
INTRODUCTION
The non-coding portion of the mammalian genome is pervasively transcribed resulting in the expression of thousands of mRNAs and long non-coding RNAs (ncRNA) [1, 2]. Although only a small number of long ncRNAs have been characterized to date they have been shown to be involved in almost every level of the gene expression program such as transcription, mRNA processing and translation [3]. Long ncRNAs are involved in the development of a variety of disorders such as cancer and neurodegenerative disorders [3, 4]. Long ncRNAs mediating regulation of tumor-suppressors and oncogenes through various mechanisms have been described [5-7]. The long ncRNAs lincRNA-p21 [8]; Pint [9]; PR-lncRNA-1 and PR-lncRNA-10 [5] are regulated by the tumor suppressor p53 and are involved in mediating its effects [6, 10]. Using a different mechanism, the long ncRNA-RB1 is co-expressed with RB1 from a bidirectional promoter and links RB1 transcription to the transcription of another tumor suppressor, Calreticulin, with effects on immunogenic cell death [7]. On the other hand, the long ncRNA ANRIL acts as an oncogene. ANRIL interacts with components of both the PRC1 and PRC2 complexes and mediates the silencing of the INK4a/ARF locus encoding tumor-suppressor genes that regulate cell cycle-progression and senescence [11]. Depletion of ANRIL leads to reduced proliferation and the long ncRNA is up-regulated in prostate cancer and leukemia further supporting an oncogenic role [12, 13].
The Myc family of proteins is a group of basic helix-loop-helixl eucine zipper transcription factors, one of the most studied classes of proteins associated to cancer. There are three members of the Myc family, c-Myc, n-Myc and l-Myc [14], all three functioning in a similar manner but with expression differences across cancers types. For instance, c-Myc is overexpressed in both blood-borne and solid tumors; n-Myc is expressed mostly in neural tumors; and l-Myc is overexpressed in small cell lung carcinomas [14-16]. Overall, many human cancers show increased expression of Myc. The transcription factor c-Myc regulates the transcription of at least 15% of the genes in the human genome with the help of its heterodimer partner MAX [17]. c-Myc is involved in many biological processes such as cell cycle, differentiation and protein synthesis [17, 18]. Since c-Myc is upregulated in many human cancers it is subject to intense investigation for cancer treatment [15, 16]. Several studies have shown that c-Myc regulates the expression of long ncRNAs, and that some of these transcripts can participate in the transcriptional functions mediated by c-Myc [19-22]. Long ncRNA-mediated regulation of translation of c-Myc has also been suggested [23, 24].
Here, we report that PARROT (proliferation associated RNA and regulator of translation) is a long ncRNA with a dynamic expression range across both transformed and normal cells that contributes to cellular proliferation in senescence and cancer and is associated to translation efficiency in HeLa cells. Using transcriptomic and proteomic approaches we identify PARROT as an upstream regulator of c-Myc. Depletion of PARROT leads to a depletion of c-Myc both at the mRNA and protein levels with subsequent effects on cellular growth and proliferation.
RESULTS
Differential promoter activity drives ncRNA expression
Long ncRNAs are often expressed at a low level, complicating the further analysis of their transcriptional regulation and biological functions. Using available data for HeLa and HEK293 cells we identified the long ncRNAs with Pol(II) at their promoter in at least one of the two cell lines, to identify actively expressed and highly abundant long ncRNAs (Figure 1A). To reduce the number of candidates and to increase the probability of identifying long ncRNAs that are functional also in non-transformed cells, we restricted the analysis to long ncRNAs suggested to be functional based on their expression in keratinocytes and responsiveness to 12-O-Tetradecanoylphorbol-13-acetate (TPA)-induced differentiation of keratinocytes [25]. This results in a set of 21 long ncRNAs that show Pol(II) binding at their promoters in at least one of the cell lines (Figure 1B). While six long ncRNAs have defined Pol(II) peaks in both cell lines, most (15) are clearly bound specifically by high levels of Pol(II) in one cell line only as shown in Figure 1C, which is recapitulated in the expression level of the long ncRNAs in the respective cell lines, reflecting the tissue-specific properties of long ncRNAs that have mostly been reported for lowly expressed long ncRNAs (Figure 1D). While the generally low expression level of long ncRNAs is believed to result from post-transcriptional regulation, we asked whether the regulation of a highly expressed long ncRNA is indeed occurring at the promoter level by cloning an upstream region of the annotated transcript into a reporter vector (Figure 1E). We observed a high induction of PARROT expression in HeLa cells (Figure 1F), while essentially no transcription occurred in HEK293 cells (Figure 1G). Further characterization of the promoter by cloning of fragments of 166, 222, 298 and 404 base pairs, respectively, upstream of the transcription start site (TSS) in the same reporter assay identified a short promoter region mediating the tissue-specific expression of PARROT (Figure 1E-1G), similar to what is often observed for protein coding genes.
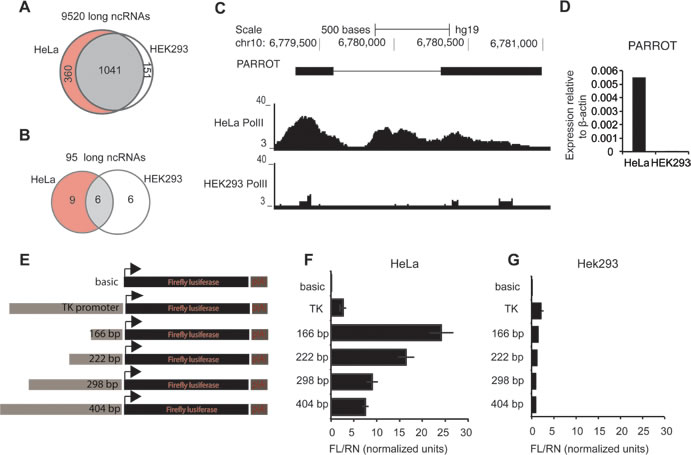
Figure 1: Differential promoter activity drives PARROT expression. A. Pol(II) association to ncRNAs in HeLa and HEK293 cells. B. Pol(II) association to ncRNAs in HeLa and HEK293 cells differentially expressed upon keratinocyte differentiation. C. Snapshot from the genome browser of Pol(II) association to PARROT in HeLa and HEK293 cell line. D. Relative expression of PARROT in HeLa and HEK293 cells measured by qPCR. (E., F., G.) PARROT promoter region was cloned upstream of the Luciferase in pGL3 vector. E. Graphical representation of the inserts used. F.-G. Luciferase reporter assay. The Firefly luciferase vectors were cotransfected with a Renilla luciferase vector (pRL-TK) for transfection control in HeLa F. and HEK293 cells G.
