
INTRODUCTION
The miR-145-5p is deregulated in several tumors [1-6]. The tumor suppressor actions of miR-145-5p comprise inhibition of cell growth and metastasis [7-9], induction of apoptosis [10] and repression of pluripotency in embryonic stem cell [11]. These actions depend on the fact that miR-145-5p targets several genes relevant to these processes, some of which, such as MDM2, linked to the TP53 pathway [9, 11-15]. TP53 is a transcriptional activator of the miR-145-5p, is implicated in the miR maturation complex and is positively regulated by the miR-145-5p [10, 13, 15, 16]. Thus the tumor-suppressor activity of the miR-145-5p is linked to the TP53 mutational/expression status [10, 16, 17].
Hepatocellular carcinoma (HCC), third most common cause of cancer-related mortality worldwide [18, 19], is associated with several chromosomal, genetic and epigenetic aberrations [3, 25-35]. Mutations in the TP53 cover only around 20% of all HCCs [20]. On the other hand lipid and glucose metabolisms are impaired in all HCCs [21-23] and HCC risk is associated with viral infections and/or metabolic disorders that promote glycolysis/lipogenesis [24].
In 50% of HCCs the miR-145-5p is down-regulated [25]. Here we show that in those HCCs with physiologic expression of the miRNA, the resistance to the pro-apoptotic miR-145/TP53 signaling depends on the over-expression of the miR-483-3p, which targets BBC3/PUMA [26]. Thus miR-483-3p could be a crucial suppressor of miR-145/TP53 signaling in the HCCs with functional TP53.
RESULTS
The miR-145-5p induces cell growth inhibition and cell death by enhancing TP53 activity in HepG2 cells
The miR-145-5p has been involved in pro-apoptotic signaling through TP53-dependent mechanisms [3, 10, 15, 27-29]. Here, to confirm this mechanism in liver cancer cells, we studied the effects of the enforced expression of miR-145-5p in HepG2 cells, a TP53 wild type hepatoblastoma cell line. Following cell transfection, we found that miR-145-5p induces a significant cell growth inhibition (p<0.05) after 72 hours (Figure 1A–1B).
In HepG2 cells we confirmed the link between the miR-145-5p and the TP53 pathway. Enforced expression of the miR-145-5p determined increased luciferase activity of the pP53-TA-luc, a TP53 responsive reporter vector (p<0001; Figure 1C) together with augmented mRNA levels of two TP53 downstream targets, CDKN1A and BBC3 (Figure 1D). Moreover, silencing of TP53 could partially rescue the effects of miR-145-5p on cell viability (Figure 1E)
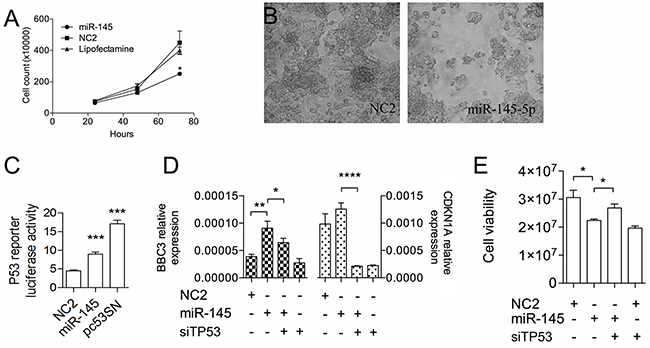
Figure 1: The miR-145-5p inhibits HepG2 cell growth by activating TP53. A. Growth kinetics of HepG2 cells transiently transfected with either miR-145-5p precursor or scramble sequence (NC2) or vehicle of transfection (Lipofectamine). B. Cell morphology of HepG2 cells at 72 hours after transfection with either miR-145-5p or NC2. C. TP53 dependent transcriptional activity measured by the TP53 responsive luciferase reporter vector, pP53-TA-luc, in HepG2 transfected with either miR-145-5p or NC2 or an expression vector carrying the human wild type TP53 cDNA (P53). Firefly luciferase activity was normalized on Renilla luciferase acitivity generated by the co-transfected vector pRL-TK. D. CDKN1A and BBC3 expression by RT-qPCR and E. Luminescent cell viability assay of HepG2 cells treated (48 hours) with miR-145-5p alone or in combination with siRNA against TP53 (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
The miR-483-3p protects HepG2 cells from miR-145-induced cell death by targeting BBC3/PUMA
To study the anti-tumor effects induced by the miR-145-5p in a subset of HCCs showing its physiological expression, we generated stable HepG2 cell clones carrying either the miR-145-5p or the control vector. Selection yielded hundreds of clones for the control vector but only 2 viable clones for the miR-145 (H8 and H9) (Figure 2A), suggesting that these clones developed resistance to the miR-145-5p constitutive expression. To identify potential interplay amongst miRNAs, we performed miRNA profiling on RNA from HepG2 cells and HepG2 H9 clone. We included in the microarray analysis cells with high expression of the miR-145-5p determined by either exogenous expression of TP53 or MDM2 silencing or Nutlin-3a treatment (Supplementary Figure S1). The analysis revealed 6 up-regulated miRNAs, with the miR-483-3p in the top list (Figure 2B). In the H9 clone the miR-483-3p exhibited a 10-fold increased expression compared to HepG2 cells. We recently reported the oncogenic activity of miR-483-3p due, at least in part, to its target PUMA [26]. Therefore we evaluated protein and mRNA levels of PUMA in the H8 and H9 clones. Both clones exhibited increased levels of BBC3 mRNA (Figure 2C), but reduced levels of PUMA protein compared to HepG2 cells (Figure 2D).
