
INTRODUCTION
The ability of cancer cells to adapt to micro-environmental tissue hypoxia is mainly mediated by hypoxia-inducible factors (HIFs), which affect every aspect of cancer progression, comprising metabolism, proliferation, inflammation, angiogenesis, metastasis and therapy resistance [1-4]. Transcriptionally active HIFs are heterodimers composed of a constitutively expressed β subunit and an oxygen labile HIF-1α or HIF-2α subunit, the stability and activity of which is regulated by prolyl-4-hydroxylase domain (PHD) and factor inhibiting HIF (FIH) enzymes [5]. Despite showing similar protein structures and having identical DNA recognition sequences, distinct - sometimes even opposite - functional roles of HIF-1α and HIF-2α in tumorigenesis have been reported [6-9]. We and others found different kinetics of hypoxic HIF-1α and HIF-2α protein induction and target gene expression [10, 11], further suggesting non-overlapping roles for these two related transcription factors.
Estrogens are steroid hormones and represent the primary female sex hormones, regulating diverse physiological processes in reproductive, mammary, cardiovascular, osseous, hepatic, and neuronal tissues [12-16]. The cellular effects of estrogens are mediated by two estrogen receptor (ER) isoforms, ERα and ERβ, which belong to the family of nuclear hormone receptors [17]. Ligand binding leads to the dissociation of heat shock proteins from the ER, which is followed by receptor dimerization and nuclear translocation. In the nucleus, the activated dimer complex binds to estrogen response elements (EREs) located within the regulatory regions of target genes [18].
Besides regulating numerous aspects of human physiology, estrogens also influence diverse pathophysiological processes, including the onset and progression of breast cancer [19]. Breast cancer is the most common cancer in women worldwide and the second most common cancer overall. Approx. 1.7 million new cases were diagnosed in 2012, which represents 12% and 25% of all new cancer cases and all cancers in women, respectively [20, 21]. The presence of elevated ERα levels in benign breast epithelium correlates with an increased risk of breast cancer, suggesting a role for ERα in breast cancer initiation [22]. 17β-estradiol (E2), the dominant circulating estrogen, regulates the growth of many breast tumors, and approx. 70% of breast cancers express ERα. Most of these ERα positive tumors depend on estrogen signaling for their growth and survival [23].
In conjunction with estrogen, hypoxia has been reported to play an important role in the development and progression of breast cancer [24-28]. In breast cancer cells, estrogen and hypoxia modulate the expression of genes involved in proliferation, differentiation, angiogenesis, metabolism and apoptosis [29-31]. Further studies revealed the presence of a cross-talk between estrogen signaling pathways and HIF-1α regulation in breast cancer [28, 32]. Estrogen has been reported to rapidly induce ERα-c-Src-PI3K interactions, which activates the PI3K/AKT/mTOR pathway and subsequent HIF-1α protein translation by phosphorylation of the p70 S6 kinase and 4EB-P1 [27]. Activation of G protein-coupled receptor GPR30 (GPER) by E2 triggers the GPER/EGFR/ERK/c-fos signaling pathway, leading to increased VEGF via HIF-1α upregulation [33]. Furthermore, ERα has been reported to directly induce HIF-1α transcription, which might modulate the anti-estrogen response in breast cancer treatment [32]. In contrast, ERβ has been reported to play an opposing role to ERα. Transcriptional activity of HIF-1 is inhibited by ERβ, which is mediated by ubiquitin-dependent degradation of HIF-β [27].
Despite the numerous studies on HIF-1α regulation by estrogen, the interaction between estrogen signaling and HIF-2α regulation is currently unknown. By immunohistochemical detection of HIF-2α in tissue microarrays of 282 invasive breast cancer cases we previously found that patients expressing high HIF-2α levels had a better overall survival rate compared to patients expressing low nuclear HIF-2α [11]. Our study was confirmed by the designation of HIF-2α in a list of genes associated with favorable outcome based on studies with different cohorts of breast cancer patients [34]. Here, we report a previously not recognized regulation of HIF-2α by estrogen, suggesting an inverse interplay between estrogen and HIF-2α signaling, which might be involved in breast cancer progression. These data provide a more complex picture of the role of HIF-2α in breast cancer, including receptor status and hormone dependent regulation.
RESULTS
E2 downregulates HIF-2α mRNA and protein levels in ERα positive but not ERα negative breast cancer cell lines
We previously reported a HIF-2 specific regulation of WISP-2 expression in breast cancer cells [35]. Because WISP-2 is a known ERα target gene [36], we aimed for the analysis of the cooperation between estrogen and oxygen signaling. Therefore, the time-dependent effect of E2 treatment on HIF signaling in MCF-7 cells was investigated. Unexpectedly, HIF-2α mRNA levels were progressively reduced with increasing time of E2 treatment (Figure 1A). Maximal inhibitory effects were reached after 12 to 24 hours, and the latter time-point was selected for all subsequent experiments, also based on our previous observation that HIF-2α protein levels in MCF-7 cells are expressed maximally after 24 hours of hypoxic exposure [11].
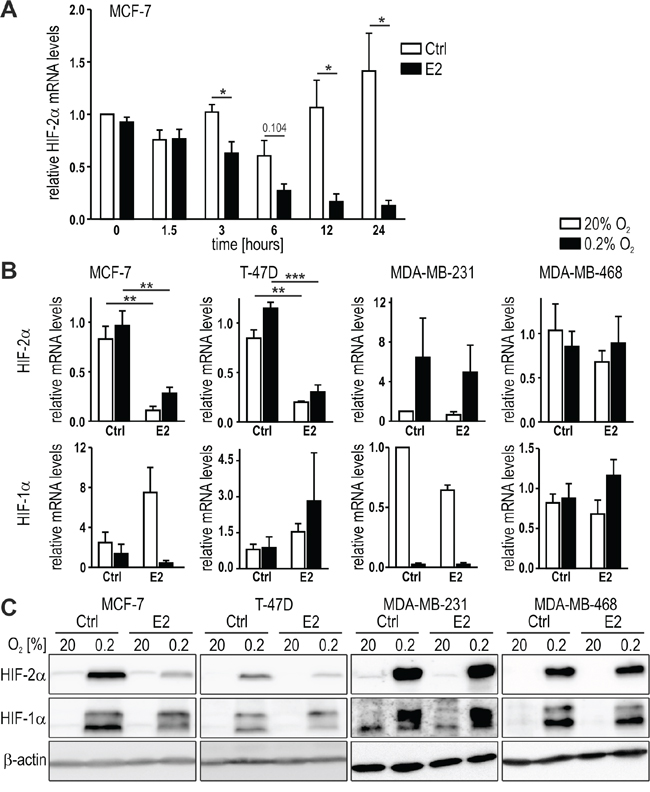
Figure 1: HIF-2α regulation by estrogen in breast cancer cell lines. A. MCF-7 cells were treated with 10 nM E2 for the indicated time periods. HIF-2α mRNA levels were quantified by RT-qPCR, corrected for the endogenous L28 mRNA levels and normalized to the starting time point. B. ERα positive (MCF-7 and T-47D) and ERα negative (MDA-MB-231 and MDA-MB-468) breast cancer cell lines were treated with 10 nM E2 for 24 hours under normoxic or hypoxic conditions. HIF-2α mRNA levels were quantified as above. Shown are mean values ± standard errors of the mean (SEM) of three to four independent experiments. For statistical evaluation, the effects of E2 treatment were compared with the ethanol solvent control (Ctrl) treatment. *P<0.05; **P<0.01; ***P<0.001. C. HIF-2α protein was detected by immunoblotting and constitutively expressed β-actin was used as loading and blotting control.
