
INTRODUCTION
Carcinogenesis is a multifaceted and complex process involving alterations in genetic or chromosomal stability [1] that disrupt the normal processes of cell growth and apoptosis progression [2] and thus propel the formation of malignant tumors [3]. Mutated genes and instable chromosomes are prone to lead to changes in cell morphology and physiology. Such aspects include cell polarity loss [4], formation of protrusions [5], adhesion alteration [6], evasion of apoptosis [7], and cell invasion and movement [8]. Furthermore, the interactions between the developing tumor cells and the extracellular matrix microenvironment can also have significant impacts upon tumor progression [9]. The detection and the diagnostic and prognostic capabilities of molecular hallmarks in malignancies has recently become a primary focus of oncology research.
The process of tumorigenesis is under the complicated but precise regulation of various factors. The indispensable roles myosins play in this process has only become apparent in recent years. Myosins constitute a diversiform superfamily of actin-dependent molecular motors which, upon the interactions with microfilaments, convert energy from ATP hydrolysis to mechanical stress [10]. Along with the initial focus upon the conventional myosinII of muscle and nonmuscle cells, there have now been at least 18 distinct classes of myosins discovered over the past twenty years [11]. The human genome contains nearly 40 different myosin-related genes, encoding 12 classes of the myosin superfamily [10]. Myosins typically consist of three functional subdomains: the head domain or NH2-terminal motor domain is required for actin binding and ATP hydrolysis and produces the primary aspects of mechanical power [11]; the neck domain, including one or more IQ motifs (consensus sequence IQXXXRGXXXR, where X is any kind of amino acid), acts to efficiently bind calmodulin and light chains [12]; and the tail domain or COOH-terminal domain, is highly divergent and class-specific and participates in cargo transport at along microfilaments. This latter tail region contains a coiled-coil ɑ-helix which promotes the dimerization of heavy chains and the formation of bipolar filaments. Other special properties of the tail region are also responsible for signal transduction and membrane interaction [12].
Previously, we have demonstrated the essential roles of the myosin superfamily in spermatogenesis and in reproductive system disease [11]. Others have also covered aspects such as myosin I’s role in in intracellular transport and acrosome biogenesis [13]; myosin II and X’s involvement in spindle assembly and karyokinesis [14]; myosin V’s association with acrosomal formation and nuclear morphogenesis [15]; myosin VI’s correlation with the unequal partitioning of both organelles and cytoskeletal components [16]; and myosin VII’s special function in the functional maintenance of Sertoli cells [17]. Furthermore, these six classes of myosins also exhibit functions in reproductive system diseases, such as in testicular tumors and prostate cancer [11]. In recent years, increasing evidence indicates that myosins play multi-functional and crucial roles during tumorigenesis. This has led to a focus upon them as potential therapeutic targets for cancer-related diseases.
The overexpression of myosins has been reported in various cancers, including colorectal cancer [18-21], prostate cancer [22-27], breast cancer [28-30], ovarian cancer [31-32], melanoma [33-34], intestinal neoplasia [35-36], gastric cancer [37-38], pancreatic cancer [39-40], anaplastic gliomas [41] and acute myeloid leukemia [42]. In each cancer type myosins seem to play different but necessary roles during tumorigenesis (Table 1). Myosin IE is a component of the actin-rich core of invadosomes where specialized cell-substrate adhesion and other associated structures display extracellular matrix degradation and tumor cell invasion [43]. Myosin II links to actin filaments and drives cancer cell motility [44]. A depletion of myosin II inhibits tumor invasion and migration [33]. The assembly dynamics of myosin II in its interactions in the process of integrin engagement have also been implicated in cell protrusion formation [45]. Positive regulation of myosin Va by Snail also seems to be integral in the migration of metastatic cancer cells [19]. The overexpression of myosin V facilitates actin assembly and cell motility whilst a deficiency invariably acts to inhibit cell spreading, cell migration and the formation of cytoskeletal architecture. This indicates the pivotal roles of myosin Va in cell motility and cytoskeleton organization [46]. Myosin VI, as an early marker of prostate cancer development [23], not only regulates DNA damage repair in response to the p53 protein (a tumor suppressor protein), but also takes part in the dissemination of ovarian cancer [31], maturation of cadherin-mediated cell adhesion during polarization [47-48] and Golgi apparatus functional maintenance [49]. Myosin VII indirectly associates with the cadherin-catenin complex through a novel transmembrane protein, vezatin, and exerts a tension stress between the actin cytoskeleton and adherens junctions, thereby strengthening cell adhesion and inhibiting tumorigenesis [50]. Myosin IX can down-regulate Rho activity and prevent actin bundle assembly during the nascent formation of cell adhesion [51]. Suppression of Myosin IX affects collective migration and causes cell scattering, two of the most critical factors of cancer cell motility [52]. Myosin X is localized at the tips of filopodia [53] and is active in filopodia formation [54]. In cancer cells, the impairing of p53 can promote increased myosin X expression levels, myosin X being responsible for cell adhesion inhibition, protrusion formation and tumor progression [55]. Various of these and other kinds of myosins and myosin-related molecules participate in other aspects of the formation and development of cancer cells. Further investigation of the many, varied and complex connections and mechanisms that exist between myosins and tumorigenesis will be necessary for future oncotherapy.
Table 1: Information of various myosins related to tumorigenesis
Myosin type |
Fuctions |
Cancer types |
Species |
Contributors |
|
Myosin I |
Recruit invadosome components to plasma membrane, transport vesicles |
Hamster |
[130] |
||
Myosin II |
Myosin II |
Cell invasion and adhesion |
Pancreatic cancer |
Human |
[39] |
Breast cancer |
Human |
||||
Prostate cancer |
Mouse |
[24] |
|||
Anaplastic gliomas |
Human |
[41] |
|||
Regulate tumor cell migration by interacting with P-cadherin |
Melanoma |
Human |
[33] |
||
Maintain cell polarization, stabilize nascent focal adhesion complexes, mediate efficient integrin-based cell migration |
Breast cancer |
Human |
[112] |
||
Promote the cell adherens junction and cadherin accumulation in response to E-cadherin, |
Human and hamster |
[146] |
|||
Myosin IIA |
Promote two-dimensional epithelial cell movement but prevent three-dimensional invasion in cancer metastasis |
Human |
[93] |
||
Link to actin filaments and take part in cancer cell motility |
Breast cancer |
Human |
[30] |
||
Regulation of actin retrograde flow |
Human and rabbit |
[127] |
|||
Myosin IIB |
Establish front–back polarity and guide organelle/nuclear orientation |
Hamster |
[112] |
||
Participate in cell migration by maintaining protrusion stability |
Mouse |
[113] |
|||
Downregulate P-cadherin and facilitate tumor cells invasiveness |
Melanoma |
Human |
[33] |
||
Myosin V |
Myosin V |
Lysosome trafficking and inheritance |
Saccharomyces cerevisiae |
[104] |
|
Trafficking of apical and basolateral proteins, regulate epithelial cell polarity |
Gastric cancer |
Human |
[38] |
||
Myosin Va |
Migration of metastatic cancer cells and cytoskeletal organization |
Colorectal cancer |
Human |
[19] |
|
Mediate the functions of Bcl-xL in tumor cell migration and spreading |
Mouse |
[102] |
|||
Myosin VI |
DNA damage repair and tumor suppression |
Human |
[82] |
||
Dissemination of cancer cells, border cell migration |
Ovarian cancer |
Drosophila (model) |
[31] |
||
Prostate cancer |
[25] |
||||
Regulate the maturation of cadherin-mediated cell adhesion during polarization |
Drosophila |
[136] |
|||
Normal material transportation and maintenance of Golgi structure and function |
Prostate cancer |
Human |
[164] |
||
Myosin VII |
Interact with cadherin-catenins complex and strengthen cell adhesion |
Human and mouse |
[50] |
||
Myosin IX |
Down-regulate Rho activity and actin bundle assembly, affect collective migration |
Human |
[52] |
||
Myosin X |
Response to impaired p53, cell adhesion inhibition, protrusion formation and tumor progression |
Breast cancer |
Human |
[55] |
|
Promote filopodia formation, metastasis development |
Primary glioblastoma |
Human |
[133] |
||
Acute lymphoblastic leukemia |
Human |
[134] |
|||
In summary, cancer-related diseases remain of global importance and the exploration of the correlative molecules and regulatory mechanisms underlying tumorigenesis has become a prime focus of oncology research. Recently, the functions of the myosin superfamily during tumor progression has become of increasing interest. Moreover, some detailed pathways and specific mechanisms related to it have been illustrated. We summarize nine distinctive pathways and myosins related up- or down-stream molecules, including MLCK, PKM2, Sds22, Cdc42, integrin-β1, syndecan-4, merlin, RhoGEF2, Rho GTPases and phytate hydrolysate, etc. Among these factors, the membrane protein integrin-β1 has gradually generated scientists’ attention. Integrin-β1 downregulates Cdc42 and TGF-β2 and results in MLCK activation. This activity can accelerate cellular proliferation and tumorigenesis. These pathways and molecules help us better understand the process of tumorigenesis.
In this review, we summarize current studies and demonstrate the integral connections between myosins and cancer. Meanwhile, we provide a prediction of the likely and logical progression of such studies related to the role of myosin in these mechanistic regulatory mechanisms and suggest some possible outcomes that may lead to new or improved medical therapies for cancer. Finally, we put forward a completely new concept of “chromomyosin” to illustrate the special functions of myosins during karyokinesis and perfect the concept of myosin superfamily.
EFFECTS OF MYOSINS ON GENETIC OR CHROMOSOMAL INSTABILITY
It is well known that carcinogenesis results from the accumulation of gene mutations related to factors controlling the growth of an organism. However, many of the related mechanisms by which such mutations are generated and function remain unknown, despite having been investigated for decades [56]. Accumulating evidence does show that underlying genetic instability is a major cause of gene mutation and acts in the inducement of tumor progression [57]. There are two kinds of such instabilities that can exist in genes. The first of these is nucleotide level instability. This is involved in a small proportion of cancers where which base insertions, deletions and substitutions occur. However, it is chromosome level instability that is the underlining instability for most cancers. This consists of a gain or loss of a whole chromosome, or the gain or loss of a large part of a chromosome. These instabilities are prone to cause elimination of normal tumor suppressor genes and other genetic impairments. These can subsequently promote uncontrolled growth characteristics and reduced survival response to cell damage. These findings provide new insights into tumorigenesis and cancer-related diseases.
Myosins have raised an interest related to tumor progression and cancer therapy because of their effects towards ongenetic and chromosomal instability. Increasing evidence suggests that myosins play essential roles in cases of cytokinesis failure [58], chromosomal and centrosomal amplification [59], multipolar spindle formation [55] and DNA microsatellite instability [36]. Identifying myosins’ specific functions and clarifying their related mechanisms have become a prime focus in oncology.