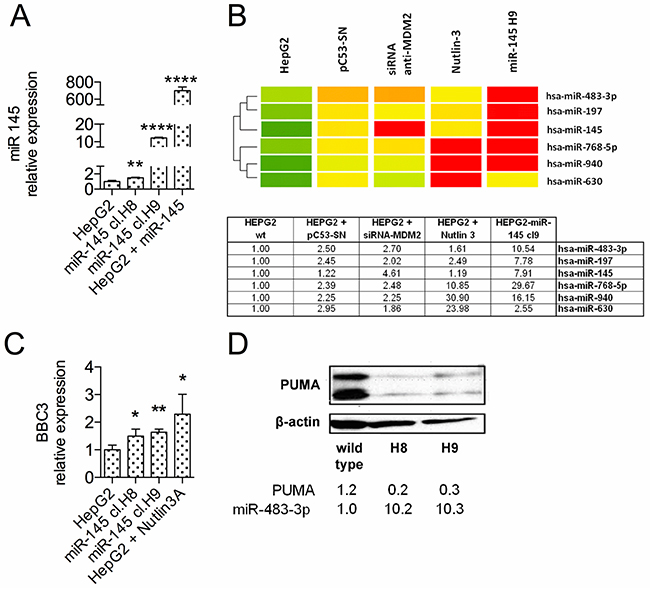
Figure 2: The miR-483-3p expression is induced in HepG2 stable clones over-expressing miR-145-5p. A. ThemiR-145-5p expression was evaluated by RT-qPCR in HepG2-miR-145 stable cell clones H8 and H9, in comparison to wild type cells and to miR-145 transiently transfected cells. H8 and H9 clones exhibited a 1.5-12 fold increase of miR-145-5p expression, compared to wild-type cells. B. Heatmap representation of the miRNAs increased in miR-145-5p over-expressing cells assessed using microRNA microarray. Fold-change analysis revealed an increased expression of 6 microRNAs both in HepG2 cells with an activated TP53 (PC53SN, siRNA anti-MDM2 and Nutlin-3) and HepG2 cells over-expressing miR-145 (miR-145 H9 stable clone) compared to HepG2 cells. C. PUMA expression was evaluated by RT-qPCR and D. by Western Blot in H8 and H9 cell clones in comparison to HepG2 cells. Nutlin-3A-treated-HepG2 cells were used as positive control in quantitative PCR analysis. PUMA expression levels were normalized according to β-actin expression in the same samples (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
To assess the role of miR-483-3p in cell protection, we transfected HepG2 cells with miR-145-5p together with an anti-miR-483-3p oligonucleotide. The combination resulted in increased cell growth inhibition (p<0.002) (Figure 3A). We also inhibited miR-483-3p expression by LNA oligonucleotide in the H9 clone. This treatment reduced cell growth up to 40% at 72 hours (p<0.0001) (Figure 3B), caused an increase of PUMA expression (Figure 3C), and induced caspase 3/7 activity (Figure 3D). A key role of PUMA was confirmed by a specific siBBC3, which abrogated the induction of caspase activity after the inhibition of miR-483-3p (Supplementary Figure S2). These results support the hypothesis that the miR-483-3p confers resistance to TP53-dependent apoptosis in HepG2 cells over-expressing the miR-145-5p.
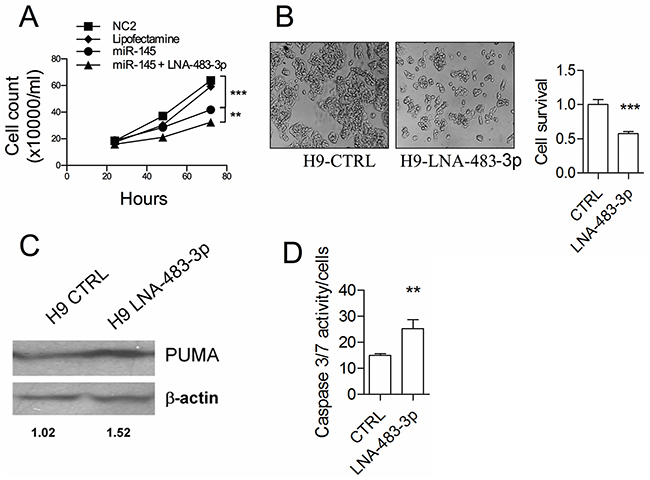
Figure 3: The miR-483-3p protects miR-145-5p induced cell death by targeting BBC3. A. Growth kinetics of HepG2 cells transiently transfected with either miR-145-5p precursor or and LNA-483-3p or scramble sequence (NC2) or vehicle of transfection (Lipofectamine). B. Cell viability by inverted microscope analysis and luminescent cell viability assay of H9 HepG2 stable clone transfected as described in A). C. BBC3 protein (PUMA) expression by western blot of the H8 and H9 HepG2 stable clones and HepG2 wild type cell line. Protein loaded was normalized by measuring the β-actin expression. Densitometric analysis is indicated. D. caspase 3/7 activity of the H9 stable clone transiently transfected with either LNA-483-3p or LNA scramble sequence (CTRL) (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
To validate these data in another liver cancer in vitro model we looked at the expression of the miR-483-3p in liver cancer cell lines harbouring either wild type (Huh-6) or mutant (SNU-449) or null (Hep3B) TP53 (Supplementary Figure S3A). We chose the HCC Hep3B cell line because of the higher expression of the miR-483-3p (Supplementary Figure S3A). Since they are TP53 null we reestablished wild type TP53 protein expression and functional activity by the pc53SN vector (Supplementary Figure S3B). In this model PUMA was not regulated by exogenous TP53 (Supplementary Figure S3B) but still targeted by miR-483-3p (Supplementary Figure S3C). AMO-483 was able to further increase the PUMA levels in presence of TP53 protein (Supplementary Figure S3C), the caspase 3/7 activity followed the same trend (Supplementary Figure S3D). The effect of the miR-483-3p in Hep3B cells TP53 null could be imputable to the regulation of PUMA by the TP73 protein instead of the TP53 (TP53 family: TP53, TP63 and TP73) [30-32].