We further tested the effects of E2 on HIF-2α in additional breast cancer cell lines, including another luminal-like ERα positive (T-47D), a basal B-like ERα negative (MDA-MB-231), and a basal A-like ERα negative (MDA-MB-468) cell line. Both, HIF-2α mRNA (Figure 1B) and hypoxically stabilized protein (Figure 1C) levels were downregulated in the ERα positive MCF-7 and T-47D but not in the ERα negative MDA-MB-231 and MDA-MB-468 cell lines. Exposure to hypoxia (0.2% O2) for 24 hours generally did not affect the HIF-2α mRNA levels, except in MDA-MB-231 cells where HIF-2α mRNA was induced. However, E2 did not alter this cell type-specific HIF-2α mRNA regulation. Neither HIF-1α mRNA (Figure 1B) nor hypoxically stabilized HIF-1α protein levels (Figure 1C) were significantly affected by 24 hours E2 treatment. These data suggest that HIF-2α is specifically downregulated by E2-ERα signaling on the mRNA level, which resulted in corresponding changes on the protein levels.
The selective estrogen receptor modulator tamoxifen prevents ERα-dependent HIF-2α downregulation
To further investigate the involvement of estrogen signaling in HIF-2α and HIF-1α regulation, MCF-7 cells were treated for 24 hours with the ERα-specific agonist propyl pyrazole triol (PPT) and with a selective agonist for GPR30 (G-1) under normoxic or hypoxic conditions. HIF-2α but not HIF-1α mRNA levels were downregulated by PPT but not by G-1 (Figure 2A). Progesterone receptor (PgR) was included as a positive control for ERα activation by PPT. Hypoxia inhibited PgR induction in MCF-7 cells, probably due to the known hypoxic degradation of ERα protein in these cells [37], as also shown below in Figure 2E. Downregulation of HIF-2α mRNA by PPT was confirmed in T-47D cells (Figure 2B). Similar data were obtained in MCF-7 cells on the protein level (Figure 2C), suggesting the involvement of ERα but not GPR30 in estrogen-mediated HIF-2α inhibition.
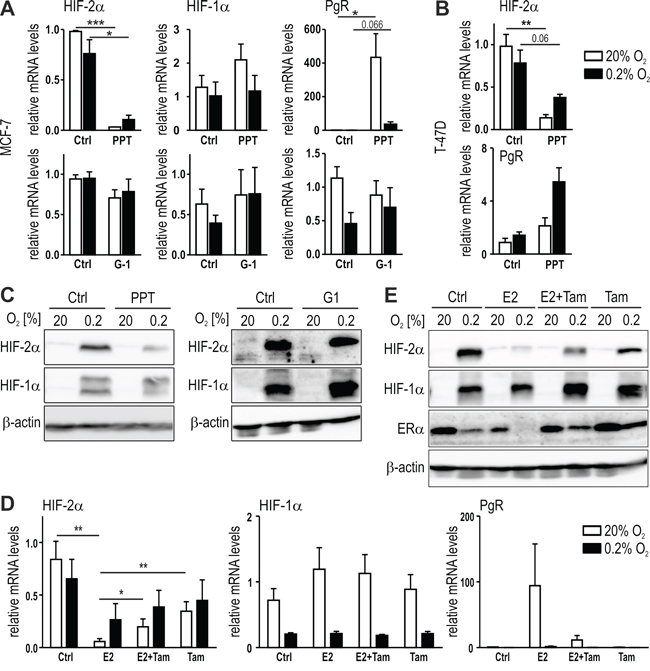
Figure 2: ERα-mediated regulation of HIF-2α by estrogen. MCF-7 (A and C) or T-47D (B) cells were treated for 24 hours with 10 nM PPT, an ERα-specific agonist with approx. 400 times higher affinity towards ERα than towards ERβ, or 1 μM G1 (MCF-7 only), a GPER-specific agonist, under normoxic or hypoxic conditions. The mRNA (A and B) and protein (C) levels were determined by RT-qPCR and immunoblotting, respectively. D and E. Tamoxifen (5 μM) was added to MCF-7 cells with or without 10 nM E2 and mRNA (D) and protein (E) levels were determined. Shown are mean mRNA values ± SEM of three independent experiments. For statistical evaluation, the effects of E2 treatment were compared with DMSO solvent control (Ctrl) treatment. *P<0.05; ***P<0.001.
The current first line therapy of ER positive breast cancer patients is the treatment with the selective estrogen receptor modulator (SERM) tamoxifen. Therefore, MCF-7 cells were treated with E2, with or without tamoxifen, under normoxic or hypoxic conditions. As shown in Figure 2D, tamoxifen at least partially prevented HIF-2α mRNA downregulation by E2, while HIF-1α mRNA levels were neither affected by E2 nor tamoxifen. Vice versa, PgR mRNA upregulation by E2 was partially prevented by tamoxifen. Corroborating the findings on the mRNA levels, tamoxifen also partially prevented the E2-mediated downregulation of the hypoxically stabilized HIF-2α protein levels (Figure 2E). The slight HIF-2α but not HIF-1α protein downregulation by tamoxifen may be explained by the similarly decreased mRNA levels. However, these findings suggest that SERMs can reverse the inhibitory effects of estrogen on HIF-2α.
