
Introduction
Head and neck squamous cell carcinomas (HNSCC) arising in the larynx, pharynx, oral cavity, paranasal sinuses and nasal cavity are among the most common types of cancers, accounting for almost 60,000 newly diagnosed cases and more than 10,000 estimated deaths per year in the United States alone [1]. The prognosis of HNSCC patients with locoregionally advanced tumors is poor as indicated by five year overall survival rates of 40-60% in recent clinical trials [2-6]. In recurrent and metastatic disease the mean overall survival does not exceed 11 months despite intensive treatment protocols [7, 8].
Cetuximab, a monoclonal antibody targeting the extracellular ligand binding domain of the epidermal growth factor receptor (EGFR), is approved for the treatment of locoregionally advanced HNSCC in combination with radiotherapy [9] and for the treatment of recurrent or metastatic disease in combination with platinum-based chemotherapy [7]. However, not all patients treated with cetuximab respond well to therapy due to primary or acquired resistance, limiting significantly the clinical benefit of this drug.
Still, the molecular mechanisms underlying clinical resistance to cetuximab in HNSCC have not yet been elucidated. In metastatic colorectal cancer (mCRC) resistance mechanisms are by far better understood and involve mutations in EGFR downstream signaling molecules such as RAS [10]. Constitutive RAS signaling is mediated by mutations that prevent GTP hydrolysis, thus locking RAS in a permanently active state, independent of EGFR engagement. For this reason, colon tumors harboring activating RAS mutations do not respond to EGFR targeting and mutational screening is therefore routinely used for patient selection prior to treatment [11, 12]. In HNSCC, however, primary RAS mutations are rather uncommon with only 4.6% of HRAS mutated tumors and their significance for this entity remains unclear [13, 14]. Just as primary resistance, acquired resistance to cetuximab represents a challenge in the treatment of both mCRC and HNSCC. A recent series of pivotal studies on mCRC suggested that acquired resistance to cetuximab may not only be mediated by selection of rare RAS mutated subclones (from predominantly RAS wildtype tumors) [15, 16] but also by acquisition of epitope-modifying EGFR mutations during cetuximab (or panitumumab) treatment [17-19]. In fact, the extracellular domain mutations R451C, S464L, G465R, K467T, I491M and S492R of the EGFR (all located in exon 12) were found in post-therapeutic tumor subclones or antibody-resistant cell lines by next-generation or sanger sequencing. These mutations abrogated antibody binding and, therefore, resulted in clinical resistance to cetuximab and/or panitumumab depending on their localization within the antibody epitopes [17, 19].
To investigate if these or related mechanisms may play a role in cetuximab resistance of HNSCC as well, we set out to scan the cetuximab-interacting ectodomain of the EGFR as well as KRAS/NRAS exons 2/3/4 and HRAS exons 2/3 for mutations in a cohort of 46 HNSCC patients by targeted next generation sequencing, 20 of these with available post-cetuximab circulating tumor DNA (ctDNA). We found that RAS mutations can be acquired in a substantial proportion of patients during cetuximab-based treatment and significantly correlate with disease progression. Future studies should quantitatively determine mutational loads that reliably predict the benefit - or lack thereof - from further cetuximab treatment in patients with acquired RAS mutations.
Results
Patients and treatment
Of the 46 cetuximab-treated patients, 13 patients (28%) were in a curative and 33 patients (72%) were in a palliative treatment setting (Table 1). Although 19 of 46 patients (41%) had HNSCC of the oropharynx, HPV-positivity was rare with 5 of 46 patients (11%). The overall response rate (complete and partial responses) with cetuximab-based treatment was only 47% implying a high rate of treatment-resistant tumors in this cohort. About two thirds (13/20) of patients with liquid biopsies had progressive disease during combination therapy with Cis- or carboplatin, 5-fluorouracil and cetuximab or cetuximab maintenance, respectively. Of the remaining seven patients without progressive disease, two patients refused cetuximab maintenance therapy, one patient died of pneumonia during combination therapy and one patient had severe bleeding complications requiring discontinuation of therapy. The median progression-free survival for patients in the liquid biopsy cohort was 4.9 months (95% CI 3.4-6.0), the median overall survival 5.2 months (95% CI 4.0-7.8).