PARROT is a spliced non-coding RNA localized to the cytoplasm
To determine the cellular distribution of PARROT we subjected HeLa cells to cellular fractionation, separating the cells into cytoplasmic, nucleoplasmic and chromatin fractions [26]. We can show the localization of the processed form of PARROT to be primarily cytoplasmic (Figure 2A) suggesting functions different from regulation of transcription as has been reported for many nuclear localized long ncRNAs. We obtained the transcript profile of PARROT from chromatin-associated RNA using RNA sequencing (RNA-seq) with random primers and from whole cells using oligod(T) primers [27]. The transcriptome profile shows a very long primary transcript associated to chromatin and a polyadenylated, two-exonic processed transcript with a cytoplasmic localization as the spliced transcript form (Figure 2B), suggesting that PARROT might have dual functions as a long ncRNA in the nucleus as the primary transcript and in the cytoplasm as the processed transcript.
To confirm that the annotation of PARROT as a non-coding RNA is correct, we cloned the full-length processed transcript and synthesized RNA in vitro from a T7 promoter. In vitro translation assay analysis did not show the presence of any peptide translated from the long ncRNA sequence (Figure 2C). The localization of PARROT in polysome profiling experiments as determined by quantitative PCR (qPCR) of individual fractions is also suggestive of a non-coding function (Figure 2D). Furthermore, we confirmed that no peptides corresponding to the sequence of PARROT are detected in proteogenomic studies [28], as well as absence of association to ribosomes in ribosome profiling studies from HeLa [29].
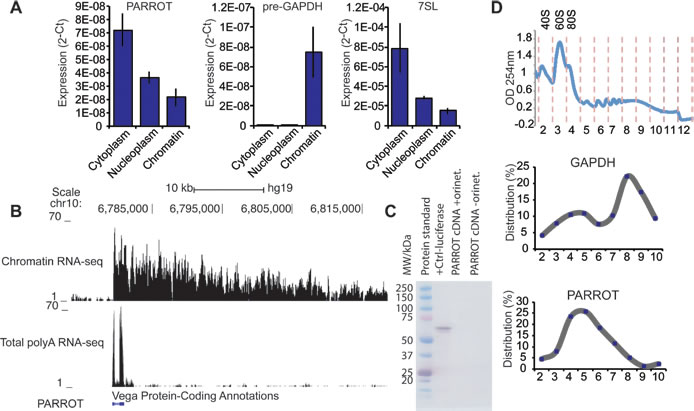
Figure 2: PARROT is primarily a cytoplasmic long non-coding RNA. A. Distribution of PARROT in the cytoplasm, nucleoplasm and chromatin. As controls for the cellular fractionation pre-gapdh and 7SL are shown. The average of 2-Ct ± s.d. are shown, n = 3. B. Snapshot from the genome browser showing the PARROT transcript in chromatin RNA-seq and polyA RNA-seq. C. In vitro translation assay. D. Polysome profile of HeLa cells. Polysomal distribution of GAPDH and PARROT RNAs was determined by isolating the RNA from each fraction collected from a 10-50% sucrose gradient. Percentage of mRNA level determined by qPCR in each fraction is shown.
PARROT regulates proliferation and translation
To assess the function of PARROT we used siRNAs for knock-down in different cell lines. Following knock-down in HeLa cells (Figure 3A) a reduction in cell growth was observed using crystal violet staining (Figure 3B) that could not be assigned to apoptosis using a number of available assays (not shown). To address effects on migration, in addition to cell growth, we used A549 cells. A549 cells display reduced migration ability following knock-down of PARROT (Figure 3C). The effect on cellular migration is comparable to knock-down of SNAI1, a known regulator of cellular migration (quantified from three independent experiments in Figure 3D) [30], demonstrating a general function of PARROT in several cell lines and effects on both cell growth and migration. Due to the cytoplasmic localization of PARROT and its association to low-molecular weight polysome profile fractions, we propose that PARROT could have a regulatory function in translation or mRNA stability. To address this, we determined the effect of knock-down on general translation using a puromycin based translation assay. We see a markedly reduced translation observed in general upon knock-down of PARROT, as determined by puromycin incorporation and western blot of the newly synthesized protein (Figure 3E). The Ponceau S staining is used as a loading control showing total protein loaded in each lane, and three independent experiments were quantified to determine statistical significance (Figure 3F).
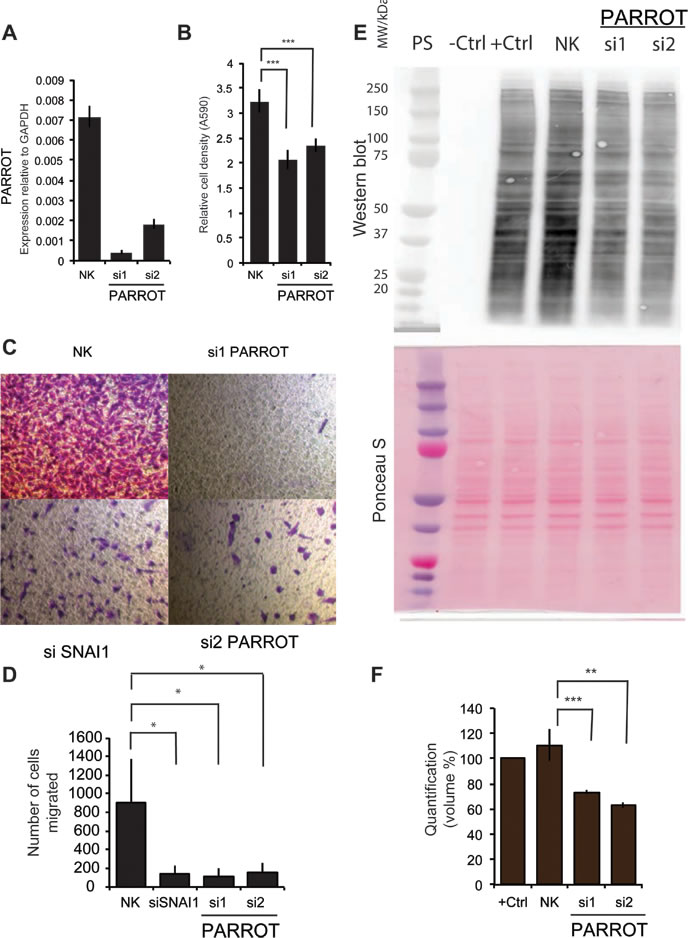
Figure 3: PARROT regulates growth and translation. A. Knock-down of PARROT in HeLa cells with two different siRNAs. As a control, HeLa cells were transfected with a non-targeting control siRNA (NK). B. Crystal violet viability assay in HeLa cells transfected with either the control siRNA (NK) or depleted of PARROT for 72h. The average ± s.d. are shown, n = 5 (si1 p = 4.87581E-06, si2 p = 1.58272E-05). C. Migration assay. A549 cells were transfected with either the control siRNA (NK), two different siRNAs against PARROT or SNAI1which was used as a positive control, and D. quantified using three independent experiments. E. Puromycin translation assay with western blot using a puromycin antibody. Shown are: Protein standard (PS), protein extracts from control HeLa cells not treated with puromycin (-ctrl), control HeLa cells treated with puromycin (+ctrl), and cells treated with puromycin and transfected with either control siRNA (NK) or two different siRNAs against PARROT for 72h. F) Ponceau S staining as the loading control and the bottom panel: quantification of puromycin incorporation in three independent experiments. (The average ± s.d. is shown; ** p < 0.01, * p < 0.05).