Chromosomal and centrosomal amplifications
An unstable genome, often related to amplified chromosome or centrosomal numbers, are invariable occurrences in all cancer cells [60]. It is these aspects that are the hallmarks malignant transformation of cells and tumorigenesis [18]. Increasing evidence suggests that these chromosome and centrosome aberrations have been also found in early premalignant lesions. This observation further supports their pivotal roles in tumorigenesis [61]. It is believed that the majority of malignant tumors in humans are associated with chromosomal instability (CIN) involving aneuploid or polyploid karyotypes and structural chromosome aberrations [62]. Underlying genomic or chromosomal instability may stimulate the accumulation of mutations and hence CIN is regarded as the initiation point of tumorigenesis [63]. CIN has been observed in various cancers, including glioblastoma multiforme [64], cervical malignancies [65], colonic adenocarcinoma [66] and other tumor-derived cell lines [67]. In addition to CIN, divisional failures also cause chromosomal and centrosomal amplifications [61]. Cytokinesis failures are prone to produce multipolar mitotic spindles. These lead to uneven chromosome segregation [68]. During cell division, centrosome aberrations also impair the positioning of the cleavage plane and the accuracy of chromosome separation. This results in multipolar spindle formation in the following phase of mitosis [2]. In addition to tumor cells, centrosome amplification and associated multipolar mitoses also takes place in the early stages of inflammation [69]. These studies indicate that the CIN and cytokinesis failures, together with the related chromosomal and centrosomal amplifications, play crucial roles in tumorigenesis and cancer development.
The detailed mechanisms of cell division failures in cancer cells are also related to myosins and their associated molecules [58]. Wu et al. [59] found that cancer cells always failed at cytokinesis because of reduced phosphorylation of the myosin regulatory light chain (MLC). This limited MLC phosphorylation level was correlated with the overexpression of myosin phosphatase (MYPT1) and decreased expression of myosin light chain kinase (MLCK). Conversely, the elevated phosphorylation of MLC markedly inhibited division failures in tested cancer cells. These results demonstrate novel relationships between molecular deficiency and cytokinesis failure and prove the important function of myosin-related proteins in tumorigenesis. Using microarray analyses, it was found that the expression of the MLCK gene was decreased in a variety of tumors, including lung, testis, brain, breast and prostate tumors [70]. The MLCK transcription and expression levels also exhibited dramatic reduction after oncogenic transformation in mesenchymal tumor cells [71] and virus-transformed chicken embryo fibroblasts [72]. Based on the above experimental results, we can put forward a reasonable regulatory pathway associated with myosin-related proteins and cytokinesis failure. At the initial stage of tumorigenesis, the expression of MLCK is inhibited, but that of MYPT1 is elevated. These two proteins then interact with MLC and suppress MLC’s phosphorylation level. This leads to cytokinesis failures and facilitates tumor progression. Taken together, these findings indicate MLCK, MYPT1 and MLC as potential agents for future cancer therapy.
DNA microsatellite instability
DNA microsatellite instability (MSI) is a form of genomic instability related to the impaired DNA mismatch repair (MMR) system in tumors [73]. MSI has become a symbol of particular tumors, including for those related to colorectal cancer [20] and intestinal neoplasia [60]. MMR-deficient cells rapidly accumulate mutations. This leads to gene instability and affects short repetitive DNA sequences. Under such circumstances the sequences containing mononucleotide microsatellites within the coding region typically tend to inactivate and cause MSI-related tumorigenesis [74]. Although most mutations act as “passengers” and do not severely affect cellular morphology and function, there are also some “driver” mutations which directly or indirectly contribute to cancer development and malignant progression.
It was reported that MYH11, a smooth muscle myosin gene, was down-expressed in MSI colorectal cancer as compared with levels in normal tissues [21]. There are two splice variants of the MYH11 gene, SM1 and SM2, and indeed, the SM2 isoform contains a repeated mononucleotide of eight cytosines (C8). This promotes MYH11 as a candidate gene of MSI-related cancers [60]. Other results have suggested that the mutated MYH11 is not involved in early tumor formation but participates in the process of MSI tumorigenesis [20]. In addition to the cases of colorectal cancer, smooth muscle myosin-related genes are also implicated in various inherited human diseases such as acute myeloid leukaemia [42], thoracic aortic aneurysm [75-76] and sarcomere and skeletal muscle diseases [35]. The specific mechanisms of the relationship between the MYH11 gene and myosins in cancer cells requires further investigation.
p53-dependent regulation
p53 is a tumor suppressor protein which can inhibit tumor progression by acting on a series of p53 target genes. Based on their diverse functions, these genes have been classified into various different categories. P21 is associated with cell cycle arrest; DDb2 and XPB mediate DNA damage and repair; Bax and Fas are involved in cell apoptosis; and VEGF functions in anti-metastasis and anti-angiogenesis [77]. In both mouse and human cells, depletion of p53 always results in cytokinesis failure [78] and spontaneous tetraploid formation [79]. Loss of p53 can also facilitate mutations related to genomic or chromosomal instability [80].
Myosin VI is often considered as a motor protein participating in organelle trafficking and the maintenance of Golgi complex [49]. However, more recently it was found to be also required for DNA damage response [81]. Jung et al. [82] suggested that myosin VI may be regulated by the p53 protein and that DNA damage would occur in a p53-dependent manner. p53 can specifically and directly bind to the myosin VI gene promoter and activate its expression. The intracellular location and functions of myosin VI are subsequently changed responsively in a p53-dependent manner. Moreover, inhibition of myosin VI can impair the integrity of the Golgi complex and suppress the activation of p53. This tends to cause DNA damage and cell apoptosis [82]. The above results demonstrate the interaction between myosin VI and the p53-dependent regulation involved in DNA damage repair and tumor suppression.
A large body of research shows that p53 depletion facilitates tumor cell invasion and metastasis development [83]. One reported mechanism related to mutant p53-induced metastasis is the accelerated accumulation of β1 integrin in the plasma membrane [84]. β1 integrin is a kind of cell adhesion receptor and is involved in filopodia formation and cell invasion [85]. In cancer cells, impaired p53 can promote increased myosin X expression levels, while suppression of endogenous mutant p53 inhibits myosin X expression and its corresponding function in cell migration. The upregulation of myosin X in depleted p53-driven malignancies is implicated in cell adhesion inhibition, protrusion formation and tumor progression [55]. This provides a clinically important invasion mechanism that may provide opportunity for therapeutic intervention.
Allelic loss at 17p, as a most frequent chromosomal deletion, often takes place in human malignancies [86]. Within the same region, some tumor suppressor loci, such as TP53, undergo alteration in most tumors. Tp53 is a typical tumor suppressor gene located at 17p13.3 which plays an important role in the regulation of normal cell morphology and function [87]. In addition, qPCR statistical analysis and genetic screening has indicated that Myo1c and Innp5k serve as the two other prominent tumor suppressor genes in the TP53 locus. These two genes are located adjacent to each other and show down-regulation in human tumors [88]. Although the detailed functions of the two candidate genes during tumorigenesis remain unknown, it is reasonable to assume their related proteins, myosin 1c and INNP5K (inositol polyphosphate-5-phosphatase K), participate in tumor suppression or have an anti-metastasis function.
ROLES OF MYOSINS IN TUMOR INVASION AND METASTASIS DEVELOPMENT
The mechanism of tumor invasion and metastasis development are commonly considered to involve the migration of neoplastic cells that have detached from the primary tumor, entered lymphatic and/or blood vessels and germinated in distant organs [4]. The molecules or other regulatory factors involved in these processes are obviously recognized as new prognostic markers for tumor formation [89]. Metastasis formation is a complex multistep process [90]. It involves invasion through the acquisition of cell motility, the degradation of extracellular matrices and basement membranes, adherence, extravasation and tumor cell proliferation [29]. Fundamentally, these physiological processes are relevant for the movement of any cells from one site to another [91]. Some treatments aiming at inhibiting tumor invasion and metastasis development have become strong candidates for further development and attention and may result in many aspects of therapeutic improvement for many cancer-related diseases.
Myosins, as defined by their roles as actin-based molecular motors that translocate along microfilaments in an ATP-dependent manner, have irreplaceable functions in many aspects of eukaryotic motility such as cell movement, cytokinesis, phagocytosis, organelle/particle trafficking, signal transduction and in the maintaining of cellular morphology [92]. In addition, there are some experimental results indicating that myosins are also required for various other cellular functions related to the formation of cancer. These include cell migration and tumor metastasis. It has been suggested that the blocking of myosin II activity by specific myosin light chain kinase inhibitors could prevent invasion by, and adhesion of, pancreatic cancer [39] and breast cancer cells [29]. Depletion of myosin II, as mediated by P-cadherin, inhibits the invasion by, and migration of, melanoma cells [33]. Myosin II also has multiple functions in the migration of colonic epithelial cells and in a number of aspects of the interplay between F-actin and cell-matrix adhesion. Previous work has demonstrated that myosin IIA can act to promote two-dimensional epithelial cell movement, as occurs in wound closure, but prevent three-dimensional invasion in cases of cancer metastasis [93]. A mutation in MYH11 (expressing the smooth-muscle myosin heavy chain) results in human colorectal cancer [20] and intestinal neoplasia formation [60]. González et al. [18] showed a positive relationship between myosin expression and tumor recurrence in colorectal cancer patients. This was associated with tumor aggressiveness and metastasis development. Positive regulation of myosin Va by Snail is also implicated in the migration of metastatic cancer cells [19].
In conclusion, based on the above experimental studies, it is suggested that a number of different myosins play various and important roles in tumor invasion and metastasis development. Thereby, it is reasonable to treat myosins as new potential therapeutic targets against tumor formation and carcinogenesis.
Myosins, cytoskeleton dynamics and cancer cell motility
The development of cancer metastasis is a multistep and complex process, the prerequisite of which is cell motility [94]. The process presumably relies on various activities relating to the cytoskeleton, actin polymerization, cell adhesion and other acto-myosin dynamics [95]. There is increasing evidence indicating that myosins, together with other actin filament and adhesion proteins, play crucial roles during cancer cell migration.
Cancer cell motility and contractile motion are relevant for the continuous structural alternations required for assembly and disassembly of the actomyosin cytoskeleton [96]. Non-muscle myosin II is a major component of actomyosin bundles [97]. Increasing evidence suggests that myosin II proteins, especially the myosin IIA isoform, link to actin filaments and take part in cancer cell motility [44]. MYBPH (myosin binding protein H) directly interplays with NMHC IIA (non-muscle myosin heavy chain IIA) and indirectly inhibits myosin IIA assembly. This leads to a reduction of cell motility [98]. Interestingly, when treated with ROCK inhibitor, myosin IIA is altered from a disassembly state to an assembly-competent state where it then suppresses the interaction between MYBPH and NMHC IIA. This indicates a higher affinity between MYBPH and assembly-competent myosin IIA. The suggestion that myosin II and its related regulatory molecules improve the cell migration and invasion stages of prostate cancer [24], breast cancer [28], pancreatic cancer [39], anaplastic gliomas [41] and melanoma [33] is becoming increasingly apparent. Myosin II may serve as a new therapeutic target for future strategies targeting the inhibition of tumor cell invasion [99-100].
The overexpression of the full-length myosin Va induces actin bundles and cell motility, but reduces the rate of initial cell spreading after plating, whilst abnormal myosin Va, such as that with a tailless structure, disrupts cells from spreading or migrating, and interferes with cytoskeletal architecture [101]. These suggest the essential roles of myosin Va in cell motility and cytoskeleton organization [46]. Lan et al. [19] found that the mRNA expression of myosin Va was highly increased in metastatic colorectal cancer cells. Suppressing myosin Va expression inhibited cell migration and metastasis capabilities. Snail, as a transcriptional repressor triggering epithelial mesenchymal transition, can bind to the E-box of the myosin Va promoter and positively promote its expression and activity. It has been demonstrated experimentally that myosin Va is required for cancer cell metastasis by affecting cell motility. This provides a novel target for Snail in its regulation of cancer migration [19].