Table 1: Patient and tumor characteristics with sequencing data of tumor samples.
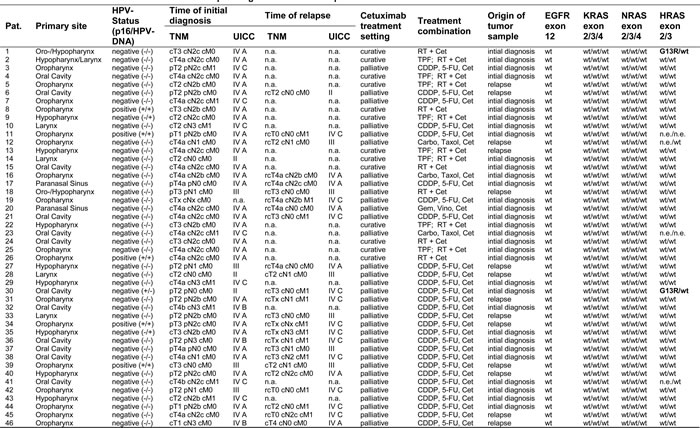
Abbreviations: HPV human papilloma virus; n.a. not applicable; RT radiotherapy; Cet cetuximab; TPF docetaxel/cisplatin/5-fluorouracil; CDDP 5-FU cis- or carboplatin/5-fluorouracil; Carbo, Taxol carboplatin/paclitaxel; Gem, Vino gemcitabine/vinorelbine; wt wildtype; n.e. not evaluable
NGS of the cetuximab-interacting EGFR ectodomain and RAS at baseline and in HNSCC cell lines
We sought to find out i) if tumor subclones expressing a mutated EGFR ectodomain or activating RAS mutations exist in HNSCC tumors before cetuximab-based treatment and ii) if such subclones emerge or expand under the selective pressure of EGFR-directed antibody treatment in this disease. We used NGS to screen EGFR exon 12, KRAS/NRAS exons 2/3/4 and HRAS exons 2/3 with a mean number of > reads per exon, ensuring that even rare mutant subclones would be detected (targeted NGS approach schematically shown in Figure 1).
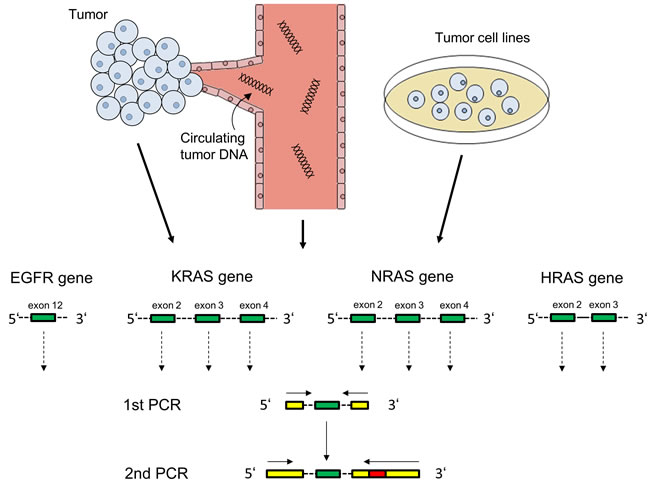
Figure 1: PCR amplification of EGFR and RAS exons for Illumina targeted next generation sequencing. EGFR exon 12, KRAS/NRAS exons 2/3/4 and HRAS exons 2/3 (green) were amplified from tumor tissue of 46 patients, post-cetuximab circulating tumor DNA of 20 patients and from 12 squamous carcinoma cell lines. Illumina-specific sequences for hybridization and sequencing (yellow) as well as patient-specific barcodes (red) were attached in a second PCR step.
None of the tumor tissue samples of all 46 patients showed evidence of mutations in the cetuximab-interacting EGFR ectodomain or KRAS/NRAS. In line with previous reports, activating HRAS mutations were found in primary tumor samples of two patients (4.3%) with one clonal (patient no. 1) and one subclonal mutation (patient no. 30), (Table 1).