PARROT is an upstream regulator of Myc
To determine the molecular mechanism by which PARROT exerts its effect on the cellular phenotype we depleted it using siRNAs and performed RNA sequencing of polyadenylated transcripts in HeLa cells. This analysis reveals that the depletion of PARROT affects the expression of 331 genes of which 181 are upregulated and 150 are downregulated. Gene ontology analysis shows that PARROT affects the expression of genes involved in cell cycle regulation, cellular growth and proliferation as well as cellular movement (Figure 4A), in accordance with the phenotype observed following knock-down of PARROT in both HeLa and A549 cells.
To further explore the effect of PARROT on protein levels we used mass spectrometry to determine abundance and differential expression of the proteome following siRNA depletion of PARROT. Here, using SILAC (stable isotope labeling by amino acids in cell culture) control cells were labeled with heavy isotopes and the cells depleted of PARROT were labeled with light isotopes. Protein extracts were mixed in equal ratios and the intensities of both heavy and light labeled peptides were measured by mass-spectrometry. Analysis reveals that depletion of PARROT affects the levels of 73 proteins of which 22 are upregulated and 51 are downregulated. Furthermore, purifying phosphoproteins and subsequent identification of differentially phosphorylated proteins by mass spectrometry reveals that depletion of PARROT affects the phosphorylation status of 264 proteins: 98 are hyper-phosphorylated and 166 are hypo-phosphorylated. Gene ontology analysis shows that PARROT affects proteins involved in cell cycle regulation, cellular growth and proliferation as well as cellular movement on the level of protein synthesis as well as phosphorylation, in agreement with the results from RNA-seq data (Figure 4B).
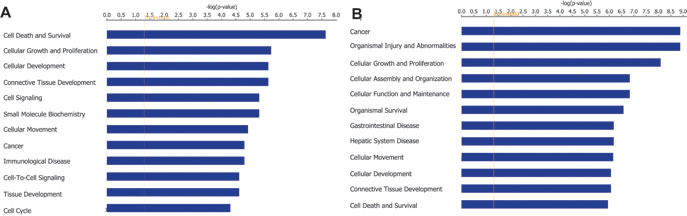
Figure 4: PARROT affects gene expression. A. Gene ontology analysis of genes differentially expressed upon knock-down of PARROT. B. Gene ontology analysis of differentially phosphorylated proteins upon knock-down of PARROT.
Incorporating all large-scale data into an Ingenuity Pathway Analysis (IPA) predicts c-Myc as a significant (p = 3.09x10-5) upstream regulator of the genes affected by the depletion of PARROT. Known Myc targets such as CDC34 (involved in cell death) [31], ROCK2 (involved in cellular adhesion, migration, apoptosis, differentiation) [32-34]; EPHA2 (migration, proliferation, adhesion, growth) [35]; RHOB (Ras family member involved in apoptosis, growth, proliferation, transformation, migration, adhesion) [36]; SLC7A5 (involved in growth and proliferation) [37]; DUSP5 (negatively regulates members of MAP kinase superfamily which are associated with cellular proliferation and differentiation) [38]; MIF (involved in proliferation, apoptosis, migration, aging and cell death) [39]; and FOSL1 (involved in proliferation, migration, motility, invasion, cell cycle progression, differentiation, apoptosis, etc.) [40, 41], are differentially expressed and/or phosphorylated following PARROT depletion (Figure 5A). We examined the direct effects of PARROT knock-down on c-Myc both at the RNA and protein levels. qPCR analysis reveals that the transcript level of c-Myc is decreased upon knock-down of PARROT (Figure 5B). In accordance with these results, the levels of both total c-Myc (Figure 5C) and phosphorylated c-Myc are decreased following knock-down of PARROT (Figure 5D). The regulation of c-Myc as an important target of PARROT is in agreement with the phenotypes in cell growth and migration observed since c-Myc is a known regulator of cellular proliferation and growth as well as of migration [17, 18].