Bcl-xL, a tumor progression factor, is involved in altering the actin cytoskeleton and promoting cancer invasiveness. Myosin Va is identified as an Bcl-xL interplaying regulatory protein which mediates the functions of Bcl-xL in tumor cell migration and spreading [102].
The mechanisms by which myosin V associates with cell motility are still controversial. It is possible that myosin V also participates in cell cytoskeletal reorganization and vesicle/organelle transportation [103]. On the one hand, in Saccharomyces cerevisiae, myosin V takes part in the movement of the vacuole/lysosome and is involved in this organelle’s inheritance during cell division. Myosin V has a unique tail structure which contains a globular carboxyl terminal domain. This specific tail is thought to regulate localization of myosin and myosin-cargo interplay by special receptors. A mutation at the tail structure of myosin V damages vacuole inheritance and prevents vacuole movement to yeast buds. Immunofluorescence shows that myosin V colocalizes with the vacuole and helps vacuole recruit its membrane to move and become the isolated structure [104]. On the other hand, cancer progression and metastasis are related to altered vacuole/lysosome trafficking and increasing expression of cathepsin, a kind of lysosomal protease [105]. Tumorigenesis triggers a series of alterations in lysosomes, including increasing the permeability of lysosomal membrane and promoting the release of cathepsins into the cytosol. These cathepsins, in turn, stimulate the generation of cell death pathways, such as p53 protein activation, growth factor deprivation and oxidative stress, etc [106-107]. Meanwhile, the cathepsins can efficiently activate caspase- and mitochondrion-independent cell programmed death, resulting in cancer cells death. As a result, we can conclude that myosin V promotes both lysosome trafficking and inheritance in cancer cells, and consequently these alterations trigger cell death pathways and allow cancer cell programmed death.
Loss of cell polarity and tissue disorganization
Cell polarity is essential for normal cellular function and physiological activity. When mammalian epithelial tumors lose cell polarity, this tends to alter their malignancy [8]. The formation of gastrointestinal tumors are always related to the abnormal cell polarity. Some growth factors, such as EGF, are synthesized in salivary glands and exist in the apical cell surface. While the EGF receptors are limited to the basolateral surfaces of non-transformed cells, so that the growth factors and their receptors interact productively only in the case of local wounding. Disruption of the adhesion junctions after loss of E-cadherin or mislocalization of the growth factor receptors to the apical surfaces of transformed cells lead to increased cell growth and division. Hence, the apical-basolateral cell polarity stimulates gastrointestinal tumorigenesis [108]. Loss of cell polarity, tissue disorganization and invasive cell growth are certainly common observations in most cancers. These affect cell adhesions, asymmetric cell division and metastasis development. Fundamentally, apical-basal polarity has two functions related to tumor suppression in epithelial cells. These are the regulation of the cell’s asymmetric division and the maintenance of the stability of the apical junction complex [109].
Recently, it was found that a deficiency of myosin V could disrupt the trafficking of apical and basolateral proteins and inhibit epithelial cell polarity in gastric cancer [138] and in other microvillus inclusion diseases [110-111]. These regulation relationships may be related to Rab and TfR proteins. In migrating cells, myosin IIB is implicated in establishing a front-back polarity and guiding organelle/nuclear orientation [112]. The localization of MTOC (the microtubule-organizing centre), centrosome, Golgi and nucleus are all the remarkable hallmarks of cell polarization. These organelles reorient near the nucleus in the direction of the protrusion [51]. The depletion of myosin II in migrating cells causes multidirectional protrusions [113] and disordered positions of MTOC, Golgi and nucleus [112], resulting in incorrect front-back polarity and impaired cell motility. Increasing evidence suggests that that myosin II serves as a master regulator of cell migration, participating in cell polarization, protrusion formation, adhesion and signaling transduction [114].
Formation of cell protrusions and cancer cell migration
To migrate and invade during cancer metastasis, the cell body needs to modify its shape and adhesion properties to interact with the extracellular matrix (ECM) and surrounding cells. The ECM microenvironment plays a crucial role in tumor cell progression. It provides a signal substrate and serves as an obstacle to the migrating cell body [9]. Cell migration involves a series of interdependent steps [5], of which the first step is the cell becoming polarized and elongating to form protrusions. Cell protrusions extend from the cells’ leading edge and attach to the ECM substrate [115]. Then the leading edge of the cell contracts and generates a traction force. This promotes the trailing edge to glide gradually forward [96]. This process results in various protrusions being formed including filopodia, pseudopodia, invadopodia and lamellipodia [116], which all contain actin filaments and other motor proteins dynamically interacting with ECM substrates. Cell extension and protrusion formation are the prerequisites for cancer cell progression [4].
Initial cell elongation and protrusion formation is driven by the actin filament assembly [117]. Subsequently, integrins interplay with actin filaments and link the contractile force of actomyosin to the extracellular environment [118]. This clusters cytoskeletal components which are then able to develop into focal contact [119]. These processes are facilitated by the action of crosslinking proteins including myosin II and α-actinin [117] and regulatory molecules involving MLCK, small G-protein RHO, ROCK (RHO-associated serine/threoninekinase)[120], FAK (focal adhesion kinase) [121] and MMPs (matrix metalloproteinases) [122].
During the formation of the leading-edge of cell protrusions, cell migration is driven by the force of actin-network expansion [95] and supported by a squeezing via myosin II-dependent contraction which is able to propel the rigid nucleus through narrow spaces [118]. Assembly of myosin II into the cell cortex is involved in stabilizing nascent focal adhesion complexes and mediating efficient integrin-based cell migration [114]. These are the crucial processes in the control of cell motility, adhesion and tissue architecture [123]. Proteomic analysis has suggested that in breast cancer cells, MHC (myosin heavy chain) phosphorylation sites were activated during integrin engagement and lamellar extension on fibronectin [30]. Although myosin II is not indispensable for the nascent formation of lamellipodium, it is truly essential to the subsequent cell’s elongation and growth [124-125]. The assembly dynamics of myosin II’s interaction with integrin engagement plays important roles in regulating the components of cancer cell migration during tissue setting [45]. Further studies are required to identify the factors and mechanisms involved in this process as well as those related to the invasion strategy of cancer cells beyond any pharmacotherapeutic intervention.
In fibroblasts, two isoforms of myosin II, myosin IIA and B, have been described. As suggested by their differing subcellular localizations, ATPase activities and contraction capabilities, they may play different roles in the regulation of the actin cytoskeleton [126]. Myosin IIB participates in cell migration by maintaining protrusion stability [113]. Myosin IIA is required for the regulation of actin retrograde flow [127]. Studies have suggested that the depletion either myosin isoforms could propel cell protrusion and prevent nascent cell adhesion [112]. Myosin IIA and myosin IIB both exist external to protrusions and promote cell elongation, signaling and the maturation of adhesions, at a distance [128]. Myosin IIA controls adhesion dynamics and myosin IIB exerts front-back polarity and substrate orientation functions [129].
Myosin I E (myo1e) is a component of the actin-rich core in the invadosome. The invadosme is a specialized structure of cell-substrate attachment which associates with ECM degradation and invasion [43]. During invadosome expansion, myo1e serves as a scaffold to recruit invadosome components to the plasma membrane and transport vesicles to the sites of new invadosome assembly. Inhibition of myo1e contributes to mislocalized invadosomes at the rosette center whereas normal myo1e expression is implicated in new invadosome formation at the periphery of the rosette [130].
Myosin X is the best known for its localization to the tips of filopodia [53] and its role in filopodia formation [54]. Myosin X overexpression promotes the formation of hundreds of filopodia in one cell and its knockdown inhibits endogenous filopodia [131]. Increasing experimental data shows that myosin X is implicated in cancer cell’s protrusions and metastasis development by transporting β1 integrins to the filopodia tips [132]. High expression levels of myosin X are also observed in primary glioblastomas [133], acute lymphoblastic leukemia [134] and pathogen infections [55].
The formation of cell extension and protrusion are the prerequisites for cancer cell migration, and understanding the process of myosins stimulating cancer cells’ movement in tissues or organs is of vital importance. Recently, myosin VI was found to have crucial functions in the dissemination of ovarian cancer. During the normal development process of the Drosophila ovary, there is a dynamic stage known as border cell migration. This stage shares a series of similarities at molecular and behavioral levels with human ovarian cancer invasion [32]. Border cell migration in the Drosophila ovary is a process in which a partial epithelial cells gradually turn into mesenchymal cells and invade the neighboring germline cells. A key gene that regulates this transition activity is slbo (slow border cells ), which encodes Drosophila C/EBP protein. C/EBP is a transcriptional activator and improves the expression content of the downstream targets, including FAK (focal adhesion kinase) and E-cadherin [135]. These factors function together in promoting cells migration and motility. Throughout ovary border cell migration, myosin VI is highly expressed in these border cells. High magnification confocal micrographs suggest that border cell myosin VI expression is specifically decreased in slbo mutant type compared with the wild type. Meanwhile, myosin VI interacts with E-cadherin and Armadillo (Drosophila β-catenin) to control cell-cell adhesion and migration. The expression of E-cadherin and Armadillo dramatically reduced in myosin VI deficient cells; myosin VI is inhibited in the cells lacking Armadillo or E-cadherin [136].
Initially, the polarized epithelial cells form specific cell-cell junctions which contain distinct membrane subdomains and adhesion molecules. When these cells gradually migration and turns into mesenchymal cells, the cell polarity inverts from apical to planar, and the adhesion proteins located in the cell-cell junctions reorganize at the protrusion of the border cells. Meanwhile, myosin VI translocates from the junctions to the membrane edge at the invasive front of the cells, thus accelerating cell migration and causing tumor invasion [23]. Emerging experimental evidence suggests myosin VI regulates border cell migration and is highly expressed in metastatic ovarian cancer but not in normal ovarian tissue. Suppressing myosin VI expression prevents cancer cell invasion and migration [31]. The border cell migration mediated by myosin VI has served as a useful paradigm for cancer cell invasion [137]. Another similar relationship between myosin VI and the spreading of cancer cells has also been observed in prostate cancer [25], where the analysis of the detailed mechanisms suggest possible myosin VI involvement in the maintenance of cell polarity, cell-cell adhesion and material transportation [48].
Cadherin-mediated cell adhesion
EMT, epithelial-to-mesenchymal transition, is a physiological process where epithelial cells lose polarity, impair cell-cell adhesion and become more invasive. EMT occurs in a variety of cell or tissue developmental processes, especially those related to tumor progression [6]. During EMT, the actin cytoskeleton undergoes structural changes from a circumferential band of filaments to stress fibers where a number of related proteins and molecules play essential roles [138]. The cell adherens junction functions in regulating EMT and correlative activities, where cadherins are the major adhesion molecules in the adherens junction.