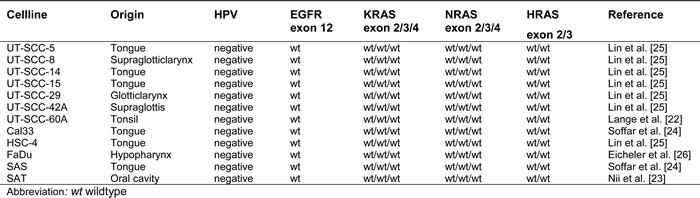
All 12 HNSCC cell lines that derived from EGFR antibody-naïve patients were unmutated for EGFR, KRAS/NRAS and HRAS (Table 2).
Table 2: Characteristics and sequencing data of squamous cell carcinoma cell lines.
NGS of the cetuximab-interacting EGFR ectodomain and RAS after cetuximab treatment
In 20 patients we obtained peripheral blood for ctDNA analysis during and after combination therapy with Cis- or carboplatin, 5-fluorouracil and cetuximab +/- cetuximab maintenance (liquid biopsy). Overall, about one third of patients acquired activating RAS mutations in the course of cetuximab-based treatment (KRAS: G12S, G13C; NRAS: Q61K, A146P; HRAS: G13R), while no EGFR ectodomain mutations were recorded (Figure 2). The emergence of activating RAS clones correlated significantly with disease progression in this cohort (Chi-square, P = 0.032). While six of 13 patients (46%) with progressive disease during cetuximab-based treatment showed evidence of acquired activating RAS mutations, none of the seven responsive patients (0%) were mutated for any of the RAS genes at any time point (Figure 2). Some of these mutations appeared early during treatment (earliest detection nine weeks after initiation of cetuximab-based treatment) and preceded clinical progression in half of the patients with the maximum time from mutation detection to clinical progression being 16 weeks in our cohort (Figure 2).
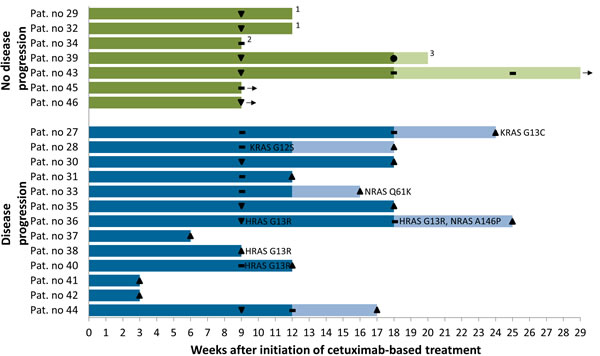
Figure 2: Swimmer plot illustrating treatment, responses and acquired mutations in liquid biopsy cohort of 20 HNSCC patients treated with cetuximab plus chemotherapy. Weeks of combination therapy with cis- or carboplatin, 5-fluorouracil and cetuximab are shown in dark colors, weeks of cetuximab maintenance in light colors. ● Complete response, ▼ partial response, ▬ stable disease, ▲ progressive disease. Activating RAS mutations are mapped at the time of their first appearance. 1Patients refused further treatment. 2Patient died of pneumonia. 3Therapy was stopped because of bleeding complications. → Ongoing treatment.
Discussion
Cetuximab-based treatment is only effective in a subset of patients with HNSCC [7]. However, little is known so far about the molecular mechanisms underlying clinical resistance and we currently lack appropriate biomarkers that could help in identifying patient subsets that are either likely or unlikely to derive benefit from this EGFR-targeted therapy or from prolonged antibody treatment in a cetuximab maintenance setting.
In this study we focused on potential modifications of the EGFR ectodomain that may interfere with antibody binding and activating mutations of RAS, which are known to confer resistance in metastatic colorectal cancer [10, 19]. While HNSCC tumors are largely negative for RAS mutations at diagnosis [14, 20] and EGFR ectodomain mutations have not been detected by conventional sequencing to date, we reasoned that potential resistance-mediating mutations could be present in rare tumor subclones before treatment (undetectable by conventional sequencing) and would subsequently be amplified under the selective pressure of EGFR-targeted antibody treatment. To be able to detect even minor subclones in a background of cells with unmutated EGFR and RAS, we used state-of-the-art targeted NGS technology for highly sensitive and specific identification of mutations in a heterogeneous tumor [21]. By comparing pre- and post-cetuximab genetic material we aimed at uncovering both primary and acquired resistance-mediating mutations. Utilizing a sequencing depth that would uncover even rare clones, none of the 46 patients included in this study showed evidence for mutations in EGFR exon 12 or KRAS/NRAS exons 2/3/4 at baseline, while two cases were HRAS mutated. About one third of cases acquired RAS mutations in the course of treatment and, interestingly, all of these cases showed progressive disease while receiving the EGFR antibody. This significant correlation suggests for the first time that activating RAS mutations represent a clinically relevant mechanism of acquired resistance in patients treated with cetuximab.