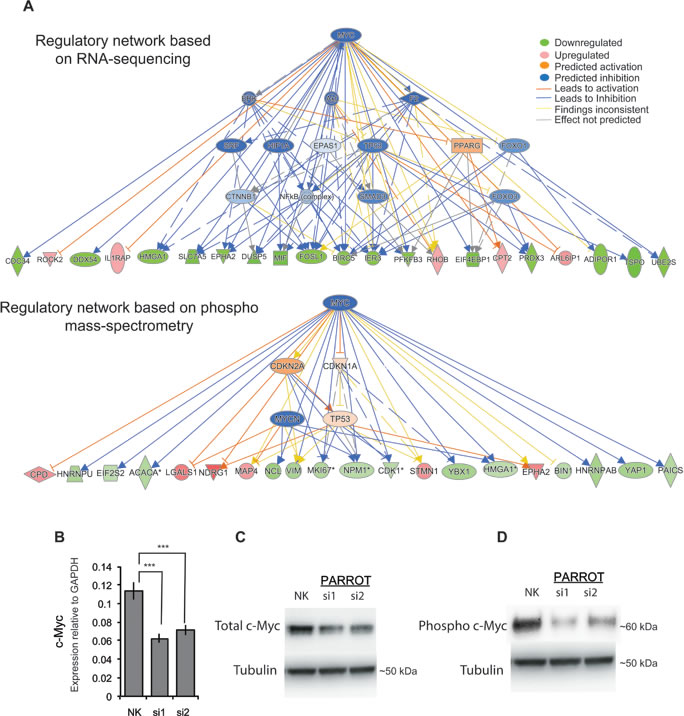
Figure 5: PARROT is an upstream regulator of c-Myc. A. Ingenuity network analysis showing Myc as the prime target of PARROT. Regulatory network based on RNA-seq data (upper panel) or phospho mass-spectrometry data (lower panel) B. qPCR showing the levels of processed c-Myc transcript in HeLa cells transfected with either the control siRNA (NK) or depleted of PARROT. The average ± s.d. are shown, n = 3 (si1 p = 0.000541914, si2 p = 0.001295592). C.-D. Western blot of total c-Myc C. or phospho-form of c-Myc D. from protein extracts of Hela cells transfected with either the control siRNA (NK) or two different siRNAs against PARROT.
PARROT is associated with proliferation in senescence and cancer
The effect on translation and the association to proliferation together with the regulation of c-Myc prompted us to look at biological scenarios where proliferation is changed. An expression analysis of PARROT across a panel of cell lines shows differential expression of PARROT between cell lines. PARROT is expressed at the highest level in human mammary epithelial cells (HMEC) (Figure 6A). HMECs are healthy, untransformed, human mammary epithelial cells that undergo replicative senescence, an irreversible arrest of cell growth, after approximately 14 passages [42]. HMECs from young, proliferative (passage 2) and senescent (passage 14) cells were analyzed for PARROT expression by RNA-sequencing (Figure 6B) and qPCR (Figure 6C). As a marker of senescence we did western blot for p16 (Figure 6D). In accordance with the RNA-seq data, qPCR analysis shows that as HMECs grow older with passaging the expression of PARROT gradually decreases (Figure 6B and 6C). Knock-down of PARROT in HMECs decreases the cell growth significantly as we also observe in HeLa cells (Figure 6E), showing that PARROT is not only involved in proliferation of transformed cells, but has an effect on cell growth in non-transformed cells also.
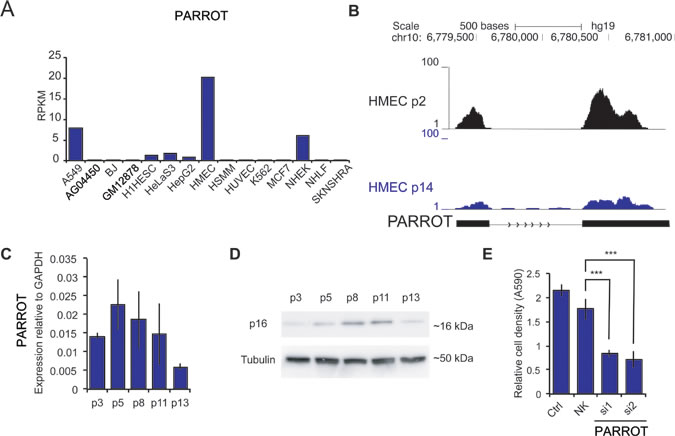
Figure 6: PARROT is differentially expressed in senescence. A. RNA-seq data showing the expression of PARROT in different cell lines. B. Snapshot from the genome browser of RNA-seq data of HMECs cells in passage 2 and in passage 14. C. qPCR of PARROT in ageing HMECs. D. p16 western-blot in ageing HMECs. In C and D p refers to passage number. E. Crystal violet viability assay of HMECs cells in passage 5 of nontransfected cells (ctrl) or cells transfected with either control siRNA (NK) or two different siRNAs against PARROT for 72h. The average ± s.d. are shown, n = 5 (si1 p = 1.09588E-12, si2 p = 2.86182E-12).
In the other end of the spectrum of proliferation, analysis of 8 paired normal and tumor samples from stomach shows that PARROT is not expressed in healthy stomach cells of stomach cancer patients. In tumor samples, on the other hand, it is highly expressed compared to normal tissue from the same patients (Figure 7A-7B). Furthermore, examination of the expression of c-Myc in these patients displays a similar trend, c-Myc is expressed at a significantly higher level in tumor samples than in normal paired tissue samples (Figure 7C). This observation supports our findings and further suggests that PARROT has an effect on proliferation that can lead to an oncogenic potential that is exerted via regulation of the c-Myc oncogene.
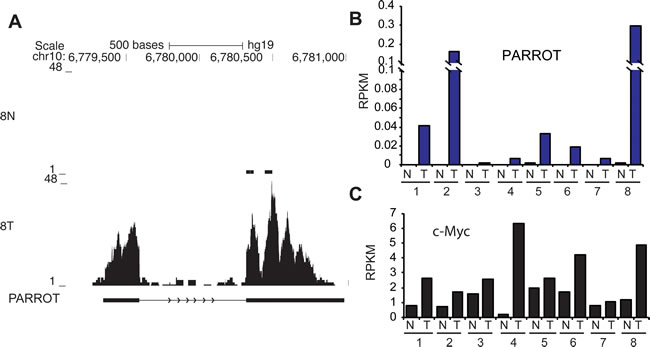
Figure 7: PARROT is upregulated in stomach cancer. A. Genome browser snapshot of RNA-seq in control or tumor stomach cancer sample. B. Expression (RNA-seq) of PARROT in control (N) and tumor (T) stomach cancer samples. C. Expression (RNA-seq) of c-Myc in control (N) and tumor (T) stomach cancer samples. B and C show expression of PARROT and Myc, respectively, as RPKM (reads per kilobase per million reads) as quantified from RNA-seq data.