Inverse correlation between HIF-2α and ERα mRNA levels in breast cancer
To complement the pharmacological E2 receptor modulation shown above, ERα was downregulated by siRNA treatment of MCF-7 cells. Knockdown efficiency was confirmed by a 96% decrease of the constitutive ERα mRNA levels as well as by a 97% decrease of the E2-induced mRNA levels of the ERα target gene PgR (Figure 3A). Both, basal and E2-inhibited HIF-2α mRNA levels were significantly increased in siERα but not siCtrl treated MCF-7 cells. While E2 strongly downregulated HIF-2α mRNA levels in siCtrl cells, the remaining slight downregulation in siERα cells was not significant (Figure 3A). Similar results were obtained in MCF-7 cells stably transfected with three independent shRNA constructs which resulted in 74-83% ERα knockdown efficiency and in a strong decrease of PgR, whereas constitutive HIF-2α mRNA levels were enhanced (Figure 3B). Probably due to the remaining ERα, blockade of the E2-mediated HIF-2α downregulation and PgR upregulation was rather inefficient in shERα compared with siERα cells (data not shown). These findings were corroborated on the protein level where hypoxically stabilized HIF-2α but not HIF-1α is clearly less downregulated by E2 in siERα than in siCtrl treated MCF-7 cells (Figure 3C).
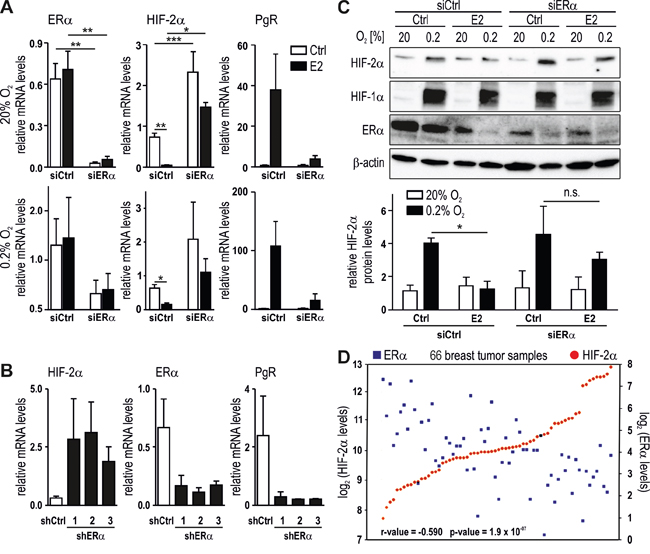
Figure 3: Reciprocal regulation of HIF-2α and ERα. ERα was knocked down in MCF-7 cells using siERα (A and C) or shERα (B) and then treated with 10 nM E2 under normoxic or hypoxic conditions. mRNA and protein levels were subsequently determined by RT-qPCR (A and B) and immunoblotting (C, upper panel), respectively. Shown are mean mRNA (A and B) and protein (C, lower panel) values ± SEM of three independent experiments. For statistical evaluation, the effects of ERα silencing were compared with siCtrl cells; the effects of E2 treatment were compared with ethanol solvent control (Ctrl) treatment. n.s., not significant; *P<0.05; **P<0.01. D. Microarray data from public databases were compiled using the R2 genomic analysis tool. Significance of the negative correlation between HIF-2α (red dots) and ERα (blue squares) mRNA levels was assessed by one-way ANOVA.
Because the results shown above suggest an inverse correlation between HIF-2α and ERα mRNA levels in breast cancer cell lines, we explored transcriptome data from various clinical breast cancer studies employing the R2 microarray analysis and visualization platform (http://hgserver1.amc.nl/cgi-bin/r2/main.cgi). A study of 66 tamoxifen-treated breast cancer patients showed a significant negative correlation between the expression of HIF-2α and ERα (Figure 3D). Several additional datasets from three different microarray studies including 136, 116 and 61 breast cancer patients, respectively, displayed a similar inverse correlation (Supplementary Figure 1).
High HIF-2α expression is a negative prognostic factor in HER2 positive breast cancer
To study the role of HIF-2α in breast tumorigenesis, two tissue microarrays containing invasive breast cancer tumor samples derived from 690 breast cancer patients with primary breast cancer were immunostained for HIF-2α. The tissue microarrays contained areas from invasive breast cancer belonging to ERα positive, HER2 positive or triple-negative cases, as described previously [38]. The signals from the invasive breast cancer samples in the tissue microarrays were scored based on the presence or absence of cytoplasmic and nuclear HIF-2α staining as exemplified in Figure 4A. When all 690 cancer cases were included, neither cytoplasmic nor nuclear HIF-2α correlated with overall survival (Figure 4B). Stratification of cytoplasmic and nuclear HIF-2α to nodal status, tumor stage and histological grade showed no correlation with overall survival (Supplementary Figure 2A-2F). Also the analysis of the 82 triple-negative breast tumor samples did not reveal any significant association between HIF-2α levels and overall survival (Supplementary Figure 2G). However, stratification of nuclear HIF-2α according to the HER2 positivity status in 38 hormone receptor positive breast cancer patients displayed a significant negative correlation with overall survival in patients with high nuclear HIF-2α (Figure 4C).
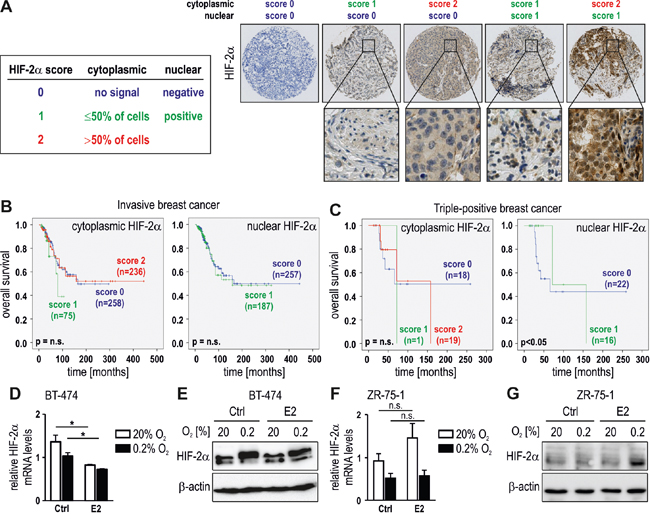
Figure 4: HIF-2α as a prognostic factor in HER2 positive breast cancer. A. Scoring system and representative images for cytoplasmic and nuclear HIF-2α staining of invasive breast cancer tissues. B. HIF-2α was scored in tissue microarrays of 608 invasive breast cancer cases (excluding triple-negative cases) and displayed in Kaplan-Meier survival curves. C. Survival curves of hormone receptor positive and HER2 overexpressing breast cancer patients according to HIF-2α scores. D to G. Triple-positive BT-474 and ZR-75-1 breast cancer cells were treated with 10 nM E2 for 24 hours under normoxic or hypoxic conditions, and mRNA (D, F) and protein (E, G) levels of HIF-2α were determined. Shown are mean mRNA values ± SEM of three independent experiments. For statistical evaluation, the effects of E2 treatment were compared with ethanol solvent control (Ctrl) treatment. *P<0.05; n.s., not significant.