Cadherin is calcium-dependent cell adhesion molecule. It strengthens the cell-cell junction through having repeated extracellular domains and linking to the actin cytoskeleton via its cytosolic domain connection to catenins [138]. The cadherin family consists of E (epithelial)-, N (neuronal)-, P (placental)-, and VE (vascular endothelial)-cadherins, named after the tissues within which they were first found. Cadherins participate in the establishment of cell polarity, cell invasion and the formation of metastasis [139]. E-cadherin is expressed in normal epithelial tissues but has lost its expression in epithelial derived cancer cells [140]. In cancer cells, downregulated E-cadherins always contribute to a decreased cell-cell adhesion, thus increasing the invasive and metastatic capabilities of cancer cells [141]. However, it is interesting that epithelial derived cancer overexpresses N-cadherin and that the upregulation of N-cadherins has the similar function to the downregulation of E-cadherin [142]. At the initial stage of tumorigenesis, cells undergo a series of physiological changes including abnormal alternation of cadherin types and quantities, a process known as cadherin switching. This process involves a switch from the expression of E-cadherin to N-cadherin [143]. Cadherins play important roles in cell adhesion, migration and cancer metastasis, and defects in cadherin function and expression are characteristic of many cancers.
Cadherins link to the actin cytoskeleton through the cytosolic domain and interact with many motor proteins including myosins, kinesins and dyneins (See Figure 1) [144]. In mammalian epithelial cells, myosin VI promotes the maturation of cadherin-mediated cell adhesion during polarization [48]. Myosin VI forms a strong biochemical complex with E-cadherin. This enables epithelial cells to form linear cohesive contacts and facilitates the integrity of the apical junctional complex. In addition to E-cadherin, vinculin also regulates the effects of myosin VI. It is these three proteins functioning together, (myosin VI, E-cadherin and vinculin), that critically mediate the morphogenesis of epithelial cell-cell contacts [145]. In Drosophila melanogaster, the protein levels of E-cadherin and Armadillo (Drosophila β-catenin) are dramatically reduced in the cells lacking myosin VI. Similarly, myosin VI is inhibited in the cells lacking E-cadherin and Armadillo. Data suggests that myosin VI is involved in border cell migrations where it stabilizes and interplays with E-cadherin and Armadillo [136]. In epithelial cells, myosin II also can be recruited and activated in response to E-cadherin, where it functions in the promotion of the cell adherens junction and cadherin accumulation [146]. Myosin Va is activated by Snail to facilitate cancer cell migration [19]. Concurrent to this, Snail has been shown to suppress normal epithelial cell adhesion by down-regulating the expression of genes encoding adherens proteins such as E-cadherin, claudins and occludin [147]. Myosin VII indirectly associates with the cadherin-catenins complex through a novel transmembrane protein, vezatin, which is a ubiquitous protein of adherens cell-cell junctions. Myosin VII is anchored by vezatin, alongside the cadherin-catenins complex. It exerts a tension force between the actin cytoskeleton and cell adherens junction, resulting in a strengthening of cell adhesion [50]. Myosin IX locally down-regulates Rho activity and the actin bundle assembly during the nascent formation of cell adhesions [148]. Depletion of Myosin IX affects collective migration and causes cellular scattering, both critical factors of cancer cell motility [52].
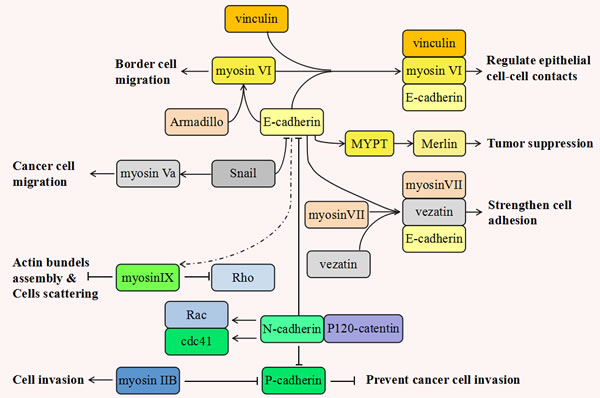
Figure 1:The inseparable relationship between various myosins and cadherins during tumorigenesis. This figure shows that the different classes of myosins and cadherins play multiple and even antagonistic roles during tumor progression. Myosin VI forms a strong biochemical complex with E-cadherin and vinculin, which critically regulates the morphogenesis of epithelial cell-cell contacts. In Drosophila melanogaster, the protein levels of E-cadherin and Armadillo (Drosophila β-catenin) have positive correlations with that of myosin VI. Myosin VI takes part in border cell migration and can stabilize and interplay with E-cadherin and Armadillois. In epithelial cells, myosin II also can be recruited and activated in response to E-cadherin where it functions in promoting cell adherens junction and cadherin accumulation. Myosin Va is activated by Snail to promote cancer cell migration. Concurrent to this, snail has been shown to suppress normal epithelial cell adhesion via the down-regulation of the expression of adherens proteins such as E-cadherin, claudins and occludin. Myosin VII links to vezatin and E-cadherin and forms a cadherin-catenins complex exerting a tension force between the actin cytoskeleton and cell adherens junctions, thus strengthening cell adhesion. Myosin IX locally down-regulates Rho activity and the actin bundle assembly during nascent formation of cell adhesions. It is reasonable to assume that E-cadherin can functionally activate myosin IX and inhibit cell scattering. E-cadherin also can activate MYPT and this leads to a “closed” active merlin, resulting in tumor suppression. Among the various cadherins, cadherin switching also takes place. This means N- cadherin gradually takes the place of other cadherins including E- cadherin and P- cadherin. Cadherin switching may be induced by P120-catenin which is involved in cadherin’s endocytosis. This results from the different cadherins competing for the binding to p120-catenin. In contrast to the actions of E- and P-cadherin, N-cadherin can maintain steady levels of phosphorylated Rac and Cdc42 thus improving cell motility and aggressiveness. It is reported that P-cadherin is downregulated in metastasizing melanomas and over-expression of P-cadherin can efficiently prevent cancer cell invasion. This process is corrected with the inhibition of myosin IIB, which contributes to tumor cells invasiveness by retracting the tail during the migratory cycle.
In human melanoma progression, the malignant melanocyte’s transformation is frequently associated with cadherin switching from E- to N- [149]. P-cadherin has also been noted to be downregulated in metastasizing melanomas and the over-expression of P-cadherin could efficiently prevent cancer cell invasion. Great progress has been made in the identification of the migration-related molecular mechanisms underlying the cutaneous melanoma metastatic process. Non-muscle myosin IIB has been discovered to downregulate P-cadherin in melanomas and myosin IIB contributes to melanoma invasiveness by triggering tail retraction during the migratory cycle [33]. The inverse relationship between myosin IIB and P-cadherin may be significant and strategic to the melanoma cell’s deeper migration into the stroma in metastasis.
Many papers have suggested that myosins function with cadherins to enhance cell adhesion and migration. Some myosins form adhesion complexes with the underlying cadherin substrates to strengthen and stabilize cell junctions. Others inter-regulate with cadherins and other membrane proteins to exert other influences. Although ample evidence supports the roles of myosins in cell adhesion and cancer invasion, detailed studies are still required to elucidate the related specific molecular mechanisms and to elucidate the other interacting proteins involved in myosin-related cancer cell migration and metastasis development.
Suppression of apoptosis
Apoptosis, a kind of programmed cell death in vertebrates, involves a series of morphological feature changes at a cellular level. These include nuclear/cytoplasmic fragmentation, cell shrinkage, formation of dense bodies and global mRNA decay [28]. Previous studies have suggested that crucial apoptotic modulators have been deregulated or inhibited in metastatic cancer cells and that the suppression of apoptosis promotes tumor invasion and metastasis development [150]. Deficient regulation of apoptosis is a general occurrence in human malignances. Cancer cells may acquire their aggressive capabilities because of these apoptosis deficiencies. The altering of cancer cells into an apoptotic status by anti-neoplastic agents has now become a major therapeutic method [151].
The high metastasis potential breast cancer , LM-MCF-7, cells display stronger anti-apoptosis abilities than normal breast cancer MCF-7 cells. This is attributed to both the upregulation of MLCK, myosin light chain kinase, and the downregulation of p-p38 (phosphorylated p38) in the LM-MCF-7 cells. Suppression of MLCK, as mediated by ML-7, could induce apoptosis in the LM-MCF-7 cells and increase the level of p-p38 [28]. Potentially, MLCK is likely to impact tumor invasion and metastasis development by mediating apoptosis and may thus serve as a novel therapeutic target for breast cancer [152].
Functional maintenance of Golgi apparatus
The Golgi apparatus, the indispensable subcellular structure primarily involved in modification, sorting and packaging of macromolecules, has been recently found to play an essential role in carcinogenesis [153]. When cells enter mitosis, the pericentriolar Golgi apparatus is regulated by mitotic signals and proceeds to fragment and disperse [154]. In this process, the Golgi apparatus mediates a series of proteins and molecules which are crucial for cell signaling, transportation and apoptosis. In addition, the reorientation of the MTOC, Golgi apparatus, and nucleus are symbols of cell polarization [155]. Studies are increasingly elucidating the important functions of Golgi in cell polarity, migration, apoptosis and mitotic spindle formation. All of these are all the necessary requirements of cancer cell invasion and metastasis development.
The interactions between myosins and Golgi apparatus always attract people’s attention. Myosin I functions in post-Golgi apparatus transportation, endocytosis and exocytosis, membrane trafficking and endo-lysosomal traffic [11]. Microtubules are the transportation sites of the trans-Golgi and they are required for transcytosis of Golgi vesicles from the basolateral membrane to the apical cytoplasm. Throughout the transport activity, myosin I and dynein interact with Golgi membranes, and cross-link Golgi to the actin filaments and the microtubules. An interesting phenomenon is that myosin I locates on all membranes in Golgi apparatus, however, dynein is found only on some small membrane fractions. The motor protein dynein promotes Golgi vesicle to translocate along microtubules to the cell cortex, and then myosin I delivers Golgi to the apical membrane along the microfilaments [13]. In addition, myosin V stimulates post-Golgi vesicle trafficking in the exocytic pathway [13]. Knock-out of myosin V gene arrests cells in an unbudded but enlarged state, with accumulation of secretory Golgi vesicles. These vesicles are prevented from transporting to the bud site and exert normal physiological functions [156]. In mammalian cells, myosin VI is responsible for promoting membrane trafficking pathways. Myosin VI is diffusely localized to endocytic and exocytic vesicle membrane, especially in Golgi apparatus [157], and myosin VI plays a series of vital roles, including maintaining steady organization of the Golgi apparatus [158], post-Golgi vesicle transportation [159] and allowing the delivery of endocytose cargoes to the recycling compartment [160].
GOLPH2 (Golgi phosphoprotein 2) and myosin VI are two candidate Golgi-associated proteins. GOLPH2 is a type-II Golgi membrane protein and serves as a serum marker for various cancers, including hepatocellular carcinomas [161] and prostate cancer [26]. Myosin VI localizes in the Golgi membranes as a peripheral membrane protein and is implicated in the normal material transportation and maintenance of the Golgi’s structure and functions [162-163]. In the fibroblasts of Snell’s waltzer mice, the absence of myosin VI causes a reduced size of the Golgi and a limited level of protein secretion [49]. Recently, it was found that both GOLPH2 and myosin VI were overexpressed in prostate cancer, where immunofluorescence showed a more obvious Golgi staining of these two proteins in cancer cells than in the adjoining normal epithelium [164]. The increasing expression level of GOLPH2 and myosin VI indicates Golgi-specific alterations related to the molecular composition in cancer cells. These alterations may aid in the deciphering of the detailed mechanisms associated with Golgi apparatus functional maintenance and cancer metastasis development. They could be targeted as novel cancer biomarkers for future therapies.
