
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the fourth most common cause of cancer-related deaths in developed countries [1]. Surgical resection remains the best chance of long-term survival for patients with apparently localized PDAC. However, despite most advanced surgical techniques and antineoplastic combination treatments for advanced PDAC, the overall 5-year survival still remains low at 6% [2]. PDAC is expected to become the second-leading cause of cancer death by 2030, due to an increasing incidence and currently limited options to improve overall survival [3]. Less than 20% of PDAC patients are amenable to a curative surgical resection due to late clinical presentation, and early and aggressive local and metastatic progression [4]. Furthermore, even among these patients, postoperative recurrence remains commonplace [5, 6], leading to a 5-year survival at less than 20% [7]. Therefore, in recent years much attention has been directed toward improving systemic treatment options for PDAC. Gemcitabine, a nucleoside analog, has limited clinical benefits but it remained a standard treatment for locally advanced and metastatic PDAC since 1997 after producing a response rate of 5% and a median survival of 5.7 months [8]. FOLFIRINOX, a combination of chemotherapy agents (oxaliplatin, irinotecan, 5-FU/leucovorin), nearly doubled the median survival of PDAC patients (11.1 months compared with 6.8 months in the gemcitabine group), but this regimen is highly toxic with serious side effects [9]. Nab-paclitaxel (NPT), a water-soluble albumin-bound paclitaxel, has recently shown efficacy against advanced PDAC [10, 11], and nab-paclitaxel in combination with gemcitabine demonstrated 8.5 months median survival compared with 6.7 months after gemcitabine alone [12]. Considering these trial-based moderate improvements in PDAC prognosis, there is an urgent requirement for novel therapeutic strategies to improve overall patient survival.
PDAC progression is a multifactorial process, with overexpression of several growth factors and their receptors including epidermal growth factor (EGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and insulin-like growth factor (IGF), being associated with the highly aggressive nature of PDAC [13, 14]. The IGF signaling axis mainly consists of ligands IGF-1, IGF-2, insulin and IGF-1 receptor (IGF-1R), insulin receptor (IR) and hybrid receptors. IGF-1R is the primary mitogenic receptor of the IGF system that is overexpressed and has increased tyrosine kinase activity in several cancer types including PDAC [14-16]. Most of the tumorigenic effects of IGF signaling involves the binding of two ligands, IGF-1 and IGF-2, to IGF-1R, leading to the activation of several downstream signal transduction pathways including the phosphatidylinositol 3’-kinase (PI3K)/AKT and mitogen-activated protein kinase (MAPK) pathways [17]. Aberrant IGF signaling stimulates proliferation, differentiation, angiogenesis, metastasis, survival, and drug resistance in many cancers [18, 19], establishing this pathway as a generally promising therapeutic target. The IGF-1R pathway inhibition has been the subject of intensive anti-cancer drug discovery efforts and currently there are more than 30 active clinical trials evaluating anti-IGF-1R targeting agents, either alone or in various combinations (www.clinicaltrial.gov).
Three main strategies were employed to target the IGF pathway: monoclonal antibodies against IGF-1R, monoclonal antibodies against IGF-1R ligands (IGF-1 and IGF-2), and IGF-1R tyrosine kinase inhibitors. IGF-1R shares significant structural homology with IR, and targeting IGF pathway with IGF-1R inhibitor antibodies is sensible as it only blocks IGF-1R induced mitogenic signaling but is not affecting IR signaling, which could lead to dysregulation of glucose homeostasis [20, 21]. IGF-1R antibodies demonstrated clinical activity in phase 2 studies in small number of patients with select tumor types including Ewing sarcoma, thymoma and thymic carcinoma [22-24]. However, many clinical trials with IGF-1R inhibitors failed to show any significant clinical benefit including hepatocellular carcinoma [25], non-small cell lung cancer [26] and breast cancer [27]. In pancreatic cancer, the combination of gemcitabine and ganitumab, a monoclonal antibody against IGF-1R, showed a significant clinical response in a phase 2 randomized trial [28], but unfortunately this combination did not show any clinical benefit in phase 3 evaluation [29]. Recently, a clinical study demonstrated a correlation between IGF-1R signaling and tumor aggressiveness in PDAC patients [15], and increased exposure to ganitumab was found to be associated with improved overall survival and progression free survival in metastatic PDAC [30]. These findings demonstrate that IGF-1R remains a valid target for PDAC therapy.
Tyrosine kinase inhibitors can more indiscriminately regulate the kinase domain activity of all IGF system receptors as their primary sequence share 84% homology in the kinase domain with near complete conservation in the ATP binding pocket [31]. BMS-754807 (Supplemental Figure S1) is a potent and reversible inhibitor of IGF-1R/IR (IC50 1.8 nM/1.7 nM), which also has more limited activity toward other kinases including Met, Aurora A/B, TrkA/B, Ron, Flt3, Lck, MK2, PKA and PKC [32, 33]. BMS-754807 has shown antitumor activity in a broad range of tumor types and it also enhanced antitumor response of other agents [33-36]. Recently, predictive biomarkers, including overexpression of IGF-1R, and KRAS and BRAF mutation status have been delineated for effectiveness of BMS-754807 [37]. In the present study, augmentation of response to nab-paclitaxel was evaluated by inhibition of IGF signaling in robust preclinical models of PDAC in an attempt to identify the efficacy of a novel therapeutic strategy.