Two major limitations of this study need to be discussed: First, this study does not formally rule out the (unlikely) possibility that the platinum/5-FU treatment (and not the EGFR-targeted antibody) may induce activating RAS mutations in the HNSCC setting. In the colon cancer setting, however, there is no evidence for the induction of activating RAS mutations by chemotherapy, while there is persuasive evidence for their induction by EGFR-targeting antibodies [15]. Since patients treated with either platinum/5-FU or cetuximab alone are rare, this question is very hard to address. The second limitation of our study refers to the fact that baseline mutational profiling was performed on primary diagnosis tumor tissue (instead of tumor tissue at recurrence) in 7/20 patients and baseline liquid biopsies were not performed. Therefore, we cannot rule out acquisition of mutations between primary diagnosis and recurrence in these seven patients. Given the overall very low RAS mutational frequency in EGFR antibody-naïve patients this point may, however, be of minor clinical relevance.
Taken together, our data suggests that i) RAS mutant subclones can only be found in a minority of HNSCC tumor samples at baseline, but emerge in a substantial proportion of patients during cetuximab treatment, ii) these mutant subclones correlate significantly with disease progression, and iii) may be detectable with state-of-the-art sequencing technology before clinical resistance occurs. Prospectively, determination of such clones may help to tailor anti-EGFR strategies warranting an evaluation in larger prospective clinical trials. More specifically, mutational loads should be defined that reliably predict a lack of response to cetuximab.
Methods - patients
Between October 2012 and January 2016, the Database of the Clinical Cancer Registry of the University Cancer Center Hamburg was screened for HNSCC patients with cetuximab-based treatment. Informed consent was obtained from a total of 46 patients for the use of their diagnostic material (tumor tissue and - in 20 cases - peripheral blood after initiation of cetuximab treatment) as approved by the institutional review board. HPV-Status was part of the routine diagnostic work-up and included a PCR for HPV-DNA and p16 immunohistochemistry. Patients were considered HPV-positive, if both HPV-DNA of a high-risk HPV type and overexpression of p16 were present. All other combinations were considered HPV-negative. All tumor samples were validated by a pathologist.
All 20 patients with available post-cetuximab peripheral blood samples were treated with a combination of cetuximab weekly (400 mg/m2 as a loading dose, followed by 250 mg/m2) and a chemotherapy regimen of cisplatin (100 mg/m² on day 1) or carboplatin (AUC 5 on day 1) plus 5-fluorouracil (1000 mg/m² on days 1-4) every three weeks for a maximum of six courses. Subject to their consent, combination therapy was followed by weekly cetuximab maintenance in patients without progressive disease. Peripheral blood samples for isolation of ctDNA were taken at interim staging after three courses of combination therapy and after completion of combination therapy / maintenance (or at progression if applicable).
materials and Methods
Cell lines and cell culture
All 12 squamos cell carcinoma cell lines derived from patients with head and neck tumors [22-25] were cultivated in Dulbecco’s Modified Eagle’s Medium (Gibco/Life Technologies, Carlsbad, USA) containing 10% fetal bovine serum (Merck & Co., Inc., Kenilworth, USA), 4 mM glutamine (Gibco/Life Technologies) and 1% Penicillin Streptomycin (Gibco/Life Technologies) and were identified using a short tandem repeat multiplex assay (Applied Biosystems/Life Technologies). UT-SCC cell lines 5, 8, 14, 15, 29, 42A and 60A were kindly provided by R. Grenman, University of Turku, Finland. The HNSCC p53-negative subline of FaDu (hypopharynx) was obtained from W. Eicheler, University of Dresden, Germany [26], and all other cell lines were kindly provided by M. Baumann, University of Dresden, Germany.