Because the ERα positive breast cancer cell lines used in Figure 1 are not overexpressing HER2, we analyzed the hormone receptor and HER2 positive breast cancer cell line BT-474. While treatment with 10 nM E2 for 24 hours downregulated HIF-2α mRNA levels (Figure 4D), the fold inhibition was less pronounced than in MCF-7 and T-47D cells (Figure 1), and cannot be detected on the protein level (Figure 4E). Whereas strongly HER2 overexpressing cell lines are quite rare, ZR-75-1 has been reported to be HER2 positive [39]. We hence repeated these experiments in ZR-75-1 cells which showed no HIF-2α downregulation by E2, neither on the mRNA (Figure 4F) nor on the protein (Figure 4G) level, confirming the results obtained in BT-474.
In conclusion, diminished downregulation of HIF-2α by E2 in HER2 high ERα positive BT-474 and ZR-75-1 cells is consistent with the correlation between high HIF-2α levels and worse survival of hormone receptor and HER2 positive breast cancer patients.
ERα-dependent HIF-2α downregulation is independent of mutual HIFα inhibition or HIF-2α mRNA stability
To address the functional mechanism underlying HIF-2α inhibition by E2/ERα, we investigated known candidate HIF-2α inhibitors. We and others previously reported the mutual inhibition of HIF-1α and HIF-2α [40, 7]. Because E2 has been shown to induce HIF-1α expression [33], we hypothesized that increased HIF-1α might subsequently decrease HIF-2α. This hypothesis was tested by adding E2 to stably HIF-1α or HIF-2α depleted MCF-7 cells. As expected, shHIF-1α MCF-7 cells express higher levels of HIF-2α mRNA, whereas shHIF-2α had no effect on HIF-1α mRNA levels in this cell line (Figure 5A). Both, PgR and ERα remained largely unaffected by either HIF-1α or HIF-2α knockdown, and E2 downregulated HIF-2α in shHIF-1α to the same extent as in shCtrl cells. Also CITED-2, a preferential HIF-2 target gene [41, 10], was still downregulated by E2 in shHIF-1α MCF-7 cells (Figure 5A). Similar results were obtained on the protein level (Figure 5B), suggesting that mutual HIFα inhibition does not play any role in E2/ERα-dependent HIF-2α downregulation.
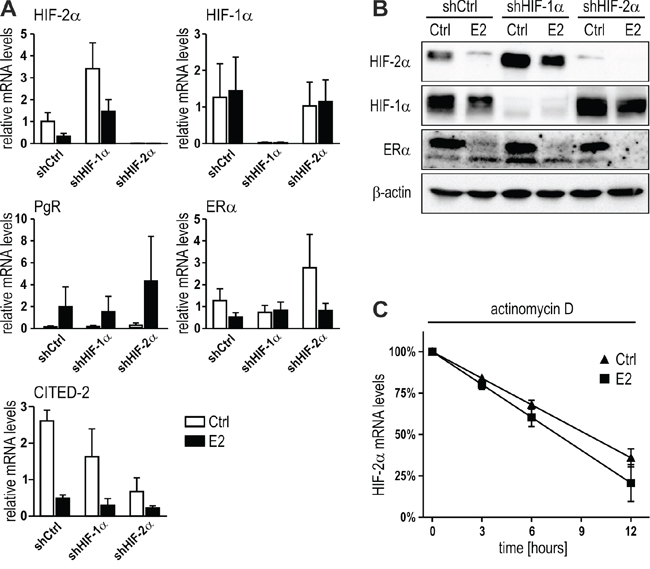
Figure 5: Mutual HIF-α inhibition or mRNA stability are not involved in E2-dependent HIF-2α regulation. A. MCF-7 shCtrl, shHIF-1α and shHIF-2α cells were treated with 10 nM E2 for 24 hours under hypoxic conditions and mRNA levels determined by RT-qPCR. Shown are mean values ± SEM of three independent experiments. The effects of E2 treatment were compared with ethanol solvent control (Ctrl) treatment. B. Immunoblotting of MCF-7 cells treated with E2 as above. C. MCF-7 cells were pre-treated with 1 μM actinomycin D for 1 hour before adding 1 nM E2. HIF-2α mRNA was quantified by RT-qPCR and normalized to the initial levels (100%). No significant difference between the linear regression slopes of the DMSO solvent control (Ctrl) and E2 treatment was found (n = 3).
Because E2/ERα-mediated HIF-2α mRNA downregulation could be due to either transcriptional repression or decreased mRNA stability, we next analyzed the decrease of HIF-2α mRNA following RNA polymerase II inhibition by actinomycin D. As shown in Figure 5C, HIF-2α mRNA decay was not significantly different in the presence of E2 compared with solvent control, suggesting that HIF-2α mRNA levels were transcriptionally decreased by E2.
Histone deacetylation is involved in HIF-2α inhibition by E2
Histone deacetylation is one of the prerequisites for chromatin remodeling and regulation of gene expression. In order to study the role of histone deacetylation in HIF-2α transcriptional regulation, MCF-7 cells were treated with E2 in the absence or presence of the class I and II mammalian histone deacetylase (HDAC) inhibitor trichostatin A (TSA). While TSA alone had no effect, it prevented HIF-2α mRNA (Figure 6A) and protein (Figure 6B) downregulation by E2, suggesting that HDACs are involved in HIF-2α regulation by E2.