RESULTS
Nab-paclitaxel and the IGF signaling inhibitor reduce the growth of PDAC xenografts
In the human subcutaneous PDAC xenografts, treatment of tumor-bearing mice with nab-paclitaxel and BMS-754807, an inhibitor of IGF signaling, caused significant antitumor effects as mono- and combination therapy of these agents demonstrated additive effects. In subcutaneous xenografts using AsPC-1 cells, nab-paclitaxel and BMS-754807 both caused a reduction in tumor growth while a combination of these two agents had additive effects (Figure 1A). Average net tumor growth, calculated by subtracting tumor volume on first therapy day from that on the last day, in different therapy groups was 248.3 mm3 in controls, 42.4 mm3 after nab-paclitaxel (p = 0.002), 93.3 mm3 after BMS-754807 (p = 0.01) and 1.9 mm3 after nab-paclitaxel plus BMS-754807 (p = 0.0002) (Figure 1B). At completion of therapy, mean tumor weight in different therapy groups was 0.33±0.19 g in controls, 0.14±0.08 g in nab-paclitaxel, 0.18±0.04 g in BMS-754807 and 0.07±0.03 g in nab-paclitaxel+BMS-754807 (Figure 1C). Furthermore, no significant change in total body weight was observed for those mice treated with nab-paclitaxel, BMS-754807 or combination (Figure 1D). In another subcutaneous PDAC xenograft experiment using Panc-1 cells, nab-paclitaxel and BMS-754807 treatment also caused a reduction in tumor growth with additive effects in combination (Figure 2A). Average net tumor growth in different therapy groups was 294.3 mm3 in controls, 23.1 mm3 after nab-paclitaxel (p = 0.002), 118.2 mm3 after BMS-754807 (p = 0.02) and -87.4 mm3 (tumor regression) after nab-paclitaxel plus BMS-754807 (p = 0.0001) (Figure 2B). Mean tumor weight in different therapy groups was: 0.30±0.06 g in controls, 0.16±0.05 g in nab-paclitaxel, 0.22±0.02 g in BMS-754807 and 0.07±0.03 g in nab-paclitaxel+BMS-754807 (Figure 2C). Also, no significant change in total body weight was observed for those mice treated with nab-paclitaxel, BMS-754807 or combination (Figure 2D).
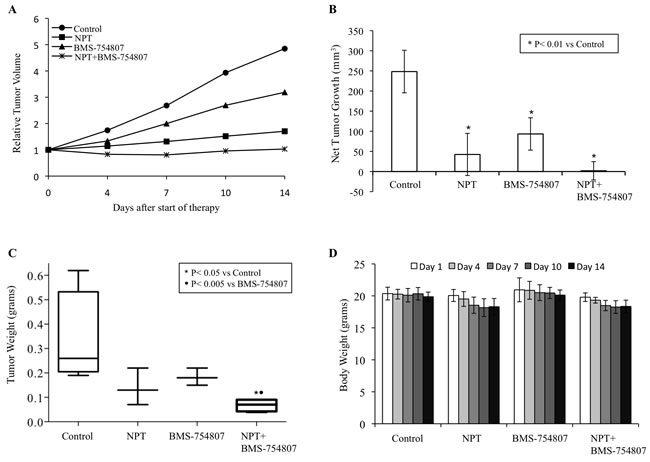
Figure 1: Antitumor activity of nab-paclitaxel and BMS-754807 in AsPC-1 tumor xenografts. AsPC-1 cells were subcutaneously injected in nude mice and treated with nab-paclitaxel and BMS-754807 for 2 weeks. Tumor volume was measured twice a week using calipers. A. Relative tumor volume is calculated by dividing the tumor volume at any time by the tumor volume at the start of treatment. B. Net tumor growth was calculated by subtracting tumor volume on the first treatment day from that on the final day. C. Mean tumor weight was calculated from final day tumor weights in each group and is presented as a box plot. Box height denotes interquartile range; horizontal line within the box denotes median; and whiskers represent minimum and maximum values. D. Mouse body weight was measured twice a week and presented as bar chart for the 2-week therapy period. Data are representative of mean values ± standard deviation from 6-8 mice per group.
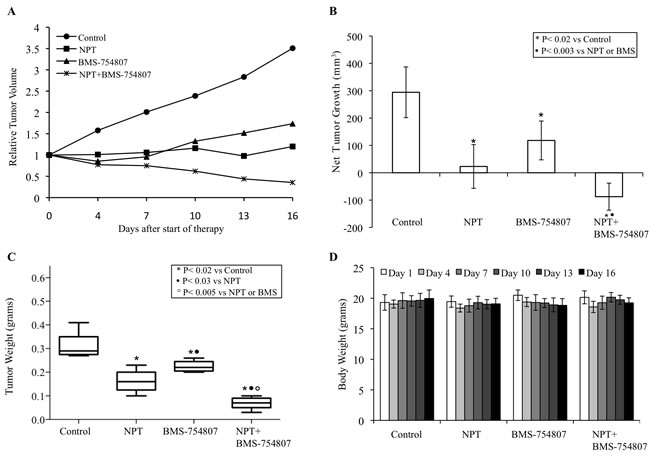
Figure 2: Antitumor activity of nab-paclitaxel and BMS-754807 in Panc-1 tumor xenografts. Panc-1 cells were subcutaneously injected in nude mice and treated with nab-paclitaxel and BMS-754807 for 2 weeks. Tumor volume was measured twice a week using calipers. A. Relative tumor volume is calculated by dividing the tumor volume at any time by the tumor volume at the start of treatment. B. Net tumor growth was calculated by subtracting tumor volume on the first treatment day from that on the final day. C. Mean tumor weight was calculated from final day tumor weights in each group and is presented as a box plot. Box height denotes interquartile range; horizontal line within the box denotes median; and whiskers represent minimum and maximum values. D. Mouse body weight was measured twice a week and presented as bar chart for the 2-week therapy period. Data are representative of mean values ± standard deviation from 6-8 mice per group.
Nab-paclitaxel and the IGF signaling inhibitor improve animal survival
In the human PDAC peritoneal dissemination model using AsPC-1 cells in NOD/SCID mice, nab-paclitaxel and BMS-754807 therapy was started two weeks after tumor cell injection and was continued for the subsequent two weeks (Figure 3A). Animal survival in different therapy groups, calculated from the start of therapy, was as follows: controls (21 days), nab-paclitaxel (40 days, a 90% increase compared with controls, p = 0.002), BMS-754807 (27 days, a 29% increase compared with controls, p = 0.01) and nab-paclitaxel+BMS-754807 (47 days, a 124% increase compared with control, p = 0.005) (Figure 3B). There was no significant change in mouse body weight during two week therapy period in all groups, indicating that there was no significant drug-related toxicity in monotherapy or combination therapy groups.