To further validate the hypothesis that miR-483-3p protects from apoptosis in a system in which the miR-145/TP53 signaling acts properly, we used SNU-449 cellular model that harbour a mutant form of TP53 (Sanger Cosmic Database) and a very low expression of the miR-483-3p (Supplementary Figure S3A). We registered no differences between the caspase activity of cells transfected with either the miR-483-3p or the control and even an induction when those cells were co-transfected with the pc53SN vector (Supplementary Figure S4).
The miR-145-5p/miR-483-3p circuitry is lost in HCCs
We searched for an association between miR-145-5p and miR-483-3p also in primary HCCs and non-neoplastic liver. We analyzed the expression of miR-145-5p and miR-483-3p on 41 RNA samples from non-neoplastic (NN) hepatic tissue, 40 cirrhotic liver samples (CL) and 1 normal liver (NL) (Supplementary Table S1); a weak but significant negative correlation between the expression of the two miRNAs was detected (ρ=-0.342, P=0.028; Figure 4A). Since enforced expression of the miR-483-3p abrogated the TP53/miR-145 pro-apoptotic loop in HepG2 cells, we searched for evidence of such mechanism in primary HCCs. By analyzing RNA samples from 21 HCC, we found a strong positive correlation between the levels of the two miRNAs (ρ=0.793, P<0.0001; Figure 4B), which supports the hypothesis that high levels of the oncogenic miR-483-3p counterbalance high expression of the pro-apoptotic miR-145-5p and that in normal cells there is a negative feedback loop between miR-145-5p and miR-483-3p that is lost in tumor cells.
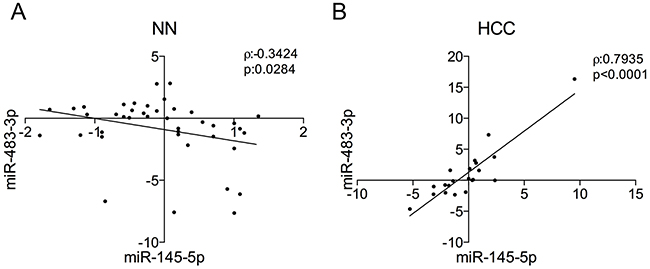
Figure 4: miR-145-5p/miR-483-3p correlations in non-neoplastic liver and hepatocellular carcinoma. The miR-145-5p and miR-483-3p expressions obtained by microarray analysis or RT-qPCR were evaluated in A. non-neoplastic liver samples (NT) and B. hepatocellular carcinomas (HCC) respectively. Spearman correlation (ρ) and correlation p value (p) are indicated.
To strengthen our data, we evaluated the spearman correlation factors in other studies that reported miRNA profiling in HCC and NN liver (ArrayExpress: E-GEOD-30297; E-TABM-866) [33, 34]. In Pineau’ study the correlation factor is negative in both NN liver (CL, n=90; NL, n=21) and HCC (n=104), but less strong in HCC than in NN liver (CL, ρ=-0.55, P<0.0001; HCC, ρ=-0.31, P:0.0013)(Supplementary Figure S5A, S5B), whereas the data from Barry et al. confirmed the loss of the negative correlation between miR-145-5p and miR-483-3p typical of NN liver (Figure 4A; Supplementary Figure S5A), by showing a positive correlation in their set of HCC sample (n=97; ρ:-0.025, P=0.8) (Supplementary Figure S5C). In vivo studies were conducted by Wang et al., who performed hepatic microRNAs profiling at very early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet (CDAA) in C57BL/6 mice [35]. We analyzed this dataset for the expression of the mmu-miR-145 and mmu-miR-483* in five mice for each diet group at each time point (6, 18, 32 and 65 weeks) and we identified a negatively correlated expression of the two miRNAs in the control group (CSAA) at all time-points, that was dampened in the CDAA treated group (Supplementary Figure S6).