MYOSIN-MEDIATED FACTORS, PATHWAYS AND MODELS DURING TUMORIGENESIS
Myosin-related molecules during tumor progression
During tumorigenesis, a variety of molecules and factors interact and perform different functions. Among them, myosin proteins and their related up- or down-stream molecules play irreplaceable roles in the regulation of genetic stability and the development of tumor metastasis. Myosins have therefore begun to be treated as new therapeutic candidates for cancer intervention. In the following sections we will list a series of detailed regulatory pathways and reveal the internal relationships between myosins and cancer formation (as summarized in Figure 2).
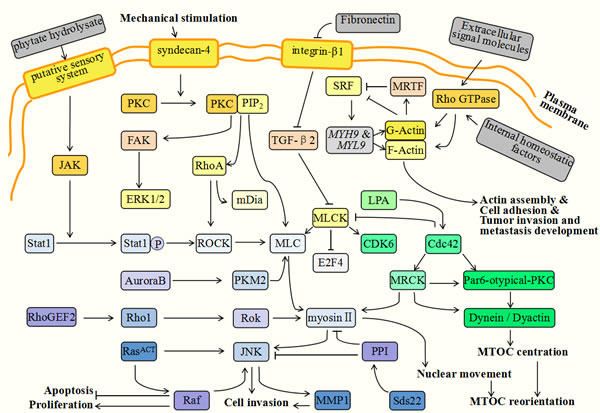
Figure 2: Myosin-related molecules during tumor progression. During tumorigenesis, various molecules and factors interact and perform different functions. Among them, myosins and their related up- or down-stream molecules, play irreplaceable roles in the regulation of tumor genetic stability and tumor metastasis development. The extracellular phytate hydrolysate takes part in the formation of a circumferential F-actin ring in colorectal cancer cells. It can stimulate the putative sensory system located in cell membranes and then activate JAK (Janus kinase) and Sata 1. Under the assistance of PKC and JNK, Sata1 induces ROCK (Rho-associated kinase) active and phosphorylates MLC. MLC and the myosin proteins are located at cell-cell contact points and the myosin contractility is responsible for actin assembly and the formation of the circumferential F-actin ring. Syndecan-4 (SDC4) is a member of HSPGs family and is over-expressed in malignant tumor cells. It can efficiently activate PKC to bind with PIP2 (phosphatidylinositol4,5-bisphosphate), and then facilitate the expression of RhoA and FAK. Activated FAK can subsequently phosphorylate ERK1/2, which is required for cell contractility and adhesion. RhoA can activate ROCK and mDia. ROCK is implicated in inhibiting MLC phosphatase and elevating MLC phosphorylation, thus promoting cell contractility and adhesion. The upregulation of integrin-β1 is induced by decreased fibronectin which can facilitate the cell’s invasion and growth. Integrin-β1 can downregulate Cdc42 and TGF-β2 and result in MLCK activation. This activity can inhibit growth suppressors, such as E2F4 (E2F transcription factor 4), and activate the cell cycle regulators, such as CDK6 (cyclin-dependent kinase 6). Rho GTPases can be activated in response to extracellular signaling molecules or internal homeostatic factors and function in cytoplasmic effectors which subsequently guide F- and G-actin assembly. The absence of G-actin can inhibit MRTFs, consequently activating SRF and its target genes, such as MYH9 and MYL9. These genes can reversely elevate the activity of actins and contribute to actin assembly, cell adhesion and tumor invasion. During cytokinesis, PKM2 is activated by Aurora B and interacts with MLC2 in the contractile ring region of mitotic cells. RhoGEF2 successively activates Rho1, Rok (Rho kinase) and myosin II. Rok and myosin II then promote JNK (Jun kinase) activation together with RasACT. JNK is responsible for the inhibition of cell differentiation and tissue growth, and acts to promote cell invasion. Myosin facilitates cell contractility, cytoskeleton reorganization and cytokinesis. These molecules interact together to regulate cooperative tumorigenesis. Sds22 is a regulatory upstream molecule of protein phosphatase 1 (PP1) which is related to the completion of mitosis. PP1 can efficiently inhibit the phosphorylation of myosin II and JNK, thus restricting tumor progression. During cell invasion, MTOC promotes reorientation and facilitates directional movements. The process of MTOC reorientation is modulated by a series of molecules involving Cdc42, myosin II, dynein and PKC. Cdc42 is a small GTPase responsible for activatePar6-atypical-PKC and MRCK (myotonic dystrophy kinase-related Cdc42 binding kinase). Subsequently, Par6-atypical-PKC activates dynein and dynactin and MRCK contributes to myosin II phosphorylation and nuclear movements.
RhoGEF2, a RhoGTP exchange factor, is an activator of Rho-family GTPases [165]. Ras is a typical oncogene which is implicated in about 30% of cancers. However, this gene is not enough to facilitate tumor progression. Recently it was found that RhoGEF2 could cooperate with activated Ras (RasACT) or an activated allele of the Ras downstream effector, Raf, and then affect normal cellular functions. This interaction may be a key initiating factor of tumorigenesis. In this regulatory pathway, RhoGEF2 successively activates Rho1, Rok (Rho kinase) and myosin II after cooperating with RasACT or Raf. Rok and myosin II then promote JNK (Jun kinase) activation together with RasACT [166]. JNK can inhibit cell differentiation and tissue growth and promote cell invasion. Myosin II is required for cell contractility, cytoskeleton reorganization and cytokinesis [167]. The above molecules interplay together to accelerate cooperative tumorigenesis.
Sds22 is a regulatory upstream molecule of Protein Phosphatase 1 (PP1), which is relevant for the completion of mitosis [168] and the regulation of epithelial polarity [169]. These two processes are both crucial aspects of malignant tumor regulation. The expression level of PP1 is invariably decreased in cancer cells and the PP1 inhibitor, okadaic acid, can promote tumor invasion and metastasis development [170]. The reasonable interpretation is that PP1 can efficiently inhibit the phosphorylation of myosin II and JNK, thus restricting tumor progression [167]. Furthermore, JNK partly mediates the expression of MMP1 (Matrix metalloprotease 1), which can facilitate cell motility [171] and the degradation of the basement membrane [172], which are both related to tumor malignancy potential [173].
Recently, it was found that phytate hydrolysate could contribute to the formation of circumferential F-actin rings in colorectal cancer cells [174]. Phytate has various biological functions involving apoptosis induction and anticancer activities [175]. It is possible that it is the hydrolysis products of phytate that may exert an anticancer function. The functions of phytate hydrolysate in F-actin ring formation is associated with the JAK-Sata1-Rock-MLC regulatory pathway [174]. Extracellular phytate hydrolysate can stimulate the putative sensory system located in the cell membranes and then activate JAK (Janus kinase) and Sata 1. Under the activation of PKC and JNK, Sata1 activates ROCK (Rho-associated kinase) and phosphorylates MLC. MLC and myosin proteins are located at cell-cell contact points and myosin contractility is responsible for actin assembly and the formation of the circumferential F-actin ring [176]. This pathway provides specific connections between myosins, phytate and corresponding anticancer functions.
HSPG (heparan sulfate proteoglycan) is a kind of polysaccharide located in the cell membrane. It participates in cell adhesion, migration and related physiological processes [177]. Syndecans are the members of the HSPG family and are overexpressed in malignant tumor cells [178]. They are considered to be contributors to tumor progression [179]. Among multiple syndecans, syndecan-4 (SDC4) can efficiently activate PKC to bind with PIP2 (phosphatidylinositol 4,5-bisphosphate) [180]. In addition, under mechanical stimulation, SDC4 also facilitates the expression of RhoA and FAK and acts as a mechanotransducer for the assembly of the actin cytoskeleton, cell contractility and motility [181]. Mechanical stress promotes RhoA activity and then activates its downstream molecules, ROCK and mDia. ROCK is implicated in inhibiting MLC phosphatase and elevating MLC phosphorylation and thus promoting cell contractility and adhesion [182]. Phosphorylated FAK can subsequently activate ERK1/2 and plays a similar role in the activation of MLC [9].
Furthermore, it has been reported that MLCK acts downstream of integrin-β1 and TGF-β2 to promote cell proliferation [27]. In prostate cancer, upregulation of integrin-β1 is induced by decreased fibronectin and can facilitate cellular invasion and growth. The activation of integrin-β1 downregulates Cdc42 and TGF-β2 and results in MLCK activation. This activity can inhibit growth suppressors, such as E2F4 (E2F transcription factor 4), and activate cell cycle regulators, such as CDK6 (cyclin-dependent kinase 6). These factors function together to accelerate cellular proliferation and tumorigenesis [27]. This indicates that TGF-β2-related mechanisms may hold potential to prevent cancers metastasis.
Pyruvate kinase M2 (PKM2) has a high expression level during tumor progression [183] and takes part in spindle assembly and chromosome segregation [184]. Its detailed mechanism is related to MLC2 two-site phosphorylation. During cytokinesis, PKM2 is activated by Aurora B and interacts with MLC2 in the contractile ring region of mitotic cells. Active MLC2 subsequently binds with ROCK2. This leads to ROCK2-dependent MLC2 phosphorylation at two sites of Y118 and S15 [183]. The activated MLC2 then contributes to the progression of cytokinesis for tumor cells [185].
The MTOC, (microtubule-organizing center), is a structure located between the nucleus and the leading edge in migrating cells [148]. During cell invasion, MTOC influences reorientation and facilitates directional movement [186]. The process of MTOC reorientation is modulated by a series of molecules involving Cdc42, myosin II, dynein and PKC [187]. Cdc42 is a small GTPase implicated in Golgi apparatus transportation and MTOC reorientation [188] which can activate Par6-atypical-PKC and MRCK (myotonic dystrophy kinase-related Cdc42 binding kinase). Subsequently, Par6-atypical-PKC activates dynein and dynactin, and MRCK contributes to myosin II phosphorylation and nuclear movements [187]. Taken together, these factors play essential roles in MTOC reorientation and cell migration.
Rho GTPases are implicated in the regulation dynamics of the cytoskeleton via multiple effectors which participate in actin-based structural assembly, cell adhesion and migration [189]. It has been recently reported that MRTFs (myocardin-related transcription factors) and SRF (serum response factor) are the downstream factors of Rho GTPases and required for cytoskeletal dynamics and tumor metastasis [190]. Rho GTPases are activated in response to extracellular signalling molecules or internal homeostatic factors and function as cytoplasmic effectors which subsequently guide F- and G-actin assembly [191]. Among them, the absence of G-actin can inhibit MRTFs, consequently activating SRF and its target genes such as MYH9 and MYL9. These target genes encode actin [192] and other cytoskeletal components including NMHCIIa and MLC2. These actin-based factors cooperatively function in cell adhesion, cell spreading and cytoskeleton contractility, thus regulating tumor invasion and metastasis development [193].