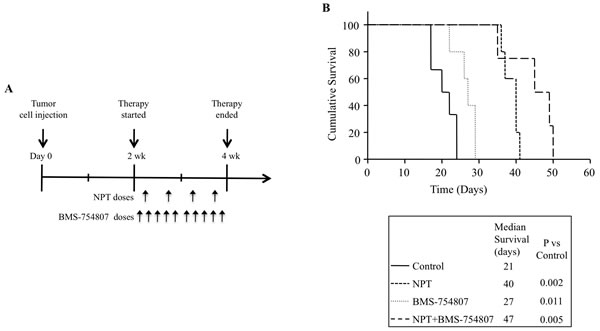
Figure 3: Improvement in animal survival by nab-paclitaxel and BMS-754807. A. Schematic representation of experimental procedure. AsPC-1 cells (0.75 x 106) were injected intraperitoneally in NOD/SCID mice and treatment started after 2 weeks with nab-paclitaxel and BMS-754807 for 2 weeks. B. The curve represents the animal survival time from the beginning of therapy. Statistical group differences in survival time were calculated using logrank testing (GraphPad Prism 6.0).
Effects of nab-paclitaxel and the IGF signaling inhibitor on intratumoral proliferation and apoptosis
Possible mechanisms for the antitumor activities of nab-paclitaxel and BMS-754807 were investigated by IHC analysis of tumor tissues obtained from subcutaneous xenograft studies after completion of two weeks of therapy.
Intratumoral proliferative activity was analyzed by Ki67 staining and this analysis demonstrated that nab-paclitaxel and BMS-754807 inhibited intratumoral cell proliferation and combination therapy group generated additive effects. Different therapy groups showed the following intratumoral proliferative indices, calculated by percentage of Ki67-positive cells over total number of cells per HPF: controls (42.5±15.2), nab-paclitaxel (24.1±4.7), BMS-754807 (19.9±10.8) and nab-paclitaxel+BMS-754807 (9.3+4.1) (Figure 4A).
Intratumoral apoptosis, measured by TUNEL assay, revealed that the nab-paclitaxel and BMS-754807 monotherapy increased intratumoral apoptosis that was further increased in the combination therapy group. The apoptotic index, calculated by number of TUNEL-positive cells over total number of cells per HPF, in these therapy groups: controls (0.02±.01), nab-paclitaxel (0.08±0.02), BMS-754807 (0.08±0.02) and nab-paclitaxel+BMS-754807 (0.15±0.05) (Figure 4B).
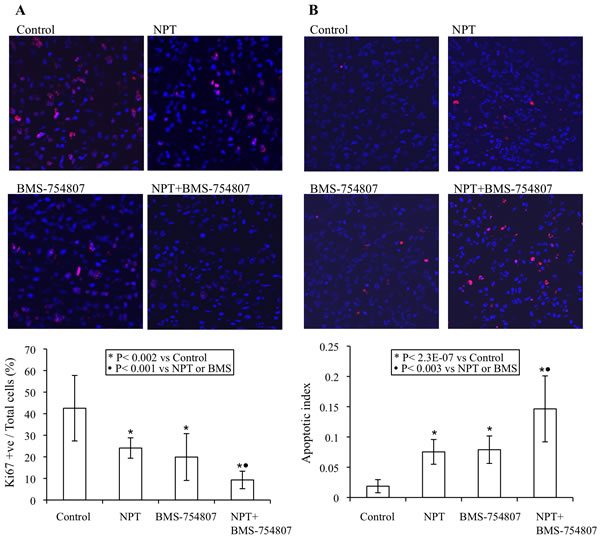
Figure 4: Mechanisms of antitumor activity of nab-paclitaxel and BMS-754807. Nude mice were subcutaneously injected with AsPC-1 cells and treated with nab-paclitaxel and BMS-754807 for 2 weeks. A. Intratumoral proliferation was measured by immunostaining tissue sections for Ki67 nuclear antigen. Ki67-positive cells were counted in five different high power fields. B. Intratumoral apoptosis was measured by staining tumor tissue section with TUNEL procedure. TUNEL-positive apoptotic cells were counted in five different high power fields. For both immunostaining experiments, slides were photographed under a fluorescent microscope and the data are expressed as the mean ± standard deviation.
Additional mechanisms of in vivo antitumor activity of nab-paclitaxel and BMS-754807 were examined by analyzing intratumoral expression of their molecular targets by Western blot analysis. BMS-754807 therapy caused a significant decrease in the expression of phospho-IGF-1R/IR, and decreased the expression of the downstream signaling proteins phospho-AKT and phospho-ERK. Furthermore, BMS-754807 treatment caused a concomitant increase in the apoptosis marker proteins cleaved caspase-3 and cleaved PARP-1 (Figure 5).
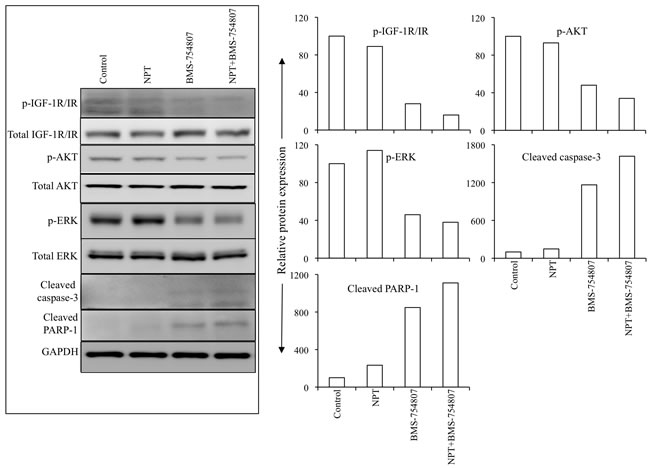
Figure 5: Nab-paclitaxel and BMS-754807 effects on the IGF signaling pathways and apoptosis-related proteins in vivo. Tumor lysates were prepared from tumor tissues obtained from AsPC-1 tumor-bearing mice and were analyzed by immunoblotting. Data are representative of pooled lysates obtained from tumor sections of at least 5 mice in each therapy group. The intensity of bands was quantitated by densitometry and is represented in the bar graph after normalizing values with corresponding total protein expression or GAPDH expression.