TP53/miR-145-5p signaling rules miR-483-3p expression and cell death dependent on the glucose concentration of the medium
Next we investigated the events that could invert the correlation between the expression of miR-483-3p and miR-145-5p in HCC cells. Given that miR-145-5p acts as an inhibitor of cellular glucose uptake in HepG2 [36] and that miR-483-3p is regulated by cellular glucose availability (A.V. unpublished dataset), we investigated if glucose could modulate the effects of the miR-145-5p on the miR-483-3p. We transfected HepG2 cells with miR-145-5p mimic in either low (1 g/L) or high glucose (4.5 g/L) DMEM. MiR-145-5p induced down regulation of miR-483-3p under low-glucose and up-regulation under high-glucose conditions (Figure 5A). This mechanism was independent of IGF2 expression since it showed an opposite trend of regulation when compared to the miR-483-3p mature expression (Supplementary Figure S7A, S7B). This suggests that glucose availability affects the regulation of the miR-483-3p by the miR-145-5p. The same results were obtained in Hep3B cells in which we restored the TP53 protein expression (Supplementary Figure S8A, S8B). Next we hypothesized that HepG2 cells cultured in high-glucose could overcome the tumor suppressive effects of miR-145-5p through the up-regulation of miR-483-3p. To verify this hypothesis we quantified the apoptotic HepG2 cells after transient transfection with miR-145-5p under either low or high glucose conditions. As expected, miR-145-5p induced apoptosis in low glucose, as shown by the increase in Annexin V-stained HepG2 cells (32.5% vs 43.4%), whereas in high-glucose the miR-145-5p decreased the number of apoptotic cells (57.9% vs 48.1%) (Figure 5B upper panel). We obtained similar results with Propidium Iodide (PI) staining (Figure 5B lower panel).
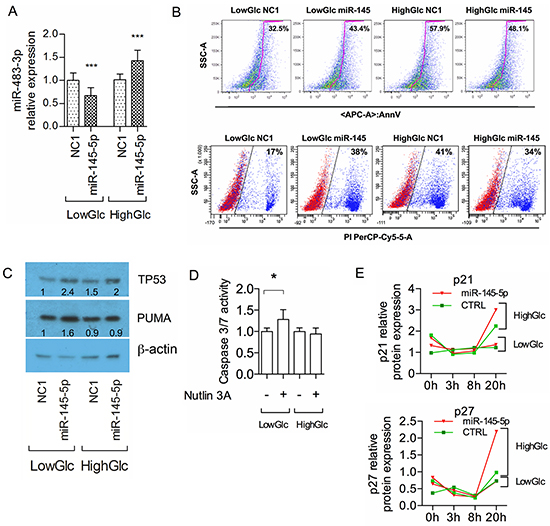
Figure 5: Glucose media concentrations rule the miR-145-5p effects on HepG2 cells. A. miR-483-3p relative expression by RT-qPCR normalized on RNU44 in HepG2 cells transfected with either miR-145-5p or negative control miR (NC1); cells were grown in either low (LowGlc) or high (HighGlc) glucose concentration (1 g/L and 4.5 g/L respectively). B. Flow cytometry analysis of annexin-V (upper panel) and propidium iodide (bottom panel) staining apoptotic/dead HepG2 cells following transfection with either miR-145-5p or control (NC1) grown for 72h in different glucose concentrations. C. TP53 and PUMA/BBC3 protein relative concentration by Western blot in HepG2 cells treated as described in B). D. Caspase 3/7 activity in HepG2 cells transfected with miR-145-5p or control (NC1) in low and high glucose concentration. E. P21 (upper panel) and P27 (bottom panel) relative protein concentration normalized on β actin protein expression by Western blot in HepG2 cells blocked with thymidine for 17 hours and transfected with miR-145-5p (red triangles) or control NC1 (green squares) in low glucose and high glucose at 0, 3, 8 and 20 hours. In the graphs of RT-qPCR and Caspase 3/7 activity the data are represented by the means and standard deviations of technical and experimental replicates. Student t test was used for the statistical analysis (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
Since we showed that miR-145-5p is able to induce TP53 activity, we tested its effect on TP53 protein levels by western blot. Cells with higher expression of the miR-145-5p revealed increased TP53 protein levels under both low and high glucose (low: +143%; high: +33%, compared to the NC1 transfections), but PUMA, target of the miR-483-3p, increased only in low-glucose (Figure 5C). This is in line with the evidence that miR-483-3p expression is lowered by miR-145-5p in low-glucose and increased in high-glucose (Figure 5A). To further support these results we indirectly induced miR-145-5p expression by activating TP53 using Nutlin-3a, an inhibitor of the E3 ubiquitin-protein ligase MDM2, under both low and high glucose. As expected, caspase 3/7 activity was slightly increased only in low glucose (Figure 5D). Induced expression of the miR-145-5p after transfection and Nutlin-3a treatment and consequent regulation of miR-483-3p were measured by RT-qPCR (Supplementary Figure S7A, S7C, S7D).
Our data show how miR-145/TP53/PUMA signaling induces apoptosis only in HepG2 cells cultured in a low–glucose; however miR-145-5p exerts a growth inhibitory effect also in high glucose, as shown in Figure 1 and 3. Since PUMA contributes to autophagy [37] we hypothesized that autophagy could be activated by the miR-145-5p/miR-483-3p regulation in HepG2 cells. By measuring the GFP-LC3-II accumulation at the autophagosomal membranes we found that miR-145-5p was able to induce autophagy in HepG2 cells under both low and high glucose (Supplementary Figure S9A–S9B). Next we investigated the effect of the miR-145-5p on cell cycle regulation. We analyzed the protein levels of p21 and p27 after enforced expression of miR-145-5p under both glucose conditions. The p21 and p27 proteins were induced by miR-145-5p ectopic expression only in HepG2 cells cultured in high glucose (Figure 5E, Supplementary Figure S9C). Overall these data suggest that the miR-145-5p is able to induce apoptosis in HepG2 cells cultured in low glucose, while it only slows down the cell cycle under high glucose.