Vestibular schwannoma (VS) is a kind of benign tumor which is derived from the internal auditory canal and the eighth cranial nerve. It is associated with hearing loss and even brainstem compression [194]. One cause of VS formation is the inactivation of the NF2 gene and its downstream molecule, merlin [195]. Merlin shares similarities to the two ERM proteins, moesin and radixin, which both participate in the connection of the actin cytoskeleton to plasma membranes [196]. Merlin has two different states: the “open” inactive state and the “closed” active state. After dephosphorylation at amino acid serine 518, merlin can be folded and activated to act as a tumor suppressor. Signals are transduced into cells by membrane-bound integrins and RTK (receptor tyrosine kinases) which then activate Rac and PAK (p21-activated kinase) [197]. Activated PAK can phosphorylate merlin and induce the “open” inactive conformation [198], thus impairing merlin’s functions of tumor suppression. Additionally, merlin also can be phosphorylated by PKA (protein kinase A), which acts not only on serine 518, but also on serine 10. Conversely, cadherin suppresses Pak activation and promotes “closed” active merlin. This similar regulatory pathway is a CD44-related mechanism. CD44 is a transmembrane receptor and anchored to the hyaluronic acid rich matrix. It plays an essential role in cell adhesion and motility [199]. CD44 can activate MYPT1 (myosin phosphatase targeting subunit 1), which acts in opposition to the functions of CPI-17 (protein kinase C-potentiated phosphatase inhibitor of 17 kDa) [200] The activated MYPT1 then leads to a “closed” active merlin, resulting in tumor suppression. The mutual transformation of these two merlin conformations is required for tumor formation or suppression.
Although a series of regulatory mechanisms have been investigated and illustrated, more candidate myosins and related molecules involved in tumorigenesis require elucidation. The interactions between all of these these factors also need to be clarified. How to utilize known mechanisms to explore unknown cancer therapeutic method is an urgent topic.
Model of myosins and cadherin-mediated cell adhesion
Earlier in this article we have illustrated the important functions of various cadherins in cell adhesion and protrusion formation. We also demonstrated the inseparable relationship between cadherins and myosins. Next, we will focus on the detailed mechanisms and models related to cadherin-mediated cell adhesion (see Figure 3).
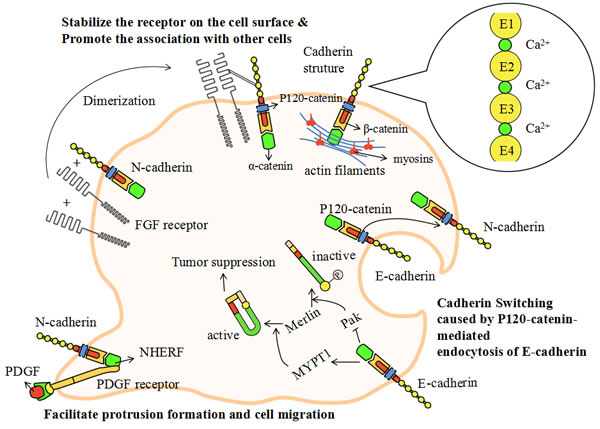
Figure 3: The model of cadherin-mediated cell adhesion. Figure 1 has revealed the inseparable relationship between cadherins and myosins. In this picture, we focus on the detailed mechanisms and models related to cadherin-mediated cell adhesion. Cadherins are single-pass transmembrane proteins which have similar structural domains. The cadherins’ extracellular domains contain five function-similarhomologous repeats (EC1-5) which are linked together by calcium ions. In the cytoplasm, cadherins connect with actin filaments by p120-catenin, β-catenin and ɑ-catenin. Cadherin switching is a process in which tumor cells alter the physiological metabolism from expressing E-cadherin to expressing N-cadherin. This is related to the endocytosis of E-cadherin via the competition for the binding of p120-catenin. N-cadherin induces the dimerization of FGF (fibroblast growth factor) receptor, which directly interacts with the EC4 of N-cadherin, stabilizing the receptor on the cell surface and promoting the association with endothelial or mesenchymal cells. NHERF is a small protein serving as a scaffold to connect N-cadherin with PDGF receptor. The PDGF receptor is located at the leading edge of migrating tumor cells. The cadherin-PDGF complex can facilitate protrusion formation and cell migration. The down-regulation of merlin is a hallmark of benign tumor formation which can be modulated by cadherins. E-cadherin can efficiently activate MYPT1 and inhibit Pak. It can then phosphorylate merlin and induce an “open” inactive conformation. Active merlin is required for tumor suppression and the inactive conformation is a signal of tumorigenesis.
Cadherins are single-pass transmembrane proteins which have similar structural domains. The cadherins’ extracellular domain contains five function-similar homologous repeats (extracellular domain1-5 or EC1-5) which are linked together by calcium ions [201]. The cytoplasmic domain directly binds to p120-catenin and β-catenin. P120-catenin is involved in cadherin endocytosis [202], which results from the competition of different cadherins to bind to p120-catenin. β-catenin binds to the cadherins near their C-terminus and acts as an intermediate junction between cadherins and ɑ-catenin. The intracellular actin cytoskeleton links to ɑ-catenin and is regulated by various cadherins [201].
When tumor cells alter the physiological metabolism from expressing E-cadherin to expressing N-cadherin, they demonstrate a highly aggressive behavior. This process is called cadherin switching. Cadherin switching can significantly affect many cellular activities including those involved in cell adhesion and migration. This process is relevant for the p120-catenin-mediated endocytosis of E-cadherin [202]. It may be reasonable to suggest that N-cadherin may act to maintain steady levels of phosphorylated Rac and Cdc42, thus improving cell motility and aggressiveness [201]. Furthermore, N-cadherin induces the dimerization of FGF (fibroblast growth factor) receptor and initiates a growth-factor signal [203]. The dimeric FGF receptor directly interacts with the EC4 of N-cadherin, stabilizing the receptor on the cell surface and promoting the association with endothelial or mesenchymal cells [204]. E-cadherin also interacts with the FGF receptor but prevents FGF-dependent signaling by virtue of restricting the mobility of the receptor [205]. There is also a small protein, named NHERF, serving as a scaffold to connect β-catenin with PDGF (platelet derived growth factor) receptor. The PDGF receptor is situated at the leading edge of migrating tumor cells and its linked complex can facilitate protrusion formation and cell migration [206].
Recently, a notable difference between transformed and untransformed epithelial cells was discovered [207]. Marginal actin cytoskeletal bundles are the typical structure at the free edges of non-transformed epithelial cells. Tangential E-cadherin, located at the cell-cell joint, causes damage in these marginal actin bundles [208]. The other remaining parts of the actin bundles form two arc structures and the generated tangential stress inhibits cell protrusion formation. This process takes place not only at the cell-cell joints but at the free edges of migrating cells [207]. However, in the transformed epithelial cells, the marginal actin bundles disappear and the inner straight actin bundles remain. Once they have formed cell-cell joints, the diminished E-cadherin-based tangential stress causes the formation of overlapping cell lamellas. In this overlapping area, radial adherens junctions assemble and exert unstable functions. The stable E-cadherin-based AJs are implicated in the integrity of the epithelial cells and the maintenance of tissue stability [209]. However, the unstable radical adherens junctions are related to straight actin bundles and centripetal myosin II [210] which contributes to impaired cell adhesion and altered motility.
The above models demonstrate the detailed mechanisms of cadherin-mediated cell adhesion. Considering the inseparable relationship between myosins and cadherins, it is reasonable to assume that myosins therefore play a pivotal role in cell adhesions and migration.
Model of myosins and cell protrusion formation
Tumor cell migration requires intracellular mechanical power to produce coordinated protrusions at the leading edge of cells. The coupling of this intracellular machinery to the extracellular environment is essential for the translocation of the cell body [45]. The formation of cell protrusions are facilitated by actin polymerization and two common protrusion structures, lamellipodia and filopodia, which display dramatically different actin reorganization and are regulated by diverse signaling mechanisms [211]. In both lamellipodia and filopodia, actin filaments take on the same polarity and elongate with the formation of the branched ends in the direction of the cell protrusions. At the roots of the actin polymers, the actin subunits are gradually released. This is responsible for the recycling of monomers and the formation of efficient protrusive power.
The lamellipodia are sheet-like cell protrusions. They are composed of a two-dimensional network of outstretched branched actin filaments [212]. The ends of actin filaments exhibit an obvious “Y” shape. Conversely, filopodia are rod-like extensions and typically contain dense bundles of about twenty actin filaments. Most protrusive cells are comprised of both filopodia and lamellipodia, thus enhancing cell migration ability [213]. The filopodia extends their roots into the lamellipodia to form a complex and cooperative structure where the lamellipodia mainly exert the mechanical power and promote locomotion and the filopodia take part in sensory reception and feedback. Moreover, it is reported that metastatic cancer cells contain more filopodia structures than non-metastatic cells [214].
A large number of proteins and molecules are involved in the formation of protrusion and play interactional roles: VASP proteins couple the ends of the actin filaments to the cell surface; capping protein and Arp2/3 regulate the capping of the actin ends; CARMIL and VASP inhibit capping and down-regulate capping proteins [211]; Arp2/3, ɑ-actinin, myosins, fascin and filamin modulate the cross-linking of the actin filaments [215]; cofilin is required for the dissociation of the filament’s roots; profilin guides the actin monomers to link to the branched ends; myosin X functions in the transportation of monomers; finally, moesin plays pivotal role in the connection of the new filament ends to the cell membrane. Other molecules have also been found related to cell protrusion formation, but their exact and detailed functions require further investigation.
Myosin X has essential functions during the formation of filopodia [53]. It was reported that the overexpression of myosin X could efficiently induce the formation of hundreds of filopodia per cell. However, the depletion of myosin X dramatically reduces endogenous filopodia [54]. At the surface of protrusions, integrin acts to anchor the cell membrane to the extracellular matrix. This is required for cell adhesion. Myosin X serves as a molecular linker between actin filaments and integrin and has crucial roles in the transportation and signaling of integrins. The tumor cells with mutated myosin X, lacking binding ability with integrin, decrease the formation of filopodia [55]. This suggests that the integrin-myosin X interaction is an irreplaceable factor during protrusion formation and cell motility (See Figure 4).
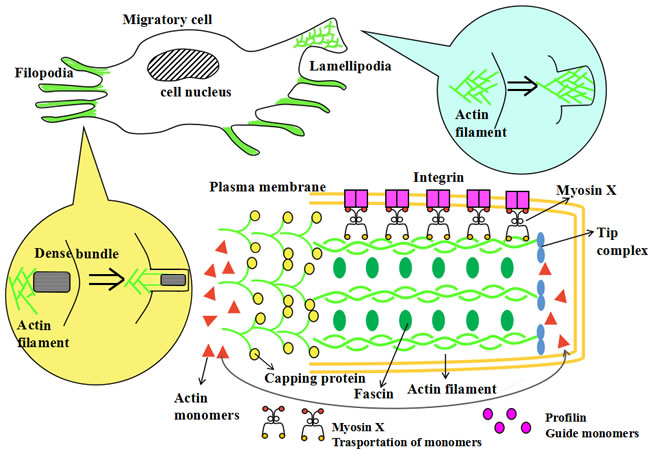
Figure 4: The model of myosin X and cell protrusions formation. The formation of cell protrusions is facilitated by actin polymerization and various related molecules. Two common protrusion structures, lamellipodia and filopodia, have differing actin reorganizations and are regulated by diverse signaling mechanisms. The lamellipodia are a sheet-like cell protrusions. They are composed of outstretched branched actin filaments with two-dimensional network. Filopodia are a rod-like extensions and typically contain a dense bundle of about 15 to 20 actin filaments. A large quantity of proteins and molecules are involved in the formation of cell protrusions and play interactional roles. Profilin guides the actin monomers to link to the branched ends and myosin X functions in the transportation of monomers. These monomers are responsible for the recycling of actin monomers and the formation of efficient protrusive power. Capping protein regulates the capping of the actin ends. Fascin modulates the cross-linking of actin filaments and stabilizes the structures and functions of the filaments. Myosin X also has an essential functions during the formation of filopodial tips. At the surface of protrusions, integrin anchors the cell membrane to the extracellular matrix, enhancing cell adhesion. Myosin X connects integrin to the actin filaments which can then facilitate the transportation and signaling of integrins. Other molecules are also found related to cell protrusion formation, but their detailed functions requires further study.