Effects of nab-paclitaxel and the IGF signaling inhibitor on PDAC cell proliferation and cellular targets in vitro
The antitumor mechanism of nab-paclitaxel and BMS-754807 was evaluated by analyzing in vitro cell viability of human PDAC epithelial cells with different mutation status. Both, nab-paclitaxel and BMS-754807, inhibited PDAC cells proliferation. The combination treatment benefits of nab-paclitaxel and BMS-754807 were observed by adding the IC25 dose of BMS-754807 with increasing doses of nab-paclitaxel. Addition of BMS-754807 decreased the nab-paclitaxel IC50 from 7.2 μM to 490 nM for AsPC-1, 430 nM to 50 nM for BxPC-3, 740 nM to 540 nM for MIA PaCa-2, and 690 nM to 280 nM for Panc-1 cell lines (Figure 6). Immunoblot analysis to determine the effect of the IGF signaling inhibitor on the IGF/receptor axis in AsPC-1 and Panc-1 cells, revealed that BMS-754807 blocked the expression of phospho-IGF-1R/IR. BMS-754807 treatment also decreased the expression of downstream signaling protein phospho-AKT but there was no significant effect on phospho-ERK expression. Nab-paclitaxel and BMS-754807 induced the expression of apoptosis-related cleaved caspase-3 protein, while combination treatment generated an additive effect (Figure 7).
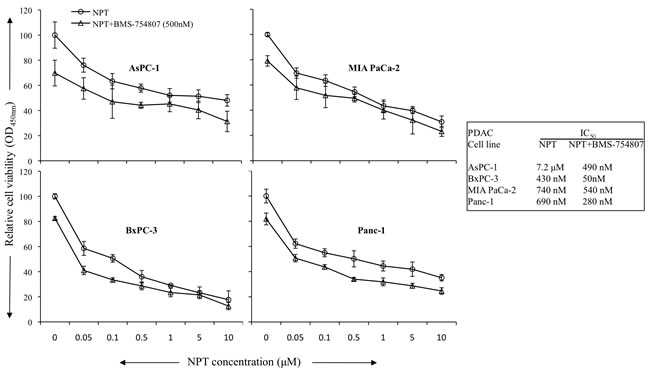
Figure 6: Addition of BMS-754807 had additive effects in combination with nab-paclitaxel to inhibit in vitro cell proliferation of PDAC cells. PDAC cells were plated on 96-well plates and treated with nab-paclitaxel (0 to 10 μM) as depicted through the open circle line. Another group of cells were treated with IC25 dose of BMS-754807 (500 μM) and nab-paclitaxel as depicted through the open triangle line. After 72 hours, 10 μl WST-1 reagent was added in each well and incubated for 2 additional hours. The absorbance at 450 nm was measured using a microplate reader. The resulting number of viable cells was calculated by measuring absorbance of color produced in each well. Data are the mean ± SD of triplicate determinations.
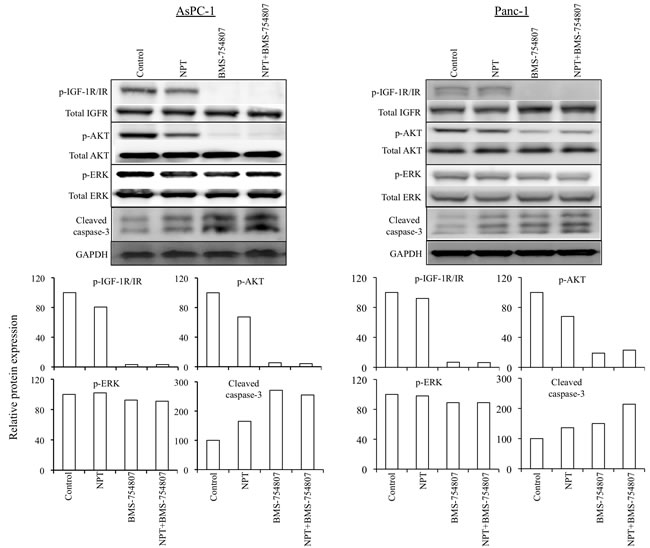
Figure 7: Nab-paclitaxel and BMS-754807 effects on the IGF signaling pathway and apoptosis-related proteins. A sub-confluent monolayer of human PDAC cells AsPC-1 and Panc-1 was treated with nab-paclitaxel (10 μM) and BMS-754807 (10 μM), either alone or in combination for 16 hours. Total cell extracts were analyzed by immunoblotting. The intensity of bands was quantitated by densitometry and is represented in the bar graph after normalizing values with corresponding total protein expression or GAPDH expression. Data are representative of two independent experiments with similar results.