Stable activation of TP53 through Nutlin-3a selects HepG2 clones with impaired miR-483/miR-145 ratio
Finally, we used Nutlin-3a (2.5 μM) under low-glucose to constitutively activate TP53 and induce resistance of HepG2 cells to death. After 30 days we counted five clones in HepG2 cells treated with Nutlin-3a. These clones were picked up and maintained in low glucose conditions for a few days; during such period only one (clone 4, cl.4) survived. After that, we re-established Nutlin-3a until the 65th day. At the same time, as a reference experiment, we treated HepG2 cells with Nutlin-3a at two concentrations (2.5 and 5 μM) for a 4 days period, sampling the RNA every day. To achieve comparability between the two experiments and determine the interplay between miR-483-3p and miR-145-5p, we took in consideration the ratio of the relative expressions of these two miRNAs (miR-483/miR-145) (Figure 6A). The HepG2 cl.4 between 50 and 65 days showed significant increases in the miR-483-3p/miR-145-5p ratio (Figure 6A, right side), contrary to what we observed after short term exposure to Nutlin-3a (Figure 6A, left side), that resulted a decrease of the miR-483-3p/miR-145-5p ratio. Our data demonstrate the ability of miR-483-3p to elude the miR-145/TP53 signaling, suggesting that this microRNA selects HCC cells that show a physiologic miR-145/TP53 signaling to nullify the tumor suppressive actions. To support this conclusion, we performed a meta-analysis correlating TP53 mutational status with miR-483-3p and miR-145-5p expression in 193 HCC samples from the TCGA dataset (http://firebrowse.org) (TP53 Mut: 60; TP53 Wt: 133). The miR-145-5p showed no difference in expression between the two groups (Figure 6C), whereas, as expected, the miR-483-3p was significantly up regulated in the TP53 Wt HCCs when compared to those harboring TP53-mutations (Figure 6B).
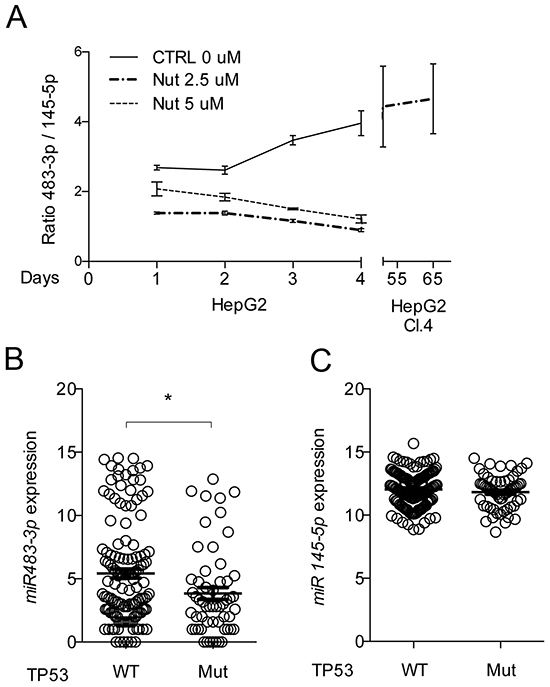
Figure 6: miR-145-5p and miR-483-3p expressions related to TP53 activity and mutational status. A. Long treatment with Nutlin-3a selects HepG2 cells with high miR-483-3p/miR-145-5p expressions rate. miR-483-3p/miR-145-5p relative expressions rate of HepG2 treated with Nutlin-3a (2.5 μM and 5 μM) and vehicle (CTRL) during four days of treatment (left side). On the right side of the graph is represented the miR-483-3p/miR-145-5p relative expressions rate of the HepG2 stable clone 4 (HepG2 Cl.4) after 51 and 60 days of treatment with Nutlin-3a. Expression level of miR-483-3p B. and miR-145-5p C. in 199 HCC samples with (n=60) or without (n=133) mutations in TP53 gene. The expression of miR-483-3p is significantly higher (p 0.01 at two-sided t-test) in TP53 WT than TP53 mutated HCC. The results shown here are based upon miRseq_mature_preprocess data generated by the TCGA Research Network: http://cancergenome.nih.gov/ and downloaded from FireBrowse portal (*: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001).