DISCUSSION
The expression level of long ncRNAs is often very low and tissue-specific, properties that make the study of their functionality challenging. We have used data for Pol(II) association to long ncRNA promoters in two well-studied cell-lines, HeLa and HEK293 cells, to identify highly expressed long ncRNA with a pattern of differential expression between the cell-lines. We restricted the set of identified long ncRNAs by overlaying with data for differential expression of long ncRNAs in keratinocytes following differentiation, to reduce the number of candidates and increase the likelihood of studying functional long ncRNAs [25]. The low expression of most long ncRNAs is believed to be a consequence of rapid degradation. Using promoter assays, we can show that the high expression of PARROT in HeLa cells is a direct consequence of a strong promoter activity, and that the absence of PARROT expression in HEK293 cells is not caused by increased degradation but simply reflects that the promoter is not active in HEK293 cells. We identify a short core promoter reflecting properties comparable to most protein coding gene promoters, and the expression of PARROT in tumor samples suggests a specific transcriptional activation causing the overexpression of PARROT.
Interestingly, PARROT is transcribed as a very long primary transcripts and only a relatively short sequence in the 5’ end is further processed by splicing to the cytoplasmic two-exon mature form, suggesting that PARROT could have both nuclear and cytoplasmic functions. This might reflect a significant difference between processing of some long ncRNAs compared to protein-coding genes.
IPA upstream pathway analysis of PARROT targets identified independently by RNA sequencing or mass spectrometry, suggested c-Myc as one of the important upstream regulators, suggesting that PARROT is an upstream regulator of c-Myc. This hypothesis is supported by a robust effect of PARROT knock-down on c-Myc both at the mRNA, protein and phosphorylation level of c-Myc.
The pronounced effects we observe on proliferation, migration and translation in HeLa, HMEC and A549 cells following knock-down of PARROT are recapitulating the phenotype that would be expected for a decreased expression or activity of c-Myc [17, 43], and suggests a general function of PARROT in mediating upstream regulation of c-Myc in both transformed and non-transformed cells.
We can demonstrate an involvement of PARROT in senescence and cell proliferation in cancer that would be required for a regulator of c-Myc. We observe an increased expression of both c-Myc and PARROT in stomach cancer samples from eight patients analyzed by RNA sequencing. PARROT is dysregulated in 4 different types of cancer: BRCA (Breast Invasive Carcinoma), KIRC (Kidney Renal Clear Cell Carcinoma), LUAD (Lung Adenocarcinoma) and LUSC (Lung Squamous Cell Carcinoma) according to the lncRNome atlas (http://tcla.fcgportal.org). PARROT has previously been found to be associated with renal and lung adenocarcinoma [44] and could therefore be an important regulatory factor of the c-Myc pathway in several cancers as well as normal cells such as HMECs.
Several studies have shown that c-Myc can regulate the expression of long ncRNAs, and that some of these ncRNAs also participate in regulating the transcription of c-Myc target genes [19, 20, 22]. The promoters of long ncRNAs that are repressed by c-Myc have been shown to be more enriched for c-Myc binding sites than repressed mRNAs, suggesting a particularly important role for long ncRNAs in the Myc signaling network [21]. These long ncRNAs can act to regulate c-Myc targets and as oncogenes themselves promoting cellular proliferation and migration [19-22].
c-Myc directly binds to the promoter of H19 activating its expression by recruiting a histone acetyltransferase. A reduction in clonogenicity and anchorage independent growth is observed upon depletion of H19 in both breast and lung cancer cells [45]. These results suggest that H19 acts downstream of c-Myc to promote tumorigenesis in breast and lung cancer cells.
Similarly, in gastric cancer c-Myc has been shown to activate the expression of colon cancer associated transcript 1 (CCAT1). Overexpression of CCAT1 leads to an increase in proliferation and migration of gastric cancer cells [46]. A longer isoform of the same transcript called CCAT1-L has been shown to regulate the expression of Myc in colon cancer. It is proposed that CCAT1-L enables the interaction between the enhancer and the c-Myc promoter through modulation of CTCF concentration thereby promoting tumorigenesis [47].
A highly expressed long RNA in gastric cancer called gastric carcinoma high expressed transcript 1 (GHET1) also regulates c-Myc. This ncRNA enhances the stability and the expression of c-Myc by cooperating with insulin-like growth factor 2 mRNA binding protein 1 (IGF2BP1) leading to an enforced physical interaction between IGF2BP1 and c-Myc RNA. Depletion of c-Myc reduces the ability of GHET1 to promote proliferation of cancer cells [48].
We observe that the spliced form of PARROT is localized to the cytoplasm. There is a decrease in c-Myc mRNA as well as protein levels upon siRNA-mediated knock-down of PARROT, suggesting a role of PARROT in regulating mRNA stability or translation of c-Myc. The long ncRNA PCAT-1 has been suggested to regulate the translation of c-Myc through disruption of miRNA binding to the Myc 3’ UTR and the long ncRNA GAS5 has been shown to bind the translation initiation factor eIF4E and specifically target c-Myc translation [22, 24]. Our findings that PARROT can orchestrate a group of genes regulated by c-Myc identifies an additional factor in this complex network with important implications for both normal and cancer cells and the understanding of the molecular mechanisms involved.
Further investigation is needed to shed light onto the mechanism by which PARROT regulates c-Myc, which could reveal important mechanistic insight into how c-Myc is regulation modulated in both untransformed and transformed cells.
MATERIALS AND METHODS
Pol(II) association analysis
Long ncRNAs differentially expressed upon keratinocyte differentiation were obtained from [25]. Publicly available ENCODE Chip-seq data for Pol(II) were used. Long ncRNAs and Pol(II) were intersected by the use of BEDTools [49].
Cell culture
HeLa, HEK293 and A549 cells were cultured in complete DMEM medium (Gibco/LifeTechnologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and penicillin-streptoMycin (Gibco). HMEC cells were cultured as previously described in a 1:1 mixture of MM4 and MCDB170 medium with of 5µg/ml of cholera toxin (Sigma), 5% AlbuMAX (Invitrogen) and 1µM oxytocin (Bachem) as described in [50]. All cells were cultured at 37 °C with 5% CO2.