DISCUSSION
Activation of the IGF signaling pathway appears to be an important mechanism for progression of several malignancies. IGF-1R overexpression has been correlated with tumor aggressiveness [43-45] while disruption of IGF-1R activation inhibited growth and motility of a wide range of cancers [46]. Additionally, the blockage of IGF-1R signaling has been shown to enhance chemotherapy response in preclinical studies of several cancer types including pancreatic and non small-cell lung cancers as well as Ewing’s sarcoma [43, 47, 48]. Since IGF-1R and IR are highly homologous, antagonist development was initially focused towards monoclonal antibodies that selectively target IGF-1R and not affect IR signaling, which could lead to dysregulation of glucose homeostasis [21]. Initial clinical studies with IGF-1R antibodies showed some responses in monotherapy and in combination with cytotoxic chemotherapy [49]. However, patients receiving antibody therapy have hyperglycemia indicating the potential of undesirable metabolic side effects of such therapy [50]. Several reports showed the involvement of stimulation of IR by insulin or IGF-2 in cancer cell progression [51, 52] suggesting that IGF-1R and IR both are therapeutic targets and inhibition of both receptors may be required for optimal tumor growth inhibition. In PDAC specifically, the IGF signaling activation has also been shown to play a major role in tumor progression [14-16]. In addition, a recent study demonstrated a correlation between IGF-1R signaling and tumor aggressiveness in PDAC patients [15]. Based on these aspects and given the recent clinical progress in PDAC cytotoxic therapy, our present study thus explored the effects of BMS-754807, a small molecule inhibitor of both the receptors IGF-1R/IR, in combination with the recently approved and more effective chemotherapeutic agent nab-paclitaxel.
In multiple PDAC epithelial cells, we observed a differential expression of phosphorylated form of IGF-1R/IR (Supplemental Figure S2), which is consistent with a previously published study [14], indicating a possible involvement of aberrant IGF signaling in PDAC progression. The IGF signaling inhibitor BMS-754807 has shown antitumor activity in multiple tumor models including epithelial, mesenchymal and hematopoietic cancer cells [33]. We observed the effect of BMS-754807 in subcutaneous xenografts using two aggressive PDAC cell lines, AsPC-1 and Panc-1. In these models, BMS-754807 inhibited tumor growth as a single agent and more importantly, caused additive effects in combination with nab-paclitaxel. Analysis of intratumoral proliferation and apoptosis appear to be correlated with tumor growth inhibition data. Evaluation of target proteins in tumor lysates showed that BMS-754807 treatment decreased levels of the activated form of IGF-1R/IR and downstream effectors AKT and ERK, and induced levels of the apoptosis related proteins cleaved caspase-3 and cleaved PARP-1, indicating that the BMS-754807 therapy is indeed managing to sufficiently affect these targets within the local tumor model. Furthermore, BMS-754807 and nab-paclitaxel demonstrated a significant additive response to improve animal survival as evaluated in a peritoneal dissemination model using AsPC-1 cells which is highly reproducible, well characterized and closely resembles the metastatic progression pattern of the clinical disease. In the present study, we have not analyzed the effect of dosing schedule of the drugs where nab-paclitaxel was given prior to BMS-754807 to evaluate nab-paclitaxel induced tumor permeability. However, in the present study dosing schedule, nab-paclitaxel (twice per week) and BMS-754807 (five times per week) were given together on day 1; therefore, for all other doses of BMS-754807, nab-paclitaxel effects on tumor permeability might already be in place.
BMS-754807 inhibited the proliferation of all PDAC cells tested; importantly, in all cell lines addition of IC25 dose of BMS-754807 decreased the IC50 values of nab-paclitaxel. Additionally, in AsPC-1 and Panc-1 PDAC epithelial cells, BMS-754807 treatment caused a decrease in phosphorylated IGF-1R/IR and its downstream signaling protein phospho-AKT, no significant change in phospho-ERK, but increased cleavage of apoptosis related caspase-3 protein. A possible explanation for BMS-754807 to block ERK phosphorylation in AsPC-1 tumor lysates but not in AsPC-1 or Panc-1 cell lines is the significant activity of stromal cells within tumors, in which ERK-phosphorylation has been shown to be blocked by BMS-754807 (own data, not shown here). A combination of these molecular signaling changes are thus likely operational in the BMS-754807 induced inhibition in cell proliferation and induction in apoptosis, and appear to represent good markers of in vivo activity testing and for clinical validation.
Active PDAC progression involves multiple distinct mechanisms including induction in cancer cell proliferation, migration, differentiation, angiogenesis and inhibition of cancer or stromal cell apoptosis. Previous studies in our lab have shown the benefits of targeting multiple pathways in different cellular compartments including epithelial, vascular and stromal elements for PDAC treatment [39, 40, 42, 53]. Nab-paclitaxel itself has antiproliferative and proapoptotic effects on tumor cells, endothelial cells and fibroblasts. The exact molecular mechanisms for the enhancement in antitumor activity of nab-paclitaxel by BMS-754807 addition is not completely clear, but additive benefits are likely caused by augmentation of antiproliferative and proapoptotic activities and possibly not just restricted to the tumor cells of epithelial origin. Because of the multifactorial nature of PDAC and limited effectiveness of standard chemotherapy agents, combination therapies targeting multiple pathways represent sensible strategies for clinical evaluation. Based on our findings the combination of nab-paclitaxel and BMS-754807 is a reasonable clinical trial option for phase II evaluation after establishing toxicity of this combination.
Of note, most of the negative clinical trials of the IGF signaling inhibitors used monoclonal antibodies specifically targeting IGF-1R and not IR [25-27]. This could have potentially provided escape mechanisms for insulin and IGF-2 signaling that have been implicated in cancer cell progression [51, 52], leading to limited clinical response. Therefore, simultaneous inhibition of IGF-1R and IR by BMS-754807 may confer a better antitumor effect. Results from our prior work with BMS-754807 and gemcitabine as single cytotoxic agent in preclinical PDAC therapy would certainly support this broader targeting approach [36]. PDAC is histologically characterized by a dense desmoplastic reaction surrounding malignant epithelial cells that plays a critical role in tumor progression, invasion, metastasis and drug-resistance by providing a mechanical barrier. Nab-paclitaxel has been proposed to disrupt the PDAC stromal architecture, causing increased perfusion and drug-delivery [12]. Our laboratory has shown that the nab-paclitaxel is the most effective single agent cytotoxic agent in comparison with gemcitabine or docetaxel [10]. In addition, nab-paclitaxel combination with targeted therapies has been shown to efficiently improve antitumor response [11, 54]. Therefore, the application of combination therapy benefits of nab-paclitaxel with BMS-754807 seems particularly logical and plausible for advanced PDAC.