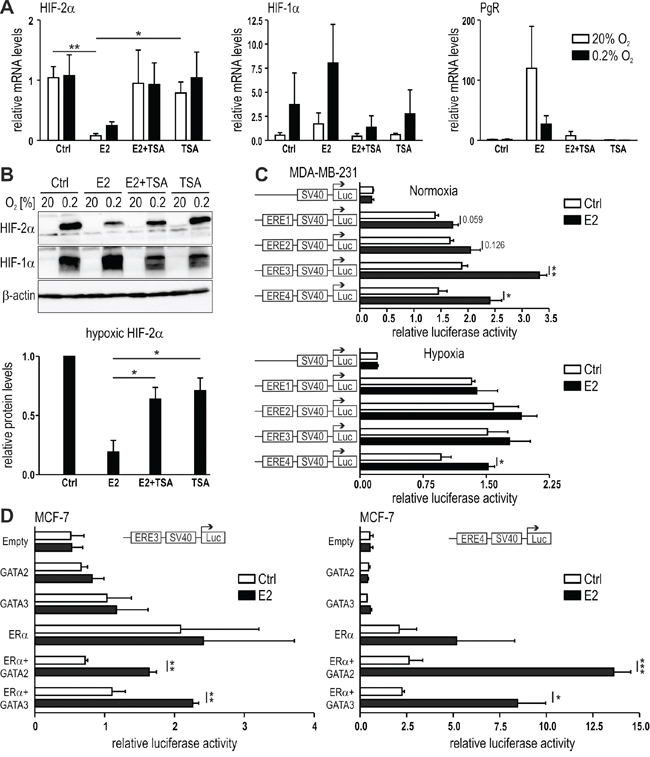
Figure 6: Role of histone deacetylation in E2-dependent HIF-2α regulation. MCF-7 cells were treated with 10 nM E2 with or without 250 nM trichostatin A (TSA), an inhibitor of HDAC. mRNA (A) and protein (B) levels were determined by RT-qPCR and immunoblotting, respectively. C. Dual luciferase reporter gene assays were performed with MDA-MB-231 cells transiently co-transfected with a constitutively expressed Renilla luciferase plasmid, an EPAS1-derived ERE1 to 4 driven firefly reporter gene plasmid, and an ERα overexpression vector. D. Luciferase reporter gene assays were performed with MCF-7 cells transiently co-transfected with a constitutively expressed Renilla luciferase plasmid, an EPAS1-derived ERE3 or ERE4 driven firefly luciferase reporter gene plasmids, and expression vectors for ERα, GATA-2 or GATA-3. Shown are mean values ± SEM of three independent experiments. For statistical mRNA evaluation (A), the effects of E2 alone were compared with DMSO solvent control (Ctrl) or with TSA or with TSA+E2 treatment. For statistical protein evaluation (B, lower panel), the values were normalized to the DMSO solvent control (Ctrl) and the effects of E2 alone were compared with TSA or with TSA+E2 treatment. For dual-luciferase reporter gene assays (C and D), three independent experiments were performed in triplicates. The relative luciferase activity was obtained by dividing the firefly luciferase values by the corresponding Renilla luciferase values. For statistical evaluation, the effects of E2 treatment were compared with ethanol solvent control (Ctrl) treatment. *P<0.05; **P<0.01; ***P<0.001.
According to published chromatin immunoprecipitation-sequencing (ChIP-seq) data [17], ERα binds to four distinct regions within the first intron of the gene encoding HIF-2α (EPAS1) upon E2 treatment (Supplementary Figure 3A). In silico analysis of the ChIP-seq information deposited in the UCSC-integrated ENCODE database revealed that these regions contained conserved estrogen response element (ERE) binding motifs. As shown in Supplementary Figure 3B, all four regions displayed robust DNaseI hypersensitivity (reflecting open chromatin) and the methylated and acetylated histone marks H3K4Me1 and H3K27Ac, respectively (reflecting active enhancers) but not H3K4Me3 (reflecting active promoters). HDAC binding may be transient and was only found in ERE2 (HDAC2). Specifically, in K562 erythroleukemia cells HDAC1/2 binding to ERE3 and HDAC1 binding to ERE4 but no HDAC binding to ERE1 and ERE2 could be detected (Supplementary Figure 3B). In T-47D breast cancer cells, ERE4 also displayed binding of ERα (data not shown). Interestingly, transcription factor (TF) ChIP-seq data further revealed the binding of GATA-2 and GATA-3, established transcriptional repressors [42-45], at ERE3 and ERE4 (Supplementary Figure 3B). Moreover, ERα also binds to an ERE within the HIF1A gene, overlapping with GATA-3 binding (data not shown).
To independently analyze the binding of ERα to the EREs 1 to 4 of the EPAS1 gene, we evaluated the potential of the ERE DNA fragments (as indicated by red bars in Supplementary Figure 3A) to regulate firefly luciferase reporter gene expression driven by the heterologous SV40 promoter. The reporter gene constructs were transiently transfected into MDA-MB-231 cells together with an ERα overexpression vector. Transfected cells were treated with E2 for 24 hours under normoxic or hypoxic conditions and the luciferase activities were determined. ERE1 and ERE2 had no effects but ERE3 and ERE4 significantly enhanced E2-induced reporter gene activity in normoxia and ERE4 in hypoxia (Figure 6C). Whereas this result using “non-chromatinised” bacterial DNA is opposing to the endogenous HIF-2α mRNA regulation by E2, it still provides further evidence for functional interaction between activated ERα and distinct EREs of the EPAS1 gene.
Plasmids containing ERE3 and ERE4 were then transfected into another breast cancer cell line (MCF-7), with or without ERα, GATA-2 or GATA-3. While co-transfection of the reporter genes together with ERα or GATA overexpression vector alone did not result in significant induction of luciferase activities upon E2 treatment, co-overexpression of ERα together with GATA-2 or GATA-3 resulted in significant E2-dependent activation of luciferase activity in normoxia and hypoxia (Figure 6D). Taken together, these results indicate that E2-activated ERα locates to at least one ERE within the EPAS1 gene and recruits several transcriptional co-factors, including GATA factors and HDACs, leading to transcriptional repression of the EPAS1 gene.
DISCUSSION
Cross-talk between estrogen signaling and hypoxia-dependent signaling pathways has previously been reported, focussing on the interactions between estrogen signaling and HIF-1α regulation [27, 32, 37, 46, 47]. In the present study, we report for the first time the association between estrogen signaling and HIF-2α regulation. Estrogen signaling is an essential component of breast cancer progression as indicated by the prevalence of ERα overexpression in breast cancer patients [48]. Hypoxia represents another major factor in breast cancer progression, and the interaction between these two signaling pathways hence is of major clinical importance [4].
