinnett-Smith J, Jacamo R, Kui R, Wang YM, Young SH, Rey O, Waldron RT and Rozengurt E. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J Biol Chem 2009; 284:13434-13445.
47. Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ, Giese NA, Zhang X, Wei L et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010; 116:2078-2088.
48. Alexander SR, Kishimoto TK and Walcheck B. Effects of selective protein kinase C inhibitors on the proteolytic down-regulation of L-selectin from chemoattractant-activated neutrophils. J Leukoc Biol 2000; 67:415-422.
49. Borge M, Remes Lenicov F, Nannini PR, de los Rios Alicandu MM, Podaza E, Ceballos A, Fernandez Grecco H, Cabrejo M, Bezares RF, Morande PE, Oppezzo P, Giordano M and Gamberale R. The expression of sphingosine-1 phosphate receptor-1 in chronic lymphocytic leukemia cells is impaired by tumor microenvironmental signals and enhanced by piceatannol and R406. J Immunol 2014; 193:3165-3174.
50. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry BA, Burger JA, Rai KR and Chiorazzi N. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia 2015.
51. Satpathy S, Wagner SA, Beli P, Gupta R, Kristiansen TA, Malinova D, Francavilla C, Tolar P, Bishop GA, Hostager BS and Choudhary C. Systems-wide analysis of BCR signalosomes and downstream phosphorylation and ubiquitylation. Mol Syst Biol 2015; 11:810.
52. Crowther-Swanepoel D, Qureshi M, Dyer MJ, Matutes E, Dearden C, Catovsky D and Houlston RS. Genetic variation in CXCR4 and risk of chronic lymphocytic leukemia. Blood 2009; 114:4843-4846.
53. Acunzo M, Romano G, Wernicke D, Balatti V, Rassenti LZ, dell’Aquila M, Kipps TJ, Pekarsky Y and Croce CM. Translocation t(2;11) in CLL cells results in CXCR4/MAML2 fusion oncogene. Blood 2014; 124:259-262.
54. Decker S, Finter J, Forde AJ, Kissel S, Schwaller J, Mack TS, Kuhn A, Gray N, Follo M, Jumaa H, Burger M, Zirlik K, Pfeifer D et al. PIM kinases are essential for chronic lymphocytic leukemia cell survival (PIM2/3) and CXCR4-mediated microenvironmental interactions (PIM1). Mol Cancer Ther 2014; 13:1231-1245.
55. Woerner BM, Warrington NM, Kung AL, Perry A and Rubin JB. Widespread CXCR4 activation in astrocytomas revealed by phospho-CXCR4-specific antibodies. Cancer Res 2005; 65:11392-11399.
56. Guinamard R, Signoret N, Ishiai M, Marsh M, Kurosaki T and Ravetch JV. B cell antigen receptor engagement inhibits stromal cell-derived factor (SDF)-1alpha chemotaxis and promotes protein kinase C (PKC)-induced internalization of CXCR4. J Exp Med 1999; 189:1461-1466.
57. Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M and Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem 2010; 285:7805-7817.
58. Majid A, Lin TT, Best G, Fishlock K, Hewamana S, Pratt G, Yallop D, Buggins AG, Wagner S, Kennedy BJ, Miall F, Hills R, Devereux S et al. CD49d is an independent prognostic marker that is associated with CXCR4 expression in CLL. Leuk Res 2011; 35:750-756.
59. Pepper C, Buggins AG, Jones CH, Walsby EJ, Forconi F, Pratt G, Devereux S, Stevenson FK and Fegan C. Phenotypic heterogeneity in IGHV-mutated CLL patients has prognostic impact and identifies a subset with increased sensitivity to BTK and PI3Kdelta inhibition. Leukemia 2015; 29:744-747.
60. Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F and Packham G. Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood 2003; 101:1087-1093.
61. Caligaris-Cappio F and Ghia P. Novel insights in chronic lymphocytic leukemia: are we getting closer to understanding the pathogenesis of the disease? J Clin Oncol 2008; 26:4497-4503.
62. Cuthill K, Zhang Y, Buggins A, Coulter E, Patten PE, Macallan D and Devereux S. In-Vivo labelling studies in patients with chronic lymphocytic leukemia studies demonstrate the existence of apparently distinct subpopulations that differ in phenotype and proliferative capacity. 57th ASH Annual Meeting 2015; abstract #615.
63. Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherzadeh A, Foxton C et al. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther 2010; 9:1136-1146.
64. Coutre SE, Barrientos JC, Brown JR, de Vos S, Furman RR, Keating MJ, Li D, O’Brien SM, Pagel JM, Poleski MH, Sharman JP, Yao NS and Zelenetz AD. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma 2015; 56:2779-2786.