Since BMS-754807 inhibits both IGF-1R and IR signaling, dysregulation of glucose homeostasis is an important concern, and therefore the advantage of its antitumor effects might have to be balanced with the potential for metabolic side effects. In our studies, there was no apparent in vivo toxicity during a 2-week treatment course, however, toxicity for long-term inhibition of the IGF-1R and IR functions remains to be elucidated. A previous study reported only a short-term increase in serum glucose or insulin levels at doses up to 12.5 mg/kg, with an average weight change of -1.5 grams at 25 mg/kg, but no mortality [33]. In our study, no significant change in mouse body weight was observed by BMS-754807 therapy, but blood glucose measurements were not obtained. In conclusion, our study indicates that the combined IGF-1R/IR signaling inhibitor, BMS-754807, can mediate mechanism-specific antitumor activity in experimental PDAC, and significantly improves nab-paclitaxel response. These findings corroborate the rationale of blocking multiple pathways of the IGF and insulin signaling, and support the potential of BMS-754807 as targeting agent for clinical PDAC therapy.
MATERIALS AND METHODS
Cell culture and reagents
Human PDAC cell lines (AsPC-1, BxPC-3, Mia PaCa-2 and Panc-1) were purchased from the American Type Culture Collection (ATCC, Rockville, MD). Cell lines were tested and authenticated by ATCC. Cells were initially grown and multiple aliquots were cryopreserved. All the cell lines were used within 6 months after reexpansion in culture. Cells were cultured in DMEM or RPMI 1640 medium (Sigma Chemical Co. St. Louis, MO) containing 10% FBS and maintained at 37°C in a humidified incubator with 5% CO2 and 95% air. BMS-754807 was purchased from Active Biochem (Maplewood, NJ). The cell proliferation reagent WST-1 was purchased from Roche Diagnostic Corporation (Indianapolis, IN).
Western blot analysis
PDAC cells were treated with nab-paclitaxel (10 μM) and BMS-754807 (10 μM), and lysed after 16 hours to prepare cell lysate for Western blotting. Tumor lysates were prepared by snap freezing tumor tissues in liquid nitrogen and stored at -80°C. These tumor tissues were suspended in lysis buffer and homogenized using the Bullet Blender Homogenizer (Next Generation, Averill Park, NY), and extracts were sonicated on ice. Proteins in supernatants were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, Hercules, CA). The membranes were incubated overnight at 4°C with the following antibodies: total IGF-1R, phospho-IGF-1R (Tyr1135/1136)/IR (Tyr1150/1151; #3024), total AKT, phospho-AKT (Ser473), total ERK1/2, phospho-ERK1/2 (Thr202/Tyr204), cleaved caspase-3, cleaved PARP-1 and GAPDH (Cell Signaling Technology, Beverly, MA). The membranes were then incubated with the corresponding HRP-conjugated secondary antibodies (Pierce Biotechnologies, Santa Cruz, CA) for 1 to 2 hour. Specific protein bands were visualized using the enhanced chemiluminescence reagent (SignalFire, Cell Signaling) with Image360 system and quantitated by densitometry.
Cell viability assay
Cell viability of PDAC lines was evaluated by the colorimetric WST-1 assay as previously described [38]. Briefly, PDAC cells (4,000 cells per well) were plated in a 96-well plate in regular growth medium. After 16 hours the medium was replaced with 2% FBS containing medium and the cells were treated with nab-paclitaxel (50 nM to 10 μM) and IC25 dose of BMS-754807 (500 nM). After 72 hours, 10 μl WST-1 reagent was added in each well followed by additional incubation for 2 hours. The absorbance was measured at 450 nm using a microplate reader.
Animal studies
All animals were housed in pathogen-free facility with access to food and water ad libitum. Animal experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) at the Indiana University School of Medicine (South Bend, IN). Female nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (4 to 6 weeks old) were subcutaneously injected with AsPC-1 cells (0.75 x 106) or Panc-1 cells (10 x 106) as previously described [11]. Two weeks after tumor cell injection, all mice had measurable tumor. Mice were then randomized (n = 6 to 8 per group) to receive PBS (control), nab-paclitaxel (10 mg/kg, twice a week) and (BMS-754807 (25 mg/kg in 100 ml PBS, 5 times a week) via intraperitoneal injection for 2 weeks. The tumor size was measured twice weekly, and tumor volume (V) was calculated using the formula V = ½ (Length x Width2). Mice were euthanized after completion of treatment, tumors were dissected and processed for histological, immunohistochemical and Western blot analysis.
Animal survival studies were performed using female NOD/SCID mice (4-6 weeks of age) as previously described [39]. Briefly, the mice were intraperitoneally injected with AsPC-1 (0.75x106) cells and two weeks after tumor cell injection, mice were randomized (n = 6 to 8 per group) to receive PBS (control), nab-paclitaxel (10 mg/kg, twice a week) or BMS-754807 (25 mg/kg in 100 ml PBS, 5 times a week) via IP injection for two weeks. Animals were euthanized when moribund according to predefined criteria [40, 41]. Animal survival was evaluated from the first day of treatment until death.
Immunohistochemistry and immunofluorescence
Standard immunohistochemistry protocol was followed to stain the tumor tissue samples, as previously described [42]. Briefly, tumor tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin, tissue sections were cut (5 μm), deparaffinized and rehydrated. The tissue sections were incubated with Ki67 antibody (1:200 dilution) followed by incubation with Cy3 (1:200 dilution) secondary antibody. Slides were mounted using mounting solution containing 4’,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA). Intratumoral proliferative index was evaluated by calculating the Ki67-positive cells from five different high-power fields (HPF) in a blinded manner. Intratumoral apoptotic activity was evaluated by staining tissue sections with “Apoptag Apoptosis Detection Kit” according to the manufacturer’s (Millipore, Temecula, CA) instructions. Fluorescence microscopy was used to detect fluorescent signals using the IX81 Olympus microscope equipped with a Hamamatsu Orca digital camera (Hamamatsu Corporation, Bridgewater, NJ) and a DSU spinning confocal unit using Slidebook software (Intelligent Imaging Innovations, Philadelphia, PA).