Luciferase assay
Promoter upstream region (166bp, 222bp, 298bp and 404 bp) sequence located upstream of the PARROT TSS were cloned upstream of the luciferase into pGL3-basic vector (Promega). Primers used for amplification of the human genomic DNA with Phusion DNA polymerase (NEB) (Forward: 404bp- 5’-TGAAGATCTCTGACCACCTGTTGAGCTGT-3’, 298bp- 5’-TGAAGATCTAGGAAATTGGTCAAGGTTGC-3’, 223bp-5’-TGAAGATCTCCACTCAGTTATTTTTGTCTCTCA-3’, 166bp- 5’-TGAAGATCTAAGCTCCCAGAAATGTCAGC-3’; Reverse: 5’-TGAAAGCTTCCACCCAGAGTCAAGGAGAC-3’) contain added BglII (NEB) and HindIII (NEB) restriction sites. Both the vector and the insert were digested with BglII and HindIII restriction enzymes to allow directional cloning.
HeLa and HEK293 cells (10,000 cells/well) were plated in 96-well white plates 24 h before transfection. Cells were co-transfected with 0.1 μg pGL3-basic-promoter constructs and 0.02 μg of pRL-TK vector (Promega) using lipofectamine2000 (Invitrogen). Luciferase activity was measured 24 h after the transfection using the Dual-Luciferase Reporter Assay System kit (Promega) according to the manufacturer’s instruction. For data analysis, Firefly luciferase activity was normalized to that of Renilla luciferase. All transfections were carried out in triplicates at least three times.
In vitro translation assay
In vitro translation assay (Promega) was performed according to the manufacturer’s instruction.
Cellular fractionation
RNA was extracted from cellular fractionsprepared from ~4 × 106 cells as described in [27].
Crystal violet viability assay
HeLa or HMEC cells (10,000 cells/well) were plated in 96-well plates 24 h before transfection with siRNAs against PARROT or control siRNAs. Three days after the transfection cells where washed with PBS and fixed with methanol for 15min after which they were washed with water and stained with 0.1% crystal violet. After 20min cells were washed with water and dried. The following day 50 μl/well of 33% acetic acid was added and the absorbance at 590nm was measured. All transfections were carried out in triplicates at least three times.
Quantitative real-time PCR
Total RNA was extracted from cells following the manufacturer’s instructions with TRIzol (Life Technologies). Reverse transcription was carried out with High Capacity RNA-to-cDNA Kit (Life Technologies) and qPCR was performed using the Fast SYBR Green Master Mix (Life Technologies) on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The relative expression was calculated by normalizing to the Actin or GAPDH expression level as control housekeeping genes.
The following primers were used: PARROT (5’-CAGAACAGAGCCACCTCCAG-3’, 5’-GCACCGTCTGTTGTTCATTC-3’), c-Myc (5’-GCTGCTTAGACGCTGGATTT-3’, 5’-CCTCCTCGTCGCAGTAGAAA-3’), GAPDH (5’-GCTCTCTGCTCCTCCTGT TC-3’, 5’-ACGACCAAATCCGTTGACTC -3’),and β-actin (5′-CGACAGGATGCAGAAGGAG-3′, 5′-GTACTTGCGCTCAGGAGGAG-3′).
RNA interference
dsiRNA oligos from IDTDNA (siRNA1 PARROT: sense 5’-GAAUGAAAGCACAGCACCAUCCUGGAA-3’, antisense 5’-CCAGGAUGGUGCUGUGCUUUCAUTC-3’; siRNA2 PARROT: sense 5’- GCUGAAUCAAGAUGCUGACUUCAGCAC-3’, antisense 5’-GCUGAAGUCAGCAUCUUGAUUCAGC-3’) were used. As a negative control we used DS NC1 (IDT DNA).Cells were transfected at a final dsiRNA concentration of 50 nM using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Cells were collected 24 h or 48h after the transfection for RNA isolation and 72 h after transfection for western blot analysis.
Sucrose gradient fractionation and RNA isolation
Cytoplasmic lysates of HeLa cells were prepared and subjected to centrifugation through linear sucrose gradients (10-50% sucrose) essentially as described previously [51]. siRNA transfected cells (3x 10cm plates per condition) were rinsed and scraped in ice-cold PBS containing 100µg/mL cycloheximide. All subsequent steps were performed in ice-cold conditions. Cells were pelleted and resuspended in extraction buffer (20 mM Tris-HCl, pH 8.0, 140 mM KCl, 0.5 mMDTT, 5 mM MgCl2, 0.5% Nonidet-P40, 0.1 mg/ml cycloheximide, and 0.5 mg/ml heparin), incubated 10 min on ice and clarified by centrifugation for 10 min at 12000g. Approximately 400µL of supernatant was layered onto a 12-ml linear sucrose gradient (10-50% sucrose (w/v) in detergent free extraction buffer) and centrifuged at 4 °C in an SW40Ti rotor (Beckman, Palo Alto, CA) at 35,000 rpm without brake for 120 min. The gradients were collected into 12 fractions (1ml each), and absorbance profiles at 260 nm were recorded (ISCO, UA-6 detector). 0.1 volume of 3 M sodium acetate (pH 5.2) and 1 volume of isopropyl alcohol were added to the probes for overnight precipitation at −20 °C. RNA was purified using total RNA isolation kit (Nucleospin RNA II, Macherey & Nagel) following the manufacturer’s protocol. RNA concentration was determined, and the samples were stored at −80 °C.
Puromycin translation assay
Three days after the knockdown 1μM puromycin was added to the cells for 30 min. Cells were harvested in RIPA buffer, sonicated and boiled for 5min at 95°C in Laemmli buffer. 15μg of protein was loaded on a 4-12% Bis-Tris gradient gel (NuPAGE Novex) and transferred onto a PVDF membrane. Membrane was blocked in 5% milk and incubated overnight at 4°C with 1μg/ml anti-puromycin antibody (Kerafast). Membrane was washed, incubated with secondary HRP conjugated antibody and developed using ECL reagent (Pierce). To make sure that the equal amount of protein from puromycin treated cells was loaded on the gel the membrane was stained with Ponceau S protein dye prior to blocking.
Migration assay
A549 cells were seeded in a 6 well plate (100000 cells/well) 4 h before transfection with siRNAs against PARROT, SnaiI or control siRNAs. 48 hours later cells were starved for 24h in a DMEM medium without FCS after which 50000 cells were added in DMEM medium without FCS to a migration chamber placed in a well of 24 well plate containing 750 μl of DMEM with FCS. 14 hours later cells where washed with PBS and cells that did not migrate were swiped. Cells that migrated were fixed with methanol for 15min after which they were washed with water and stained with 0.1% crystal violet. After 20min cells were washed with water, dried and number of migrating cells quantified.