DISCUSSION
The expression of the miR-145-5p is generally down-regulated in HCC and correlated to tumor grade (Edmondos grade) [3, 27], but it is not uncommon to find HCCs that show normal expression of the miR-145-5p. We relied on HepG2 cell clones stably expressing miR-145-5p to investigate the possible processes of resistance to cell death induced by TP53/miR-145-5p signaling in this particular HCC subset. These HepG2 clones showed up-regulation of 5 microRNAs related to TP53 activity: miR-483-3p, miR-197, miR-768-5p, mir-940 and miR-630. Interestingly 4 out of these 5 deregulated miRNAs are involved in resistance to apoptosis: miR-483-3p is known to target BBC3/PUMA in HCC and CRC [26, 38]; miR-197 is deregulated in lung cancers harbouring wild type TP53 [39]; miR-940 and miR-630 are up-regulated in cisplatin-resistant lung and head and neck cancers [40-42]. We focused on the most significantly up-regulated miRNA, the miR-483-3p. We demonstrated that the over-expression of miR-483-3p is an important factor to overcome the pro-apoptotic effects of miR-145-5p. We also identified a negative correlation between the miR-145-5p and the miR-483-3p in non-neoplastic liver that became less evident, lost or positive in HCCs. This suggests the presence of a regulative loop between these two microRNAs: in physiological conditions miR-145/TP53 signaling results in the inhibition of miR-483-3p (negative correlation), whereas in neoplastic conditions miR-145-5p selects cells that show higher expression of miR-483-3p (positive correlation). The expression and activity of the miR-145-5p is associated to TP53 [10, 15, 16] whereas miR-483-3p is correlated to β–catenin nuclear activity [38]. Since mutations in TP53 and β–catenin are mutually exclusive in HCC [20], we speculate that the HCCs that show TP53 mutations, and consequent down regulation of PUMA, p21 and other TP53 targets, do not need the up-regulation of miR-483-3p to reduce the apoptotic rate. On the contrary, as confirmed by our data, the HCCs with wild-type TP53 select cells with higher expression of the miR-483-3p. We found that, on the basis of glucose availability, the miR-145-5p represses (low glucose) or induces (high glucose) the expression of the miR-483-3p. We propose that increased glucose uptake in the progression from pre-neoplastic to neoplastic liver cells and the metabolic shift from aerobic to anaerobic metabolism (Warburg’s effect), typical of cancer [43, 44], affect the physiologically negative regulation of miR-483-3p and the miR-145/TP53 axis, dampening the anti-tumoral effect of this pathway and facilitating tumor development. Thus, to exclude resistance via miR-483-3p upregulation, the possible therapeutic development of miR-145-5p mimics should comprise strategies directed also to the concurrent inhibition of the miR-483-3p.
MATERIALS AND METHODS
Cell lines and in vitro assays
Hepatoblastoma (HepG2, Huh-6) and hepatocarcinoma (Hep3B, SNU-449) cell lines are from ATCC (UK). HepG2 cells harbour a wild type TP53 gene, while Hep3B are null for the TP53 gene (www-p53.iarc.fr). They were cultured in IMEM/DMEM supplemented with 10% fetal calf serum. For in vitro cell growth assays, cells were seeded in 24-well plates at a density of 70,000cells/well. The transfection was performed by Lipofectamine2000 protocol (Invitrogen, CA, USA) with a final concentration of 100 nM for either miR-145 precursor or negative control molecule (Ambion negative control #2). Four replicates of each condition were counted at each time point (0, 24, 48 and 72 hours). Time 0 was the time of transfection. siRNA-PUMA (Dharmacon) and siRNA-p53 (Dharmacon) were transfected at final concentration of 100 nM. To test the effect of Nutlin-3a on miR-145 expression, we treated cells for 48 hours at a final concentration of 5 μM. The number of cells was counted using hemocytometer chamber. Cell viability was analyzed by using CellTiter- GloTM Luminescent Cell Viability Assay (Promega, WI, USA), and, apoptosis by using Caspase- Glo™ 3/7 Assay (Promega, WI, USA)
MicroRNA precursor molecules and siRNAs
Synthetic microRNA precursor molecules and negative controls (Ambion negative control #2) were purchased form Ambion (Austin, TX). The synthetic oligos were dissolved in nuclease free water to a stock concentration of 50μM. Small interfering RNA (siRNA) against TP53 and PUMA were purchased from Dharmacon (Thermo Scientific, MA, USA). Anti-miR-483-3p LNA oligonucleotide was purchased from Exiqon (Vedbaek, Denmark).
TP53 expression and reporter vectors
The reporter vector pp53-TA-Luc (Clontech Carlsbad, CA, USA) was used to quantify TP53 transcriptional activity. The vector contains a TP53 responsive element located upstream the TATA box from the herpes simplex virus timidine kinase promoter (pTA). Downstream of pTA is the firefly luciferase reporter gene (Luc). The pRL-TK vector contains the herpes simplex virus thymidine kinase promoter to provide low to moderate levels of Renilla luciferase expression in co-transfected mammalian cells. The vector was used as an internal control reporter in combination with pp53-TA-Luc reporter vector. Luciferase activity was measured using a dual luciferase kit (Promega, WI, USA) and quantified at a luminometer (Turner, Biosystems, Sunnyvale, CA, USA). Each condition was assayed in four replicates in two independent experiments. The TP53 mammalian expression vectors pc53SN, which carries a human wild type TP53 cDNA were a kind gift of Dr. Arnold Levine and were previously described [45].
RNA isolation, retrotranscription and quantitative PCR
The RNA purification by Trizol was performed according to manufaturer’s indications (Invitrogen, Carlsband, CA, USA). For mature microRNA quantification we performed a Taqman Real time PCR, using miR-145-5p, miR-483-3p, RNU6B and RNU44 probes (Applied Biosystems, Foster City, CA). 25 ng of total RNA was retrotranscribed using the specific stemloop primers. For BBC3/PUMA, CDKN1A/p21 and TP53 and quantification we used Taqman assays from Applied Biosystems (Foster City, CA). IGF2 expression were quantified by using UPL technology (Roche) (UPL: #40; U40_IGF2_F: acaccctccagttcgtctgt; U40_IGF2_R: gaaacagcactcctcaacga). First, 0.5-1 ug of total RNA was retrotranscribed by SuperScript II (Invitrogen, Carlsband, CA, USA) with random hexamers; then, PCR amplification was performed using Taqman primers and probes as indicated by manufacturer (Applied Biosystems, Foster City, CA). microRNAs expressions were normalized to RNU6B or RNU44 expressions, whereas mRNAs were normalized on ACTB expression. The delta Ct method was used to calculate the relative abundance.