Statistical analysis
Statistical analysis for in vivo tumor growth studies was performed by one-way ANOVA for multiple group comparison and Student’s t-test for the individual group comparison. Survival study statistics were performed using logrank group comparison (GraphPad Prism 6.0). P values less than 0.05 were considered statistically significant. In vitro cell proliferation data are expressed as mean ± standard deviation. Statistical significance was analyzed by the two-tailed Student’s t-test using GraphPad Prism 6.0 Software (GraphPad Software, San Diego, CA) for individual group comparisons.
ACKNOWLEDGMENTS
This work was financially supported in part by the Department of Surgery, Indiana University School of Medicine funds and by 5R01HL114977 (MAS) from NIH.
CONFLICTS OF INTEREST
The authors indicate no conflicts of interest.
REFRENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
2. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
3. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM and Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913-2921.
4. Paulson AS, Tran Cao HS, Tempero MA and Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013; 144:1316-1326.
5. Butturini G, Stocken DD, Wente MN, Jeekel H, Klinkenbijl JH, Bakkevold KE, Takada T, Amano H, Dervenis C, Bassi C, Buchler MW and Neoptolemos JP. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg. 2008; 143:75-83; discussion 83.
6. Wilkowski R, Thoma M, Bruns C, Duhmke E and Heinemann V. Combined chemoradiotherapy for isolated local recurrence after primary resection of pancreatic cancer. JOP. 2006; 7:34-40.
7. Hartwig W, Werner J, Jager D, Debus J and Buchler MW. Improvement of surgical results for pancreatic cancer. Lancet Oncol. 2013; 14:e476-485.
8. Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD and Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997; 15:2403-2413.
9. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364:1817-1825.
10. Awasthi N, Zhang C, Schwarz AM, Hinz S, Wang C, Williams NS, Schwarz MA and Schwarz RE. Comparative benefits of Nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer. Carcinogenesis. 2013; 34:2361-2369.
11. Awasthi N, Zhang C, Schwarz AM, Hinz S, Schwarz MA and Schwarz RE. Enhancement of nab-paclitaxel antitumor activity through addition of multitargeting antiangiogenic agents in experimental pancreatic cancer. Mol Cancer Ther. 2014; 13:1032-1043.
12. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691-1703.
13. Ozawa F, Friess H, Tempia-Caliera A, Kleeff J and Buchler MW. Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen. 2001; 21:27-44.
14. Bergmann U, Funatomi H, Yokoyama M, Beger HG and Korc M. Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 1995; 55:2007-2011.
15. Hirakawa T, Yashiro M, Murata A, Hirata K, Kimura K, Amano R, Yamada N, Nakata B and Hirakawa K. IGF-1 receptor and IGF binding protein-3 might predict prognosis of patients with resectable pancreatic cancer. BMC Cancer. 2013; 13:392.
16. Pollak MN, Schernhammer ES and Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004; 4:505-518.
17. Zha J and Lackner MR. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clin Cancer Res. 2010; 16:2512-2517.
18. Samani AA, Yakar S, LeRoith D and Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007; 28:20-47.
19. Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L and Luu HH. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015; 2:13-25.
20. Beltran PJ, Mitchell P, Chung YA, Cajulis E, Lu J, Belmontes B, Ho J, Tsai MM, Zhu M, Vonderfecht S, Baserga R, Kendall R, Radinsky R and Calzone FJ. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol Cancer Ther. 2009; 8:1095-1105.
21. Ii M, Li H, Adachi Y, Yamamoto H, Ohashi H, Taniguchi H, Arimura Y, Carbone DP, Imai K and Shinomura Y. The efficacy of IGF-I receptor monoclonal antibody against human gastrointestinal carcinomas is independent of k-ras mutation status. Clin Cancer Res. 2011; 17:5048-5059.
22. Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, Toner GC, Maki RG, Meyers PA, Chugh R, Ganjoo KN, Schuetze SM, Juergens H, Leahy MG, Geoerger B, Benjamin RS, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011; 29:4541-4547.
23. Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, Benedetto PW, Friberg G, Deng H, McCaffery I, Leitch I, Badola S, Chang S, Zhu M and Tolcher A. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012; 30:1849-1856.
24. Rajan A, Riely GJ, Carter CA, Thomas A, Khozin S, Bergagnini I and al. e. Phase II study of cixutumumab (IMC-A12) in thymic malignancies. J Clin Oncol. 2012; 30:7033.
25. Abou-Alfa GK, Gansukh B, Chou JF and al. e. Phase II study of cixutumumab (IMC-A12, NSC742460; c) in hepatocellular carcinoma (HCC). J Clin Oncol. 2011; 29.
26. Jassem J, Langer CJ, Karp DD and al. e. Randomized, open label, phase III trial of figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin in patients with non-small cell lung cancer (NSCLC). J Clin Oncol. 2010; 28.
27. Robertson JF, Ferrero JM, Bourgeois H, Kennecke H, de Boer RH, Jacot W, McGreivy J, Suzuki S, Zhu M, McCaffery I, Loh E, Gansert JL and Kaufman PA. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013; 14:228-235.
28. Kindler HL, Richards DA, Stephenson J and al. e. A placebo-controlled, randomized phase II study on conatumumab (C) or AMG 479 (A) or placebo (P) plus gemcitabine (G) in patients (pts) with metastatic pancreatic cancer (mPC). J Clin Oncol. 2010; 28.
29. Fuchs CS, Azevedo S, Okusaka T, Van Laethem JL, Lipton LR, Riess H, Szczylik C, Moore MJ, Peeters M, Bodoky G, Ikeda M, Melichar B, Nemecek R, Ohkawa S, Swieboda-Sadlej A, Tjulandin SA, et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trial. Ann Oncol. 2015; 26:921-927.