A NEW CONCEPT OF “CHROMOMYOSIN”
The conventional myosin (myosin II) plays an essential role in muscle contraction and actin-dependent motility. In the cytoplasm of the cardiomyocyte, myosins convert energy from ATP hydrolysis to mechanical stress upon interaction with microfilaments. Cytoplasmic microfilaments are the binding sites and motion trails of myosins. Mutated myosins, lacking binding ability with microfilaments, aren’t able to function normally. However, to date, more than seven classes of the myosin superfamily (including myosin I, II, V, VI, X, XVI and XVIII) have been discovered in the nucleus. It is well known that microfilaments don’t diffusely exist in the nucleus, and just keep transient and reversible appearances [216-217], so how do these nuclear myosins exert functions effectively?
Our previous experiments show the co-localization between myosin Va and the nucleus in spermatocytes of Eriocheir sinensis [15]. Immunofluorescent localization analysis indicates in the presence of spermatid nuclear membrane, myosin Va enters nucleus and displays specific spatial distribution, which is similar to the distribution of KIFC1 in nucleus (named chromokinesin). Compared with the functions of chromokinesin, including chromosome condensation and spindle organization [218], we propose a hypothesis that myosin Va plays a “chromomyosin” role in nucleus. Increasing evidence suggests that other myosins participate in spindle assembly and positioning, karyokinesis, cytokinesis and other processes involved in mitosis or meiosis [14]. The mitotic spindle, which contains a dynamic array of microtubules and associated proteins, is required for normal chromosomal separation during mitosis [219]. At least three different classes of myosins have been identified to localize to the mitotic spindle and play essential roles. Depletion of myosin II is prone to impair centrosome separation and spindle assembly [220]. Myosin X is correlated with the positioning of the spindle microtubules and the maintenance of normal spindle length in Xenopus eggs [221]. Moreover, myosin I interacts with kinesin and efficiently improves the transportation of the spindle apparatus [222]. It is also reported that the ablation of myosin II in mouse atrial myocytes and embryonic cardiacmyocytes results in cell karyokinesis defects. This indicates the pivotal functions of myosin II in normal karyokinesis. Cytokinesis is the final stage of mitosis in which the cytoplasm is partitioned into two daughter cells. Myosin II and cytoplasmic actin are found to be located at the contractile ring and provide the motor function required for its contraction. Two structurally different myosin IIs have been identified due to their special roles during cytokinesis: Myo2p initiates the formation of the contractile ring; and Myp2p functions later to improve cytokinesis efficiency [223]. Furthermore, myosin II is also found to participate in the formation of the cleavage furrow and in chromosomal movement [224].
Furthermore, several nuclear myosins (myosin I, II, V, VI, XVI and XVIII) act with nuclear actin and regulate certain genes transcription or cell cycle [225-226]. Myosin I is regarded as the mediator of ribosomal RNA genes transcription. Actin and myosin I are both located in the specific position in the nucleolus, and the distribution of myosin I protein is closely connected to the transcription process. Meanwhile, myosin I and its related actin are responsible for the maturation of rRNA and maintenance of nucleolar structure [227]. Myosin II, as a protein first reported by scientists [228], serves as another core transcription factor to mediate the assembly of precursor complex for gene transcription. The phosphorylation or dephosphorylation of nuclear myosin light chain triggers the relative sliding between myosin II and actin, resulting in separation or connection between RNA polymerase II holoenzyme and DNA, and then decrease or increase the expression of target gene [229]. Myosin Va is found to respond the transcriptional inhibition and promote nuclear compartmentalization. The inhibition of transcription in HeLa cells caused by actinomycin D motivate myosin Va phosphorylation and cause the redistribution of myosin Va to nucleoli [230]. Nevertheless, myosin Vb is present in nucleolus, and this protein interacts with actin, RNA polymerase I and newly transcribed ribosomal RNA [231]. The roles of myosin Vb in transcription process have attracted the interest of scientists. Myosin VI is detected in the mammalian cells’ nucleus, in which myosin VI co-localizes with RNA polymerase II and newly formed mRNA. On the basis of Chromatin immunoprecipitation assays, myosin VI is recruited to the promoter and activates the transcription of target genes. In the meantime, the depletion of myosin VI protein dramatically decreases the content of steady-state target mRNAs [232]. Myosin XVI is a special myosin which prominently localizes in the nucleus, especially in the nucleolus [233]. Myosin XVI interacts with stress-induced nuclear actin rods and is directed to localize at nuclear compartment. Overexpression of myosin XVI efficiently delays S-phase progression during cell cycle and prevents cell proliferation [234]. Immunolocalization results demonstrate that myosin XVIII is distributed to cytoplasm of undifferentiated myoblasts; however, after differentiation to myotubes, a part of myosin XVIII enter cell nuclei. The similar phenomena are observed in primary cardiomyocytes and skeletal muscle cells [235].
The above evidence suggests the crucial function of nucleus-related myosins. In the absence of microfilaments, myosins efficiently regulate spindle assembly, drive chromosomal movement, control mitotic dynamics and mediate gene transcription. Although we have no idea how these myosins function, we do know that these unconventional myosins play important roles in keeping normal cell division and chromosomal inheritance. Based on these viewpoints, we creatively put forward a completely new concept of “chromomyosin”. We define chromomyosin as another kind of unconventional myosin which interacts with nuclear actin, utilizes the energy from ATP hydrolysis and converts it to mechanical stress, however, unlike the other myosins, chromomyosin simply exists in the nuclear matrix and doesn’t interact with cytoplasmic microfilaments. During cell mitosis and meiosis, chromomyosin participates in spindle assembly and positioning, karyokinesis, cytokinesis and other physiological processes related to cell division. This new concept impacts the traditional ideas that myosins are just the actin-based molecular motors and translocate along cytoplasmic actin microfilaments.
Nonetheless, another question is how chromomyosins translocate and walk in the nucleus. Although this problem is still controversial and has not been solved, we can still get some inspiration from existing research. In interphase nuclei, individual chromosomes are assembled within chromosome territories, which are the nuclear space of these chromosomes. These chromosome territories show nonrandom and radical positioning, which is associated with active motor proteins which includes nuclear myosin I [236]. Once actin binds to the head domain of myosin I, the complex directs myosin I’s tail to link to the nuclear entity. Then myosin I conveys the nuclear entity along highly dynamic paths of nuclear actin [237]. On the other hand, during the process of myosins regulating transcription, the tail of myosin binds to the target DNA and nuclear actin is bound to polymerase. Due to the intimate interplay between myosin and actin, polymerase is led to the target DNA and activates the initiation of transcription [225].
The way how myosins enter nucleus and function as chromomyosins are still unfathomed. Some research suggests that it is the COOH-terminal tail domain, which often represents class-specific functions for various myosins, that directs chromomyosins localization and recognition on the cell nucleus [233, 238]. Other kinds of myosins, such as myosin XVI, are expressed only in nucleus and mainly locate in the nucleolus. Interestingly, some DNA replication stresses (DNA damage, deoxynucleotides insufficient or suppression of DNA polymerases) direct myosin XVI to export to the cytoplasm [233].
There are three main differences between chromokinesin and chromomyosin: First, all chromokinesins belong to two kinesin subfamilies, kinesin IV and kinesin X. These two kinesin family proteins all possess nuclear localization signals and DNA binding activity, which can explain their diffuse nuclear distribution [239]. While, the chromomyosins contain at least seven classes of myosins (including myosin I, II, V, VI, X, XVI and XVIII), which are important for spindle assembly, chromosomal movement, gene transcription, cell cycle regulation and mitotic/meiotic dynamics. Second, chromokinesins function during interphase and links to the chromosome arms during cell division. These motor proteins are responsible for chromosome segregation, normal cytokinesis and maintenance of genome stability [239]. The chromomyosins not only play a similar role with chromokinesins during interphase and cell division stage, but also take part in special genes’ transcription and maturation of RNA. Third, during mitosis and meiosis, chromokinesins translocate chromosome arms along the microtubules [240]. As for chromomyosins, although they don’t walk or carry cargos along the cytoplasmic microfilaments, they still interact with nuclear actin and exert specific functions; however, the polymerization status of nuclear actin is still unknown.
Chromomyosin can be described as the special and distinct myosins functioning in nucleus and directly interplaying with chromosomes. This novel concept will aid in the clarification and interpretation of the irreplaceable roles of myosins in cytokinesis and maintenance of nuclear stability. We believe that in rapidly dividing cancer cells, chromomyosin shoulders a number of indispensable functions. Studying the functions of chromomyosin during tumorigenesis may be an interesting and important topic for future research.
CONCLUSIONS AND PERSPECTIVES
In recent years, studies have increasingly demonstrated that the myosin superfamily plays a pivotal role during tumorigenesis and cancer-related diseases. At the current research level, some questions remain ambiguous and require further investigation. For example, the actomyosin cytoskeleton has fundamental but poorly understood roles in cell adhesion and protrusion formation. The detailed functions and regulations of this remain to be clarified. In addition, the molecular mechanisms defining the different modes of cancer cell motility and migration remain unelucidated. However, one thing we can confirm is that various myosins have different but interactional functions in the formation and development of tumor cells.
Tumorigenesis is a complex and multistep process. It primarily results from the accumulation of gene mutations related to an organism’s growth and development. It is now widely accepted that genetic and chromosomal instabilities are major causes of gene mutations and potential tumor progression. Subsequently, malignant cells detach from the primary tumor and enter metastasis and invasion stages. To facilitate this, a series of physiological and metabolic processes require alteration. These include the loss of cell polarity and tissue disorganization, cell protrusion formation, cell adhesion damage and the suppression of apoptosis. Seven classes of the myosin superfamily, including myosin I, II, V, VI, VII, IX and X, have been shown to participate in these processes during tumorigenesis. In addition, we have illustrated a variety of myosin-related factors, pathways and mechanisms during tumor progression, and put forward two models relevant for the tumor cell’s motility and migration. Finally, a completely new concept of “chromomyosin” has been presented to interpret the special functions of myosins during karyokinesis and optimization of the myosin superfamily.
Although abundant research efforts have been conducted in the exploration of the myosins’ functions during tumorigenesis, the current understanding is far from sufficient or complete. The following two aspects, in particular, require further examination. Firstly, although we have illustrated plenty of myosin-mediated factors and pathways, there are still some unknown mechanisms associated with tumorigenesis, such as myosin-related chromosomal and centrosomal amplifications, that needs to be elucidated. Future studies may focus on a panoramic view of the myosin interaction network. Secondly, the ultimate target of researching cancer-related molecules is finding the possible candidates for future cancer therapy. Accumulating evidence has proved that myosins play irreplaceable roles in the formation and development of cancer. Therefore, how to combine these myosins-related studies with medical therapy is a key next stage focus.