In this study, we observed an ERα-dependent downregulation of HIF-2α mRNA and protein levels by E2. Cell lines with different ERα status, pharmacological and RNA interference experiments confirmed the requirement of ERα for the E2 effects on HIF-2α. Higher constitutive expression of HIF-2α both on the mRNA and protein levels in ERα depleted MCF-7 was phenocopied in microarray data of breast cancer patients with different ERα levels. This observation suggests a constitutive ERα-dependent suppression of HIF-2α expression, which is strengthened by hormonal ERα activation. Of note, the E2-induced HIF-2α repression was almost completely abrogated in hormone receptor and HER2 triple-positive cells. While it is currently unclear how HER2 interferes with HIF-2α regulation, HER2 signalling is known to induce HIF-1α by PI3K/Akt/mTOR signalling [49-51], and a similar mechanism might also overcome E2-mediated HIF-2α repression.
The ERα utilizes multiple mechanisms to either induce or suppress transcription of its target genes, which include direct binding of ligand-activated receptor to the DNA at the EREs, followed by recruitment of transcriptional co-regulators [52, 53]. Also an indirect modulation via sequestration of general transcriptional components has been suggested [54]. ERα activation is usually assumed to be associated with increased gene expression, however in fact almost 70% of the genes regulated by E2 are down-regulated in MCF-7 [55]. Our observation of HIF-2α down-regulation by E2 is thus consistent with the majority of genes being downregulated upon E2 treatment of MCF-7 cells.
Currently, we have no definitive explanation for the mechanism by which ERα inhibits HIF-2α. The state of histone acetylation is a predictor of gene activity, and HDACs are known to repress gene expression by modulating the conformational state of the chromatin. Furthermore, HDACs have been reported to be involved in tumorigenesis [56, 57]. Estrogen-mediated repression of the cell cycle inhibitor Reprimo (RPRM) required the interactions between ERα, HDAC7 and FoxA1 [58]. In addition, expression of the gene encoding human uridine diphosphate glucuronosyltransferase (UGT1A) was inhibited in the presence of ERα. ChIP assays further demonstrated the recruitment of ERα, HDAC1 and HDAC2 to the xenobiotic response elements of UGT1A promoters during gene repression [59]. Moreover, tamoxifen-bound ERα has been reported to recruit HDAC to silence gene transcription of ERα targets [60]. Interestingly, tamoxifen treatment of MCF-7 cells has recently been shown to significantly increase HDAC1 binding on the ERE of HIF-1α [32]. In our study, treatment of MCF-7 cells with E2 and TSA abolished ERα-dependent HIF-2α regulation. We identified several EREs within the regulatory region of the EPAS1 gene, and ERE4 interacts with ERα in both MCF-7 and T-47D cells. ERE4 can also interact with GATA-2 and GATA-3, consistent with recent studies revealing substantial enrichment of GATA3 binding to ERα occupied DNA regions [61, 62]. Furthermore, besides inducing gene expression, GATAs have been reported to also exert repressive functions [42-45]. Luciferase reporter gene assays demonstrated a functional interaction between GATA-2/3 and the EPAS1 ERE4, suggesting a role of ERE4 in ERα-dependent HIF-2α regulation.
In conclusion, hormone activation may lead to enhanced ERα dimerization, binding to EPAS1 ERE4, recruitment of transcriptional co-factors, including GATAs and HDACs, and repression of gene expression. The lack of ERα in triple-negative breast cancer cells allows for constitutively higher HIF-2α and prevents estrogen-mediated HIF-2α downregulation seen in ERα positive breast cancer cells. This model is consistent with the inverse correlation between ERα and HIF-2α mRNA levels, which we observed in several breast cancer gene array studies.
Intriguingly, the current study using 690 breast cancer tissue samples could only partially confirm the previously reported positive correlation between HIF-2α protein levels and overall patient survival [11]. In the previous study, we analyzed a smaller cohort of 282 invasive breast cancer cases containing mainly ERα positive and HER2 negative luminal A/B samples, with only 13 cases being HER2 positive [11]. In the current study, we used a different and much larger cohort, including 90 HER2 positive cases and 82 triple-negative samples. Moreover, different antibodies, pretreatment protocols and the nuclear vs. cytoplasmic signal scoring scheme applied in the current study may have contributed to the incomplete overlap between the results of the two studies. The correlation between high nuclear HIF-2α and shortened overall survival in the HER2 and hormone receptor positive patient sub-cohort may be due to the attenuated downregulation of HIF-2α upon E2 treatment in HER2 positive breast cancer cells.
MATERIALS AND METHODS
Reagents
E2 (17β-estradiol), PPT (propyl pyrazole triol), tamoxifen, fulvestrant and actinomycin D were purchased from Sigma-Aldrich (St Louis, MO, USA). G-1 was purchased from Tocris Biosciene (Bristol, UK). Reagents were dissolved in ethanol or dimethyl sulfoxide (DMSO). Antibodies against the following proteins were used: HIF-1α (BD Transduction Laboratories, Allschwil, Switzerland), HIF-2α (immunoblotting: Abnova Corporation, Taiwan; immunohistochemistry: Abcam, Cambridge, UK), ERα (Santa Cruz Biotechnology, Dallas, TX, USA), and β-actin (Sigma-Aldrich).
Cell culture and treatments
The human breast cancer cell lines MCF-7, T-47D, MDA-MB-231, MDA-MB-468, ZR-75-1 and BT-474 were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich). Before experiments, the cells were maintained in phenol red-free DMEM supplemented with 10% charcoal-treated FCS (Gibco, Thermo Fischer Scientific, Waltham, MA, USA) for 1 to 2 days. Cells were treated with E2 or vehicle control (0.1% ethanol or 0.1% DMSO) alone or in combination with other ligands for 24 hours under normoxic and hypoxic conditions. Hypoxic experiments were performed at 0.2% oxygen and 5% CO2 in a gas-controlled glove box (Invivo2 400, Baker Ruskinn, Bridgend, UK) as described previously [63].
mRNA and protein detection and quantification
Total cellular RNA was extracted as previously described [11]. Total RNA (2 μg) was reverse transcribed (RT) using AffinityScript reverse transcriptase (Agilent, Santa Clara, CA, USA) and complementary DNA (cDNA) levels were estimated by quantitative polymerase chain reaction (qPCR) using the primers listed in Supplementary Table 1A and a SYBR® Green qPCR reagent kit (Sigma-Aldrich) in a MX3000P light cycler (Agilent). Transcript levels were calculated by comparison with a calibrated standard and expressed as ratios relative to ribosomal protein L28 mRNA levels. Immunoblotting, signal imaging and quantification were performed as previously reported [64]. Values were normalized to a β-actin loading control. Breast cancer tissue microarray analysis was performed as previously described [38].