Preparation of genomic DNA from tumor tissue and HNSCC cell lines
Formalin fixed paraffin embedded tumor tissue was deparaffinized by xylene and ethanol. After digestion with proteinase K at 56 °C overnight, genomic DNA was isolated with the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany). Genomic DNA from EGFR positive cell lines was extracted using NucleoSpin Tissue XS kit (Macherey-Nagel, Düren, Germany). Quantity and quality of DNA was evaluated using a Nanodrop spectrophotometer ND-1000 (Thermo Fisher Scientific Inc., Wilmington, USA).
Isolation of circulating tumor DNA (ctDNA) from blood
Blood samples were centrifuged at 1200 x g for 10 min within two hours after blood draw. ctDNA was isolated from the serum using the QIAamp Circulating Nucleic Acid Kit (Qiagen).
PCR amplification of EGFR and RAS exons for Illumina targeted next generation sequencing
In two consecutive PCR reactions, EGFR exon 12, KRAS/NRAS exons 2/3/4 and HRAS exons 2/3 were amplified from tumor or ctDNA and adapters for next-generation sequencing (NGS) were attached (schematically shown in Figure 1). The primers for the first reaction annealed with exon-flanking intron regions (dotted lines) and contained Illumina-compatible adapters (yellow) for later hybridization of amplicons to the Illumina flow cell and for sequencing primer annealing. A second PCR reaction was performed to extend the Illumina adapter sequences and to add a 6 or 7 nucleotide barcode (red) allowing to match each sequence during data analysis to a certain patient / cell line and time point. All primers are shown in Supplementary Table S1.
The PCR was performed using Phusion HS II (Thermo Fisher Scientific Inc.). Amplicons were purified after agarose gel electrophoresis using the NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel).
Illumina sequencing and data analysis
All amplicons were sequenced with a 500-cycle single indexed (8 nucleotides) paired-end run on a MiSeq (Illumina, San Diego, USA). Overlapping paired reads were merged using the software FLASH (v1.2.6) [27]. The format of the merged reads was subsequently converted to FASTA while non-overlapping reads were excluded from further analysis. Usearch (v6.0.307) [28] was employed to dereplicate and cluster the merged reads. Sequences observed less than 30 times were discarded and the remaining sequences were clustered according to their similarity with reference EGFR and RAS exon sequences (Supplementary Table S2). For each cluster of similar sequences, MAFFT (v7.045b) [29] was used to calculate a multiple sequence alignment.
Statistics
IBM® SPSS® version 22 (IBM, New York, USA) was used for statistical analysis. Contingency tables were calculated and compared using the Pearson Chi-square test. A p-value < 0.05 was considered significant.
Acknowledgments
We thank Adam Grundhoff, Malik Alawi and Daniela Indenbirken for assistance with next-generation sequencing and Christopher Ford for proofreading the manuscript.
conflicts of interest
The authors have declared no conflicts of interest.
Funding
This work was supported by Deutsche Forschungsgemeinschaft [BI 1711/1-1 to M.B.]. MB holds a professorship supported by the Hubertus Wald foundation, Hamburg.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2015; 65:5-29.
2. Budach V, Becker ET, Boehmer D, Badakhshi H, Jahn U, Wernecke KD, Stromberger C. Concurrent hyperfractionated accelerated radiotherapy with 5-FU and once weekly cisplatin in locally advanced head and neck cancer. The 10-year results of a prospective phase II trial. Strahlentherapie Onkologie. 2014; 190:250-255.
3. Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, Youssoufian H, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet oncology. 2010; 11:21-28.
4. Forastiere AA, Zhang Q, Weber RS, Maor MH, Goepfert H, Pajak TF, Morrison W, Glisson B, Trotti A, Ridge JA, Thorstad W, Wagner H, Ensley JF et al. Long-term results of RTOG 91-11: a comparison of three nonsurgical treatment strategies to preserve the larynx in patients with locally advanced larynx cancer. Journal of clinical oncology. 2013 31:845-852.