Western blot
Cells were harvested in RIPA buffer, sonicated and boiled for 5min at 95°C in Laemmli buffer. 15μg of protein was loaded on a 4-12% Bis-Tris gradient gel (NuPAGE Novex) and transferred onto a PVDF membrane. Membrane was blocked in 5% milk and incubated overnight at 4°C with total c-Myc (Santa Cruz Biotechnology sc-40), phosphor-c-Myc (Abcamab32029)or p16 (Santa Cruz Biotechnology sc-468) antibody. Membrane was washed, incubated with secondary HRP conjugated antibody and developed using ECL reagent (Pierce).
RNA sequencing
Total RNA was extracted using TRIzol (Life Technologies). RNAseq libraries were prepared with the TruSeq RNA Sample Preparation Kit v2. Sequencing was performed on a HiSeq 2000 instrument (Illumina, San Diego, CA, USA) using paired-end sequencing (2 × 50 bp). Data was analyzed with the TRUP pipeline [52]. Differential expression analysis was preformed in edgeR Bioconductor package [53]. The cutoff for differentially expressed genes was set at FDR < 0.05. The RNA-seq data are deposited in GEO with the accession number GSE69842.
Mass-spectrometry
HeLa cells used for MS were grown in SILAC medium.They were lysed and digested by trypsin under denaturing conditions as previously reported [55]. Peptides were purified with C18 columns and fractionated by a strong cation exchange (SCX, Polysulfoethyl A, 200 x 9.4 mm, 5µm 200Å, PolyC, Columbia, MA) column, 5% of resulting peptides in each of the 11 fractions were used for proteome profiling. The remaining peptides were further enriched for phospho peptides by TiO2 (GL Sciences, Japan) columns [55]. Each sample fraction was dissolved in 3 µL of 5% ACN and 2% FA and analyzed by LC-MS/MS. LC−MS/MS was carried out by nanoflow reverse phase liquid chromatography (Dionex Ultimate 3000, Thermo Scientific, Waltham, MA) coupled online to a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Scientific) as described previously [54]. Raw MS data were processed with MaxQuant v1.4.1.2 [56] and searched against the human proteome database UniProtKB with 81.194 entries, released in 02/2012. A false discovery rate (FDR) of 0.01 for proteins and peptides and a minimum peptide length of 7 amino acids were required. A maximum of two missed cleavages was allowed for the tryptic digest. Following SILAC modifications were used: 13C615N4-arginine and 13C615N2-lysine. Cysteine carbamidomethylation was set as fixed modification, while N-terminal acetylation and methionine oxidation were set as variable modifications in all runs, phosphorylation of serine, threonine and tyrosine were set as variable modifications in the according enriched fractions.In order to filter for differentially regulated proteins or phosphoproteins we set the cutoff for the ratio of light (knock-down treatment) to heavy (control) to be > 1.2 for upregulated and < 0.8 for downregulated. Additionally, we also required that the ratio of knock-down/control to be > 1.2 or < 0.8 compared to the ratio of control knock-down(NK)/control.
Gene ontology analysis
The functional analysis and networks were generated through the use of QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity).
Statistics
Statistical analysis was performed using two-tailed Student’s T-test.
Acknowledgments
We thank Ruping Sun for analyzing the RNA sequencing data and Adrian Bracken for sharing the HMEC RNA sequencing data. Work in the author’s laboratories is funded by the German Ministry for Research and Education and the Alexander von Humboldt Foundation through the Sofja Kovalevskaja Award to U.A.Ø. and is supported by the Max Planck Society.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, et al. Landscape of transcription in human cells. Nature. 2012; 489:101-108.
2. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775-1789.
3. Ulitsky I and Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013; 154:26-46.
4. Vucicevic D, Schrewe H and Orom UA. Molecular mechanisms of long ncRNAs in neurological disorders. Front Genet. 2014; 5:48.
5. Sanchez Y, Segura V, Marin-Bejar O, Athie A, Marchese FP, Gonzalez J, Bujanda L, Guo S, Matheu A and Huarte M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat Commun. 2014; 5:5812.
6. Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, de Laat W and Agami R. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013; 49:524-535.
7. Musahl AS, Huang X, Rusakiewicz S, Ntini E, Marsico A, Kroemer G, Kepp O and Orom UA. A long non-coding RNA links calreticulin-mediated immunogenic cell removal to RB1 transcription. Oncogene. 2015; 34:5046-54.
8. Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T and Rinn JL. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010; 142:409-419.
9. Marin-Bejar O, Marchese FP, Athie A, Sanchez Y, Gonzalez J, Segura V, Huang L, Moreno I, Navarro A, Monzo M, Garcia-Foncillas J, Rinn JL, Guo S and Huarte M. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013; 14:R104.
10. Leveille N, Melo CA, Rooijers K, Diaz-Lagares A, Melo SA, Korkmaz G, Lopes R, Akbari Moqadam F, Maia AR, Wijchers PJ, Geeven G, den Boer ML, Kalluri R, de Laat W, Esteller M and Agami R. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat Commun. 2015; 6:6520.
11. Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ and Zhou MM. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010; 38:662-674.
12. Pasmant E, Sabbagh A, Vidaud M and Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. Faseb J. 2011; 25:444-448.
13. Huarte M and Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet. 2010; 19:R152-161.
14. Dang CV. MYC on the path to cancer. Cell. 2012; 149:22-35.
15. Dang CV, Le A and Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009; 15:6479-6483.
16. Chen BJ, Wu YL, Tanaka Y and Zhang W. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int J Biol Sci. 2014; 10:1084-1096.
17. van Riggelen J, Yetil A and Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010; 10:301-309.
18. Meyer KD, Donner AJ, Knuesel MT, York AG, Espinosa JM and Taatjes DJ. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. Embo J. 2008; 27:1447-1457.
19. Doose G, Haake A, Bernhart SH, Lopez C, Duggimpudi S, Wojciech F, Bergmann AK, Borkhardt A, Burkhardt B, Claviez A, Dimitrova L, Haas S, Hoell JI, Hummel M, Karsch D, Klapper W, et al. MINCR is a MYC-induced lncRNA able to modulate MYC’s transcriptional network in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 2015; 112:E5261-5270.