Plasmid construction and reporter gene assays
DNA fragments containing the EPAS1-derived EREs were generated by PCR using the primers listed in Supplementary Table 1B, and cloned into the pGL3-promoter luciferase reporter vector (Promega Corporation, Madison, WI, USA). A cDNA encoding human ERα cloned into the pCMV5 mammalian expression vector was kindly provided by A. Odermatt (Basel, Switzerland). Human GATA-2, GATA-3, and GATA-4 cloned into pcDNA3.1 were kindly provided by C. Dame (Berlin, Germany). Dual luciferase reporter gene assays were performed as described previously [64].
In silico expression data
The R2 database (http://hgserver1.amc.nl/cgi-bin/r2/main.cgi) was searched using the across datasets and 2D gene overview options for correlations between EPAS1 and ESR1 in Affymetrix HG-U133plus2.0 based expression profiles normalized using MAS5. The following GEO IDs were employed: GSE29431 (r-value=-0.590, p-value=1.9e-07, 66 samples), GSE28844 (r-value=-0.349 p-value=5.8e-03, 61 samples), GSE12093 (r-value=-0.438 p-value=9.9e-08, 136 samples) and GSE5462 (r-value=-0.313 p-value=6.2e-04, 116 samples).
Statistical analysis
If not indicated otherwise, unpaired Student’s t-tests were applied. Differences between two values at the P<0.05 level were considered to be statistically significant.
ACKNOWLEDGMENTS
We thank P. Spielmann for excellent technical assistance; A. Odermatt and C. Dame for providing plasmids; and A. Fitsche for the TMA immunohistochemistry.
FUNDING
This project was supported by the Swiss National Science Foundation grant 31003A_146203 and the KFSP Tumor Oxygenation of the University of Zurich. K.G. was supported by a grant from the Vontobel-Stiftung (Switzerland).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest in this work.
REFERENCES
1. Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000; 157: 411-421.
2. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004; 4: 891-899.
3. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008; 8: 967-975.
4. Semenza GL. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim Biophys Acta. 2016; 1863: 382-391.
5. Wenger RH, Hoogewijs D. Regulated oxygen sensing by protein hydroxylation in renal erythropoietin-producing cells. Am J Physiol Renal Physiol. 2010; 298: F1287-1296.
6. Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, Nilsson H, Pietras A, Vallon-Christersson J, Borg A, Gradin K, Poellinger L, Pahlman S. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell. 2006; 10: 413-423.
7. Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J Biol Chem. 2006; 281: 23482-23491.
8. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010; 29: 625-634.
9. Pahlman S, Lund LR, Jogi A. Differential HIF-1α and HIF-2α expression in mammary epithelial cells during fat pad invasion, lactation, and involution. PLoS One. 2015; 10: e0125771.
10. Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006; 66: 5641-5647.
11. Stiehl DP, Bordoli MR, Abreu-Rodríguez I, Wollenick K, Schraml P, Gradin K, Poellinger L, Kristiansen G, Wenger RH. Non-canonical HIF-2α function drives autonomous breast cancer cell growth via an AREG-EGFR/ErbB4 autocrine loop. Oncogene. 2012; 31: 2283-2297.
12. Findlay JK, Liew SH, Simpson ER, Korach KS. Estrogen signaling in the regulation of female reproductive functions. Handbook of experimental pharmacology. 2010: 29-35.
13. Gillies GE, McArthur S. Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicines. Pharmacol Rev. 2010; 62: 155-198.
14. Murphy E. Estrogen signaling and cardiovascular disease. Circ Res. 2011; 109: 687-696.
15. Stingl J. Estrogen and progesterone in normal mammary gland development and in cancer. Hormones & cancer. 2011; 2: 85-90.
16. Manolagas SC, O'Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013; 9: 699-712.
17. Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG. ChIP-Seq of ERα and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009; 28: 1418-1428.
18. Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA. Sequence requirements for estrogen receptor binding to estrogen response elements. J Biol Chem. 1998; 273: 29321-29330.
19. Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006; 354: 270-282.
20. Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer. 2013; 132: 1133-1145.
21. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136: E359-386.
22. Ali S, Coombes RC. Estrogen receptor α in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia. 2000; 5: 271-281.
23. Lappano R, Santolla MF, Pupo M, Sinicropi MS, Caruso A, Rosano C, Maggiolini M. MIBE acts as antagonist ligand of both estrogen receptor α and GPER in breast cancer cells. Breast Cancer Res. 2012; 14: R12.
24. Coradini D, Pellizzaro C, Speranza A, Daidone MG. Hypoxia and estrogen receptor profile influence the responsiveness of human breast cancer cells to estradiol and antiestrogens. Cell Mol Life Sci. 2004; 61: 76-82.
25. Mukundan H, Kanagy NL, Resta TC. 17-β estradiol attenuates hypoxic induction of HIF-1α and erythropoietin in Hep3B cells. J Cardiovasc Pharmacol. 2004; 44: 93-100.
26. Yi JM, Kwon HY, Cho JY, Lee YJ. Estrogen and hypoxia regulate estrogen receptor α in a synergistic manner. Biochem Biophys Res Commun. 2009; 378: 842-846.
27. Lim W, Park Y, Cho J, Park C, Park J, Park YK, Park H, Lee Y. Estrogen receptor β inhibits transcriptional activity of hypoxia inducible factor-1 through the downregulation of arylhydrocarbon receptor nuclear translocator. Breast Cancer Res. 2011; 13: R32.
28. Sudhagar S, Sathya S, Lakshmi BS. Rapid non-genomic signalling by 17β-oestradiol through c-Src involves mTOR-dependent expression of HIF-1α in breast cancer cells. Br J Cancer. 2011; 105: 953-960.
29. Cooper C, Liu GY, Niu YL, Santos S, Murphy LC, Watson PH. Intermittent hypoxia induces proteasome-dependent down-regulation of estrogen receptor α in human breast carcinoma. Clin Cancer Res. 2004; 10: 8720-8727.
30. Seifeddine R, Dreiem A, Tomkiewicz C, Fulchignoni-Lataud MC, Brito I, Danan JL, Favaudon V, Barouki R, Massaad-Massade L. Hypoxia and estrogen co-operate to regulate gene expression in T-47D human breast cancer cells. J Steroid Biochem Mol Biol. 2007; 104: 169-179.