30. Lu J, Deng H, Tang R and al. e. Exposure-response (E-R) analysis to facilitate phase III (P3) dose selection for AMG 479 (A479) in combination with gemcitabine (G) to treat metastatic pancreatic cancer (mPC). J Clin Oncol. 2011; 29.
31. Munshi S, Hall DL, Kornienko M, Darke PL and Kuo LC. Structure of apo, unactivated insulin-like growth factor-1 receptor kinase at 1.5 A resolution. Acta Crystallogr D Biol Crystallogr. 2003; 59:1725-1730.
32. Wittman M, Carboni J, Attar R, Balasubramanian B, Balimane P, Brassil P, Beaulieu F, Chang C, Clarke W, Dell J, Eummer J, Frennesson D, Gottardis M, Greer A, Hansel S, Hurlburt W, et al. Discovery of a (1H-benzoimidazol-2-yl)-1H-pyridin-2-one (BMS-536924) inhibitor of insulin-like growth factor I receptor kinase with in vivo antitumor activity. J Med Chem. 2005; 48:5639-5643.
33. Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, Discenza L, Menard K, Li A, Trainor G, Vyas D, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther. 2009; 8:3341-3349.
34. Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, Fargnoli J, Graf Finckenstein F, Gottardis MM and Carboni JM. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010; 70:7221-7231.
35. Dayyani F, Parikh NU, Varkaris AS, Song JH, Moorthy S, Chatterji T, Maity SN, Wolfe AR, Carboni JM, Gottardis MM, Logothetis CJ and Gallick GE. Combined Inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS One. 2012; 7:e51189.
36. Awasthi N, Zhang C, Ruan W, Schwarz MA and Schwarz RE. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther. 2012; 11:2644-2653.
37. Huang F, Chang H, Greer A, Hillerman S, Reeves KA, Hurlburt W, Cogswell J, Patel D, Qi Z, Fairchild C, Ryseck RP, Wong TW, Finckenstein FG, Jackson J and Carboni JM. IRS2 copy number gain, KRAS and BRAF mutation status as predictive biomarkers for response to the IGF-1R/IR inhibitor BMS-754807 in colorectal cancer cell lines. Mol Cancer Ther. 2015; 14:620-630.
38. Awasthi N, Schwarz MA, Verma V, Cappiello C and Schwarz RE. Endothelial monocyte activating polypeptide II interferes with VEGF-induced proangiogenic signaling. Lab Invest. 2009; 89:38-46.
39. Schwarz RE, Awasthi N, Konduri S, Cafasso D and Schwarz MA. EMAP II-based antiangiogenic-antiendothelial in vivo combination therapy of pancreatic cancer. Ann Surg Oncol. 2010; 17:1442-1452.
40. Awasthi N, Yen PL, Schwarz MA and Schwarz RE. The efficacy of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy and antiangiogenic response in pancreatic cancer. J Cell Biochem. 2012; 113:784-791.
41. Awasthi N, Kirane A, Schwarz MA, Toombs JE, Brekken RA and Schwarz RE. Smac mimetic-derived augmentation of chemotherapeutic response in experimental pancreatic cancer. BMC Cancer. 2011; 11:15.
42. Awasthi N, Zhang C, Ruan W, Schwarz MA and Schwarz RE. Evaluation of poly-mechanistic antiangiogenic combinations to enhance cytotoxic therapy response in pancreatic cancer. PLoS One. 2012; 7:e38477.
43. Min Y, Adachi Y, Yamamoto H, Ito H, Itoh F, Lee CT, Nadaf S, Carbone DP and Imai K. Genetic blockade of the insulin-like growth factor-I receptor: a promising strategy for human pancreatic cancer. Cancer Res. 2003; 63:6432-6441.
44. Min Y, Adachi Y, Yamamoto H, Imsumran A, Arimura Y, Endo T, Hinoda Y, Lee CT, Nadaf S, Carbone DP and Imai K. Insulin-like growth factor I receptor blockade enhances chemotherapy and radiation responses and inhibits tumour growth in human gastric cancer xenografts. Gut. 2005; 54:591-600.
45. Imsumran A, Adachi Y, Yamamoto H, Li R, Wang Y, Min Y, Piao W, Nosho K, Arimura Y, Shinomura Y, Hosokawa M, Lee CT, Carbone DP and Imai K. Insulin-like growth factor-I receptor as a marker for prognosis and a therapeutic target in human esophageal squamous cell carcinoma. Carcinogenesis. 2007; 28:947-956.
46. Sachdev D and Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007; 6:1-12.
47. Morgillo F, Kim WY, Kim ES, Ciardiello F, Hong WK and Lee HY. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007; 13:2795-2803.
48. Martins AS, Mackintosh C, Martin DH, Campos M, Hernandez T, Ordonez JL and de Alava E. Insulin-like growth factor I receptor pathway inhibition by ADW742, alone or in combination with imatinib, doxorubicin, or vincristine, is a novel therapeutic approach in Ewing tumor. Clin Cancer Res. 2006; 12:3532-3540.
49. Gualberto A and Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene. 2009; 28:3009-3021.
50. Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, Yap TA, Roberts ML, Sharma A, Gualberto A, Adjei AA and de Bono JS. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007; 13:5834-5840.
51. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A and Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999; 19:3278-3288.
52. Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, Sbraccia P, Goldfine ID, Vigneri R and Belfiore A. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999; 18:2471-2479.
53. Awasthi N, Schwarz MA and Schwarz RE. Combination effects of bortezomib with gemcitabine and EMAP II in experimental pancreatic cancer. Cancer Biol Ther. 2010; 10:99-107.
54. Volk LD, Flister MJ, Bivens CM, Stutzman A, Desai N, Trieu V and Ran S. Nab-paclitaxel efficacy in the orthotopic model of human breast cancer is significantly enhanced by concurrent anti-vascular endothelial growth factor A therapy. Neoplasia. 2008; 10:613-623.