20. Kim T, Cui R, Jeon YJ, Fadda P, Alder H and Croce CM. MYC-repressed long noncoding RNAs antagonize MYC-induced cell proliferation and cell cycle progression. Oncotarget. 2015; 6:18780-18789. doi:10.18632/oncotarget.3909.
21. Winkle M, van den Berg A, Tayari M, Sietzema J, Terpstra M, Kortman G, de Jong D, Visser L, Diepstra A, Kok K and Kluiver J. Long noncoding RNAs as a novel component of the Myc transcriptional network. Faseb J. 2015; 29:2338-2346.
22. Hung CL, Wang LY, Yu YL, Chen HW, Srivastava S, Petrovics G and Kung HJ. A long noncoding RNA connects c-Myc to tumor metabolism. Proc Natl Acad Sci U S A. 2014; 111:18697-18702.
23. Prensner JR, Chen W, Han S, Iyer MK, Cao Q, Kothari V, Evans JR, Knudsen KE, Paulsen MT, Ljungman M, Lawrence TS, Chinnaiyan AM and Feng FY. The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia. 2014; 16:900-908.
24. Hu G, Lou Z and Gupta M. The long non-coding RNA GAS5 cooperates with the eukaryotic translation initiation factor 4E to regulate c-Myc translation. PLoS One. 2014; 9:e107016.
25. Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R and Shiekhattar R. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010; 143:46-58.
26. Conrad T and Orom UA. Insight into miRNA biogenesis with RNA sequencing. Oncotarget. 2015; 6:26546-26547. doi: 10.18632/oncotarget.5264.
27. Conrad T, Marsico A, Gehre M and Orom UA. Microprocessor Activity Controls Differential miRNA Biogenesis In Vivo. Cell Rep. 2014; 9:542-54.
28. Lipovich L, Dachet F, Cai J, Bagla S, Balan K, Jia H and Loeb JA. Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics. 2012; 192:1133-1148.
29. Ingolia NT, Brar GA, Rouskin S, McGeachy AM and Weissman JS. Genome-wide annotation and quantitation of translation by ribosome profiling. Curr Protoc Mol Biol. 2013; Chapter 4:Unit 4 18.
30. Barrallo-Gimeno A and Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005; 132:3151-3161.
31. Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, Tayber O, Hather G, Liu R, Narayanan U, Milhollen MA and Lightcap ES. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013; 73:225-234.
32. Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, Matsuura Y and Kaibuchi K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol. 1999; 145:347-361.
33. Piazzolla D, Meissl K, Kucerova L, Rubiolo C and Baccarini M. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J Cell Biol. 2005; 171:1013-1022.
34. Li B, Antonyak MA, Zhang J and Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012; 31:4740-4749.
35. Surawska H, Ma PC and Salgia R. The role of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev. 2004; 15:419-433.
36. Prendergast GC. Actin’ up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001; 1:162-168.
37. Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG and Fu XD. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011; 474:390-394.
38. Nunes-Xavier CE, Tarrega C, Cejudo-Marin R, Frijhoff J, Sandin A, Ostman A and Pulido R. Differential up-regulation of MAP kinase phosphatases MKP3/DUSP6 and DUSP5 by Ets2 and c-Jun converge in the control of the growth arrest versus proliferation response of MCF-7 breast cancer cells to phorbol ester. J Biol Chem. 2010; 285:26417-26430.
39. Nishihira J. Macrophage migration inhibitory factor (MIF): its essential role in the immune system and cell growth. J Interferon Cytokine Res. 2000; 20:751-762.
40. Galvagni F, Orlandini M and Oliviero S. Role of the AP-1 transcription factor FOSL1 in endothelial cells adhesion and migration. Cell Adh Migr. 2013; 7:408-411.
41. Adiseshaiah P, Lindner DJ, Kalvakolanu DV and Reddy SP. FRA-1 proto-oncogene induces lung epithelial cell invasion and anchorage-independent growth in vitro, but is insufficient to promote tumor growth in vivo. Cancer Res. 2007; 67:6204-6211.
42. Bertram C and Hass R. Cellular senescence of human mammary epithelial cells (HMEC) is associated with an altered MMP-7/HB-EGF signaling and increased formation of elastin-like structures. Mech Ageing Dev. 2009; 130:657-669.
43. Cappellen D, Schlange T, Bauer M, Maurer F and Hynes NE. Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO Rep. 2007; 8:70-76.
44. Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, Poliakov A, Cao X, Dhanasekaran SM, Wu YM, Robinson DR, Beer DG, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015; 47:199-208.
45. Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS and Penn LZ. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006; 66:5330-5337.
46. Yang F, Xue X, Bi J, Zheng L, Zhi K, Gu Y and Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol. 2013; 139:437-445.
47. Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z, Zhang S, Wang HB, Ge J, Lu X, Yang L and Chen LL. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014; 24:513-531.
48. Yang F, Xue X, Zheng L, Bi J, Zhou Y, Zhi K, Gu Y and Fang G. Long non-coding RNA GHET1 promotes gastric carcinoma cell proliferation by increasing c-Myc mRNA stability. Febs J. 2014; 281:802-813.
49. Quinlan AR and Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841-842.
50. Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ and Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009; 69:7557-7568.
51. Dhamija S, Doerrie A, Winzen R, Dittrich-Breiholz O, Taghipour A, Kuehne N, Kracht M and Holtmann H. IL-1-induced post-transcriptional mechanisms target overlapping translational silencing and destabilizing elements in IkappaBzeta mRNA. J Biol Chem. 2010; 285:29165-29178.
52. Fernandez-Cuesta L, Sun R, Menon R, George J, Lorenz S, Meza-Zepeda LA, Peifer M, Plenker D, Heuckmann JM, Leenders F, Zander T, Dahmen I, Koker M, Schottle J, Ullrich RT, Altmuller J, et al. Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biol. 2015; 16:7.
53. Robinson MD, McCarthy DJ and Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139-140.
54. Gielisch I and Meierhofer D. Metabolome and Proteome Profiling of Complex I Deficiency Induced by Rotenone. Journal of proteome research. 2014.
55. Meierhofer D, Weidner C, Hartmann L, Mayr JA, Han CT, Schroeder FC and Sauer S. Protein sets define disease states and predict in vivo effects of drug treatment. Molecular & cellular proteomics. 2013; 12:1965-1979.
56. Cox J and Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008; 26:1367-1372.