31. George AL, Rajoria S, Suriano R, Mittleman A, Tiwari RK. Hypoxia and estrogen are functionally equivalent in breast cancer-endothelial cell interdependence. Mol Cancer. 2012; 11: 80.
32. Yang J, AlTahan A, Jones DT, Buffa FM, Bridges E, Interiano RB, Qu C, Vogt N, Li JL, Baban D, Ragoussis J, Nicholson R, Davidoff AM et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc Natl Acad Sci USA. 2015; 112: 15172-15177.
33. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, Maggiolini M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014; 74: 4053-4064.
34. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481: 389-393.
35. Fuady JH, Bordoli MR, Abreu-Rodríguez I, Kristiansen G, Hoogewijs D, Stiehl DP, Wenger RH. Hypoxia-inducible factor-mediated induction of WISP-2 contributes to attenuated progression of breast cancer. Hypoxia. 2014; 2: 23-33.
36. Banerjee S, Saxena N, Sengupta K, Tawfik O, Mayo MS, Banerjee SK. WISP-2 gene in human breast cancer: estrogen and progesterone inducible expression and regulation of tumor cell proliferation. Neoplasia. 2003; 5: 63-73.
37. Chen M, Xiao D, Hu XQ, Dasgupta C, Yang S, Zhang L. Hypoxia represses ER-α expression and inhibits estrogen-induced regulation of Ca2+-activated K+ channel activity and myogenic tone in ovine uterine arteries: causal role of DNA methylation. Hypertension. 2015; 66: 44-51.
38. Pomp V, Leo C, Mauracher A, Korol D, Guo W, Varga Z. Differential expression of epithelial-mesenchymal transition and stem cell markers in intrinsic subtypes of breast cancer. Breast Cancer Res Treat. 2015.
39. Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, Tang P. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast cancer (Auckl). 2010; 4: 35-41.
40. Carroll VA, Ashcroft M. Role of hypoxia-inducible factor (HIF)-1α versus HIF-2α in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 2006; 66: 6264-6270.
41. Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene up-regulation by hypoxia-inducible factor-1α and hypoxia-inducible factor-2α in HEK293T cells. Cancer Res. 2005; 65: 3299-3306.
42. Imagawa S, Yamamoto M, Miura Y. GATA transcription factors negatively regulate erythropoietin gene expression. Acta Haematol. 1996; 95: 248-256.
43. Imagawa S, Yamamoto M, Miura Y. Negative regulation of the erythropoietin gene expression by the GATA transcription factors. Blood. 1997; 89: 1430-1439.
44. Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, Bresnick EH. GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc Natl Acad Sci USA. 2003; 100: 8811-8816.
45. Xiong Y, Castro E, Yagi R, Zhu J, Lesourne R, Love PE, Feigenbaum L, Bosselut R. Thpok-independent repression of Runx3 by Gata3 during CD4+ T-cell differentiation in the thymus. Eur J Immunol. 2013; 43: 918-928.
46. Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Miyamoto K, Tando T, Morioka H, Matsumoto M et al. HIF1α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc Natl Acad Sci USA. 2013; 110: 16568-16573.
47. Xu D, Niu W, Luo Y, Zhang B, Liu M, Dong H, Liu Y, Li Z. Endogenous estrogen attenuates hypoxia-induced pulmonary hypertension by inhibiting pulmonary arterial vasoconstriction and pulmonary arterial smooth muscle cells proliferation. Int J Med Sci. 2013; 10: 771-781.
48. Ferguson AT, Davidson NE. Regulation of estrogen receptor α function in breast cancer. Crit Rev Oncog. 1997; 8: 29-46.
49. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001; 21: 3995-4004.
50. Li YM, Zhou BP, Deng J, Pan Y, Hay N, Hung MC. A hypoxia-independent hypoxia-inducible factor-1 activation pathway induced by phosphatidylinositol-3 kinase/Akt in HER2 overexpressing cells. Cancer Res. 2005; 65: 3257-3263.
51. Kazi AA, Gilani RA, Schech AJ, Chumsri S, Sabnis G, Shah P, Goloubeva O, Kronsberg S, Brodie AH. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. 2014; 16: R15.
52. Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000; 74: 311-317.
53. Safe S. Transcriptional activation of genes by 17 β-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001; 62: 231-252.
54. Harnish DC, Scicchitano MS, Adelman SJ, Lyttle CR, Karathanasis SK. The role of CBP in estrogen receptor cross-talk with nuclear factor-κB in HepG2 cells. Endocrinology. 2000; 141: 3403-3411.
55. Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003; 144: 4562-4574.
56. Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004; 112: 26-32.
57. Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008; 14: 1669-1677.
58. Malik S, Jiang S, Garee JP, Verdin E, Lee AV, O'Malley BW, Zhang M, Belaguli NS, Oesterreich S. Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol Cell Biol. 2010; 30: 399-412.
59. Kalthoff S, Winkler A, Freiberg N, Manns MP, Strassburg CP. Gender matters: estrogen receptor α (ERα) and histone deacetylase (HDAC) 1 and 2 control the gender-specific transcriptional regulation of human uridine diphosphate glucuronosyltransferases genes (UGT1A). J Hepatol. 2013; 59: 797-804.
60. Huang HJ, Norris JD, McDonnell DP. Identification of a negative regulatory surface within estrogen receptor α provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol. 2002; 16: 1778-1792.
61. Osmanbeyoglu HU, Lu KN, Oesterreich S, Day RS, Benos PV, Coronnello C, Lu X. Estrogen represses gene expression through reconfiguring chromatin structures. Nucl Acids Res. 2013; 41: 8061-8071.
62. Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013; 23: 12-22.
63. Storti F, Santambrogio S, Crowther L, Otto T, Abreu-Rodríguez I, Kaufmann M, Hu CJ, Dame C, Fandrey J, Wenger RH, Hoogewijs D. A novel distal upstream hypoxia response element regulating oxygen-dependent erythropoietin gene expression. Haematologica. 2014; 99: e45-e48.
64. Schörg A, Santambrogio S, Platt JL, Schödel J, Lindenmeyer MT, Cohen CD, Schrödter K, Mole DR, Wenger RH, Hoogewijs D. Destruction of a distal hypoxia response element abolishes trans-activation of the PAG1 gene mediated by HIF-independent chromatin looping. Nucl Acids Res. 2015; 43: 5810-5823.