Western blotting and antibodies
Cells were collected by trypsin-EDTA and dissolved in NP40 lysis buffer (0.5% NP40, 250mM NaCl, 50mM Hepes, 5mM EDTA and 0.5mM EGTA) freshly supplemented with Complete inhibitor (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail 1&2 (Roche, Mannheim, Germany). The following antibodies were used for the detection of p53 (polyclonal p53 Antibody #9282 Cell Signaling, Danvers, MA, 1:1000), CDKN1A/p21 (clone CP74, P1484 Sigma-Aldrich, St. Louis, MO, 1:200), PUMA (polyclonal PUMA Antibody #4976 Cell Signaling, Danvers, MA, 1:1000) and β-actin (clone AC-40, A4700 Sigma Aldrich, St. Louis, MO, 1:1000).
Human microRNA microarray detection and data analysis
MiRNA expression was investigated using the Agilent Human miRNA microarray v.2 (#G4470B, Agilent Technologies). This microarray consists of 60-mer DNA probes synthesized in situ and contains 15,000 features which represent 723 human microRNAs, sourced from the Sanger miRBASE database (Release 10.1). One-hundred ngs of total RNA were employed in each experiment. RNA labeling and hybridization were performed in accordance to manufacturer’s indications. Agilent scanner and the Feature Extraction 10.5 software (Agilent Technologies) were used to obtain the microarray rawdata. Microarray results were analyzed by using the GeneSpring GX 10 software (Agilent Technologies). Data transformation was applied to set all the negative raw values at 1.0, followed by a Quantile and on-gene median normalization. Fold-change analysis was used to identify the microRNAs activated both by TP53 (pc53SN, siRNA anti-MDM2 and Nutlin-3a) and miR-145-5p (miR-145 H9 stable clone).
Statistical analysis
The Student’s t test was used to compare average values between groups of samples (such as miRNA expression data, number of proliferating cells, etc). All reported p-values were calculated assuming groups with unequal variance.
ACKNOWLEDGMENTS
We thank Elvira d’Annunzio and Augusto Bevilacqua for excellent technical and management assistance. RV is supported by Marie Curie Career Integration Grants (GA-2011-303735). This work was supported by grants from the Marie Curie Career Integration Grant to AV (GA-2011-303987), the Italian Ministry for Health to AV (GR-2011-02350699) and the Italian Association for Cancer Research (AIRC) to MN and AV (MFAG2011-12142).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005; 65:7065-7070.
2. Bandres E, Cubedo E, Agirre X, Malumbres R, Zarate R, Ramirez N, Abajo A, Navarro A, Moreno I, Monzo M and Garcia-Foncillas J. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer. 2006; 5:29.
3. Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, Croce CM, Bolondi L and Negrini M. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007; 67:6092-6099.
4. Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, Calin GA, Menard S and Croce CM. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007; 67:8699-8707.
5. Ozen M, Creighton CJ, Ozdemir M and Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008; 27:1788-1793.
6. Liu X, Sempere LF, Galimberti F, Freemantle SJ, Black C, Dragnev KH, Ma Y, Fiering S, Memoli V, Li H, DiRenzo J, Korc M, Cole CN, Bak M, Kauppinen S and Dmitrovsky E. Uncovering growth-suppressive MicroRNAs in lung cancer. Clin Cancer Res. 2009; 15:1177-1183.
7. Cho WC, Chow AS and Au JS. Restoration of tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung adenocarcinoma patients with epidermal growth factor receptor mutation. Eur J Cancer. 2009; 45:2197-2206.
8. Wang S, Bian C, Yang Z, Bo Y, Li J, Zeng L, Zhou H and Zhao RC. miR-145 inhibits breast cancer cell growth through RTKN. Int J Oncol. 2009; 34:1461-1466.
9. Sachdeva M and Mo YY. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010; 70:378-387.
10. Spizzo R, Nicoloso MS, Lupini L, Lu Y, Fogarty J, Rossi S, Zagatti B, Fabbri M, Veronese A, Liu X, Davuluri R, Croce CM, Mills G, Negrini M and Calin GA. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-alpha in human breast cancer cells. Cell Death Differ. 2010; 17:246-254.
11. Xu N, Papagiannakopoulos T, Pan G, Thomson JA and Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009; 137:647-658.
12. Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, DeAngelis T and Baserga R. Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J Biol Chem. 2007; 282:32582-32590.
13. Sachdeva M and Mo YY. p53 and c-myc How does the cell balance “yin” and “yang”? Cell Cycle. 2009; 8:1303-1303.
14. Cho WC, Chow AS and Au JS. MiR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR and NUDT1. RNA Biol. 2011; 8:125-131.
15. Zhang J, Sun Q, Zhang Z, Ge S, Han ZG and Chen WT. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene. 2013; 32:61-69.
16. Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S and Miyazono K. Modulation of microRNA processing by p53. Nature. 2009; 460:529-533.
17. Suh SO, Chen Y, Zaman MS, Hirata H, Yamamura S, Shahryari V, Liu J, Tabatabai ZL, Kakar S, Deng G, Tanaka Y and Dahiya R. MicroRNA-145 is regulated by DNA methylation and p53 gene mutation in prostate cancer. Carcinogenesis. 2011; 32:772-778.
18. Ferlay J, Shin HR, Bray F, Forman D, Mathers C and Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010; 127:2893-2917.
19. El-Serag HB and Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007; 132:2557-2576.
20. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012; 44:694-698.
21. Gao S, Li A, Liu F, Chen F, Williams M, Zhang C, Kelley Z, Wu CL, Luo R and Xiao H. NCOA5 haploinsufficiency results in glucose intolerance and subsequent hepatocellular carcinoma. Cancer Cell. 2013; 24:725-737.
22. Huang Q, Tan YX, Yin PY, Ye GZ, Gao P, Lu X, Wang HY and Xu GW. Metabolic Characterization of Hepatocellular Carcinoma Using Nontargeted Tissue Metabolomics. Cancer Research. 2013; 73:4992-5002.
23. Karagozian R, Derdak Z and Baffy G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism. 2014; 63:607-617.
24. Michelotti GA, Machado MV and Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013; 10:656-665.
25. Law PT, Ching AK, Chan AW, Wong QW, Wong CK, To KF and Wong N. MiR-145 modulates multiple components of the insulin-like growth factor pathway in hepatocellular carcinoma. Carcinogenesis. 2012; 33:1134-1141.
26. Veronese A, Lupini L, Consiglio J, Visone R, Ferracin M, Fornari F, Zanesi N, Alder H, D’Elia G, Gramantieri L, Bolondi L, Lanza G, Querzoli P, Angioni A, Croce CM and Negrini M. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010; 70:3140-3149.
27. Noh JH, Chang YG, Kim MG, Jung KH, Kim JK, Bae HJ, Eun JW, Shen Q, Kim SJ, Kwon SH, Park WS, Lee JY and Nam SW. MiR-145 functions as a tumor suppressor by directly targeting histone deacetylase 2 in liver cancer. Cancer Lett. 2013; 335:455-462.
28. Boominathan L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev. 2010; 29:613-639.
29. Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K and Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009; 106:3207-3212.
30. Kim JS, Lee JM, Chwae YJ, Kim YH, Lee JH, Kim K, Lee TH, Kim SJ and Park JH. Cisplatin-induced apoptosis in Hep3B cells: mitochondria-dependent and -independent pathways. Biochem Pharmacol. 2004; 67:1459-1468.
31. Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C and Vousden KH. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004; 279:8076-8083.
32. Ray RM, Bhattacharya S and Johnson LR. Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73- and E2F1-mediated expression of PUMA and Siva-1. Apoptosis. 2011; 16:35-44.
33. Barry CT, D’Souza M, McCall M, Safadjou S, Ryan C, Kashyap R, Marroquin C, Orloff M, Almudevar A and Godfrey TE. Micro RNA expression profiles as adjunctive data to assess the risk of hepatocellular carcinoma recurrence after liver transplantation. Am J Transplant. 2012; 12:428-437.
34. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM and Dejean A. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010; 107:264-269.
35. Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T, Schmittgen TD, Croce C, Ghoshal K and Jacob ST. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009; 50:1152-1161.
36. Wen F, Yang Y, Jin D, Sun J, Yu X and Yang Z. MiRNA-145 is involved in the development of resistin-induced insulin resistance in HepG2 cells. Biochem Biophys Res Commun. 2014; 445:517-523.
37. Yee KS, Wilkinson S, James J, Ryan KM and Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009; 16:1135-1145.
38. Veronese A, Visone R, Consiglio J, Acunzo M, Lupini L, Kim T, Ferracin M, Lovat F, Miotto E, Balatti V, D’Abundo L, Gramantieri L, Bolondi L, Pekarsky Y, Perrotti D, Negrini M, et al. Mutated beta-catenin evades a microRNA-dependent regulatory loop. Proc Natl Acad Sci U S A. 2011; 108:4840-4845.
39. Fiori ME, Barbini C, Haas TL, Marroncelli N, Patrizii M, Biffoni M and De Maria R. Antitumor effect of miR-197 targeting in p53 wild-type lung cancer. Cell Death Differ. 2014; 21:774-782.
40. Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, Sidransky D and Ratovitski E. Phospho-DeltaNp63alpha is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ. 2011; 18:1220-1230.
41. Wang Q, Shi S, He W, Padilla MT, Zhang L, Wang X, Zhang B and Lin Y. Retaining MKP1 expression and attenuating JNK-mediated apoptosis by RIP1 for cisplatin resistance through miR-940 inhibition. Oncotarget. 2014; 5:1304-1314. doi: 10.18632/oncotarget.1798.
42. Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, Servant N, Paccard C, Hupe P, Robert T, Ripoche H, Lazar V, Harel-Bellan A, Dessen P, Barillot E and Kroemer G. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010; 70:1793-1803.
43. Vander Heiden MG, Cantley LC and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-1033.
44. Ward PS and Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012; 21:297-308.
45. Haldar S, Negrini M, Monne M, Sabbioni S and Croce CM. Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res. 1994; 54:2095-2097.