5. Posner MR, Hershock DM, Blajman CR, Mickiewicz E, Winquist E, Gorbounova V, Tjulandin S, Shin DM, Cullen K, Ervin TJ, Murphy BA, Raez LE, Cohen RB et al. Cisplatin and fluorouracil alone or with docetaxel in head and neck cancer. The New England journal of medicine. 2007; 357:1705-1715.
6. Vermorken JB, Remenar E, van Herpen C, Gorlia T, Mesia R, Degardin M, Stewart JS, Jelic S, Betka J, Preiss JH, van den Weyngaert D, Awada A, Cupissol D et al. Cisplatin, fluorouracil, and docetaxel in unresectable head and neck cancer. The New England journal of medicine. 2007; 357:1695-1704.
7. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. The New England journal of medicine. 2008; 359:1116-1127.
8. Vermorken JB, Stohlmacher-Williams J, Davidenko I, Licitra L, Winquist E, Villanueva C, Foa P, Rottey S, Skladowski K, Tahara M, Pai VR, Faivre S, Blajman CR et al. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): an open-label phase 3 randomised trial. Lancet oncology. 2013; 14:697-710.
9. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2006; 354:567-578.
10. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of clinical oncology. 2008; 26:5705-5712.
11. Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer research. 2006; 66:3992-3995.
12. Lievre A, Laurent-Puig P. Genetics: Predictive value of KRAS mutations in chemoresistant CRC. Nature reviews clinical oncology. 2009; 6:306-307.
13. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nature reviews clinical oncology. 2010; 7:493-507.
14. Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer discovery. 2013; 3:761-769.
15. Diaz LA, Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537-540.
16. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486:532-536.
17. Arena S, Bellosillo B, Siravegna G, Martinez A, Canadas I, Lazzari L, Ferruz N, Russo M, Misale S, Gonzalez I, Iglesias M, Gavilan E, Corti G et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clinical cancer research. 2015; 21:2157-2166.
18. Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Seshagiri S, Serrano S et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nature medicine. 2012; 18:221-223.
19. Braig F, Marz M, Schieferdecker A, Schulte A, Voigt M, Stein A, Grob T, Alawi M, Indenbirken D, Kriegs M, Engel E, Vanhoefer U, Grundhoff A et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget. 2015; 6:12035-12047. doi: 10.18632/oncotarget.3574.
20. Bissada E, Abboud O, Abou Chacra Z, Guertin L, Weng X, Nguyen-Tan PF, Tabet JC, Thibaudeau E, Lambert L, Audet ML, Fortin B, Soulieres D. Prevalence of K-RAS Codons 12 and 13 Mutations in Locally Advanced Head and Neck Squamous Cell Carcinoma and Impact on Clinical Outcomes. International journal of otolaryngology. 2013; 2013:848021.
21. Samuel N, Hudson TJ. Translating genomics to the clinic: implications of cancer heterogeneity. Clinical chemistry. 2012; 59:127-137.
22. Lange MJ, Lasiter JC, Misfeldt ML. Toll-like receptors in tonsillar epithelial cells. International journal of pediatric otorhinolaryngology. 2009; 73:613-621.
23. Nii M, Kayada Y, Yoshiga K, Takada K, Okamoto T, Yanagihara K. Suppression of metastasis by tissue inhibitor of metalloproteinase-1 in a newly established human oral squamous cell carcinoma cell line. International journal of oncology. 2000; 16:119-124.
24. Soffar A, Storch K, Aleem E, Cordes N. CDK2 knockdown enhances head and neck cancer cell radiosensitivity. International journal of radiation biology. 2013; 89:523-531.
25. Lin CJ, Grandis JR, Carey TE, Gollin SM, Whiteside TL, Koch WM, Ferris RL, Lai SY. Head and neck squamous cell carcinoma cell lines: established models and rationale for selection. Head & Neck. 2007; 29:163-188.
26. Eicheler W, Zips D, Dorfler A, Grenman R, Baumann M. Splicing mutations in TP53 in human squamous cell carcinoma lines influence immunohistochemical detection. Journal of histochemistry & cytochemistry. 2002; 50:197-204.
27. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011; 27:2957-2963.
28. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26:2460-2461.
29. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research. 2002; 30:3059-3066.