Abbreviations
CDK6, cyclin-dependent kinase 6; CIN, chromosomal instability; CPI-17, protein kinase C-potentiated phosphatase inhibitor of 17 kDa; E2F4, E2F transcription factor 4; EMT, epithelial-to-mesenchymal transition; FAK, focal adhesion kinase; FGF, fibroblast growth factor; GOLPH2, Golgi phosphoprotein 2; HSPG, heparan sulfate proteoglycan; INNP5K, inositol polyphosphate-5-phosphatase K; JAK, Janus kinase; JNK, Jun kinase; MHC, myosin heavy chain; MLC, myosin regulatory light chain; MLCK, myosin light chain kinase; MMPs, matrix metalloproteinases; MRCK, myotonic dystrophy kinase-related Cdc42 binding kinase; MRTFs, myocardin-related transcription factors; MYBPH, myosin binding protein H; MYPT1, myosin phosphatase; NMHC IIA, non-muscle myosin heavy chain IIA; PAK, p21-activated kinase; PDGF, platelet derived growth factor; PIP2, phosphatidylinositol 4,5-bisphosphate; PKA, protein kinase A; PKM2, Pyruvate kinase M2; PP1, Protein Phosphatase 1; p-p38, phosphorylated p38; RhoGEF2, RhoGTP exchange factor; ROCK, RHO-associated serine/threoninekinase; Rok, Rho kinase; RTK, receptor tyrosine kinases; SDC4, syndecan-4; SRF, serum response factor;
ACKNOWLEDGMENTS
The authors are grateful to all the members of the Sperm Laboratory in Zhejiang University in their provision of valuable discussion. This project was supported in part by the National Natural Science Foundation of China (Nos. 41276151 and 31572603).
Author contributions
Y.-R. Li and W.-X. Yang conceived of and authored the manuscript.
CONFLICTs OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review.
REFERENCES
1. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998; 396:643-649.
2. Sluder G, Nordberg JJ. The good, the bad and the ugly:the practical consequences of centrosome amplification. Curr Opin Cell Biol. 2004; 16:49-54.
3. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70.
4. Friedl P, Wolf K. Tumour-cell invasion and migration:diversity and escape mechanisms. Nat Rev Cancer. 2003; 3:362-374.
5. Friedl P, Bröcker EB. The biology of cell locomotion within three-dimensional extracellular matrix.Cell. Mol. Life Sci. 2000; 57:41-64.
6. Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia.Cancer Res 2005; 65:5996-6001.
7. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet 1993; 9:138-141.
8. Bilder D. Epithelial polarity and proliferation control:links from the Drosophila neoplastic tumor suppressors. Genes Dev 2004; 18:1909-1925.
9. Huang CP, Cheng CM, Su HL. Lin YW.Syndecan-4 Promotes Epithelial Tumor Cells Spreading and Regulates the Turnover of PKC Activity under Mechanical Stimulation on the Elastomeric Substrates. Cell Physiol Biochem. 2015; 36:1291-1304.
10. Krendel M, Mooseker MS. Myosins:tails (and heads) of functional diversity. Physiology (Bethesda) 2005; 20:239-251.
11. Li YR, Yang WX. Myosin superfamily: The multi-functional and irreplaceable factors in spermatogenesis and testicular tumors. Gene. 2016; 576:195-207.
12. Mooseker MS, Cheney RE. Unconventional mysins. Annu. Rev. Cell Dev. Biol. 1995; 11:633-675.
13. Fath KR, Trimbur GM, Burgess DR. Molecular motors are differentially distributed on Golgi membranes from polarized epithelial cells. J. Cell Biol 1994; 126:661-675.
14. Ma XF, Jana SS, Conti MA, Kawamoto S, Claycomb W, Adelstein RS. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol. Biol. Cell 210; 21:3952-3962.
15. Sun X, He Y, Hou L, Yang WX. Myosin Va participates in acrosomal formation and nuclear morphogenesis during spermatogenesis of Chinese mitten crab Eriocheir sinensis. PLoS One 2010; 5:e12738.
16. Kelleher JF, Mandell MA, Moulder G, Hill KL, L’Hernault SW, Barstead R. Titus MA. Myosin VI is required for asymmetric segregation of cellular. Curr. Biol. 2000; 10:1489-1496.
17. Velichkova M, Guttman J, Warren C, Eng L, Kline K, Vogl AW, Hasson T. A human homologue of Drosophila kelch associates with myosin-VIIa in specialized adhesion junctions. Cell Motil. Cytoskeleton. 2002; 51:147-164.
18. González L, Eiró N, González-Reyes S, Andicoechea A, González LO, García-Muñiz JL, Vizoso FJ. Clinical significance of myosin in colorectal cancer. Annals of Diagnostic Pathology 2012; 16:260-266.
19. Lan L, Han H, Zuo H, Chen Z, Du Y, Zhao W, Gu J, Zhang Z. Upregulation of myosin Va by Snail is involved in cancer cell migration and metastasis. Int J Cancer 2010; 126:53-64.
20. Vickaryous N, Polanco-Echeverry G, Morrow S, Suraweera N, Thomas H, Tomlinson I, Silver A. Smooth-muscle myosin mutations in hereditary non-polyposis colorectal cancer syndrome.Br J Cancer. 2008; 99:1726-1728.
21. Kruhoffer M, Jensen JL, Laiho P, Dyrskjøt L, Salovaara R, Arango D, Birkenkamp-Demtroder K, Sørensen FB, Christensen LL, Buhl L, Mecklin JP, Järvinen H, Thykjaer T, et al. Gene expression signatures for colorectal cancer microsatellite status and HNPCC.Br J Cancer. 2005; 92:2240-2248.
22. Loikkanen I, Toljamo K, Hirvikoski P, Väisänen T, Paavonen TK, Vaarala MH. Myosin VI is a modulator of androgen-dependent gene expression. Oncol Rep 2009; 22:991-995.
23. Knudsen B. Migrating with myosin VI.Am J Pathol 2006; 169:1523-1526.
24. Tohtong R, Phattarasakul K, Jiraviriyakul A, Sutthiphongchai T. Dependence of metastatic cancer cell invasion on MLCK-catalyzed phosphorylation of myosin regulatory light chain. Prostate Cancer Prostatic Dis. 2003; 6:212-216.
25. Dunn TA, Chen S, Faith DA, Hicks JL, Platz EA, Chen Y, Ewing CM, Sauvageot J, Isaacs WB, De Marzo AM, Luo J, A Novel Role of Myosin VI in Human Prostate Cancer. Am J Pathol. 2006; 169:1843-1854.
26. Laxman B, Morris DS, Yu J, Siddiqui J, Cao J, Mehra R, Lonigro RJ, Tsodikov A, Wei JT, Tomlins SA, Chinnaiyan AM. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res. 2008; 8:645-649.
27. Ruppender N, Larson S, Lakely B, Kollath L, Brown L, Coleman I, Coleman R, Nguyen H, Nelson PS, Corey E, Snyder LA, Vessella RL, Morrissey C, Lam HM. Cellular Adhesion Promotes Prostate Cancer Cells Escape from Dormancy. PLoS One. 2015; 10:e0130565.
28. Cui WJ, Liu Y, Zhou XL, Wang FZ, Zhang XD, Ye LH. Myosin light chain kinase is responsible for high proliferative ability of breast cancer cells via antiapoptosis involving p38 pathway. Acta Pharmacol Sin 2010; 31:725-732.
29. Zhou X, Liu Y, You J, Zhang H, Zhang X, Ye L. Myosin light-chain kinase contributes to the proliferation and migration of breast cancer cells through cross-talk with activated ERK1/2. Cancer Res. 2007; 67:5691-5698.
30. Betapudi V, Gokulrangan G, Chance MR, Egelhoff TT. A proteomic study of myosin II motor proteins during tumor cell migration. J Mol Biol. 2011; 407:673-686.
31. Yoshida H, Cheng W, Hun J, Montell D, Geisbrecht E, Rosen D, Liu J, Naora H. Lessons from border cell migration in the Drosophila ovary: A role for myosin VI in dissemination of human ovarian cancer. Proc Natl Acad Sci USA 2004; 101:8144-8149.
32. Bai J, Uehara Y, Montell DJ. Regulation of invasive cell behavior by taiman, a Drosophila protein related to AIB1, a steroid receptor coactivator amplified in breast cancer. Cell. 2000; 103:1047-1058.
33. Jacobs K, Van Gele M, Forsyth R, Brochez L, Vanhoecke B, De Wever O, Bracke M. P-cadherin counteracts myosin II-B function:implications in melanoma progression. Mol Cancer 2010; 9:255.
34. Li H, Zhou F, Wang H, Lin D, Chen G, Zuo X, Sun L, Zhang X, Yang S. Knockdown of myosin VI by lentivirus-mediated short hairpin RNA suppresses proliferation of melanoma. Mol Med Rep. 2015; 12:6801-6806.
35. Laing NG, Nowak KJ. When contractile proteins go bad:the sarcomere and skeletal muscle disease.Bioessays 2005; 27:809-822.
36. Alhopuro P, Phichith D, Tuupanen S, Sammalkorpi H, Nybondas M, Saharinen J, Robinson JP, Yang Z, Chen LQ, Orntoft T, Mecklin JP, Järvinen H, Eng C, et al. Unregulated smooth-muscle myosin in human intestinal neoplasia.Proc Natl Acad Sci U S A. 2008; 105:5513-5518.
37. Pessina P, Conti V, Pacelli F, Rosa F, Doglietto GB, Brunelli S, Bossola M. Skeletal muscle of gastric cancer patients expresses genes involved in muscle regeneration. Oncol Rep 2010; 24:741-745.
38. Dong W, Chen X, Chen P, Yue D, Zhu L, Fan Q. Inactivation of MYO5B promotes invasion and motility in gastric cancer cells. Diq Dis Sci. 2012; 57:1247-1252.
39. Kaneko K, Satoh K, Masamune A, Satoh A, Shimosegawa T. Myosin light chain kinase inhibitors can block invasion and adhesion of human pancreatic cancer cell lines. Pancreas 2002; 24:34-41.
40. Roy I, McAllister DM, Gorse E, Dixon K, Piper CT, Zimmerman NP, Getschman AE, Tsai S, Engle DD, Evans DB, Volkman BF, Kalyanaraman B, Dwinell MB. Pancreatic Cancer Cell Migration and Metastasis Is Regulated by Chemokine-Biased Agonism and Bioenergetic Signaling. Cancer research. 2015; 75:3529-3542.
41. Gillespie GY, Soroceanu L, Manning TJ, Gladson CL, Rosenfeld SS. Glioma migration can be blocked by nontoxic inhibitors of myosin II. Cancer Res. 1999; 59: 2076-2082.
42. Liu P, Tarlé SA, Hajra A, Claxton DF, Marlton P, Freedman M, Siciliano MJ, Collins FS. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993; 261:1041-1044.
43. Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia:characteristics, formation and function. Nat Rev Mol Cell Biol 2011, 12:413-426.
44. Huang Y, Arora P, McCulloch CA, Vogel WF. The collagen receptor DDR1 regulates cell spreading and motility by associating with myosin IIA, J. Cell Sci. 2009; 122:1637-1646.
45. Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res 30; 8:629-642.
46. Wang FS, Wolenski JS, Cheney RE, Mooseker MS, Jay DG. Function of myosin-V in filopodial extention of neuronal growth con. Science. 1996; 273:660-663.
47. Mangold S, Norwood SJ, Yap AS, Collins BM. The juxtamembrane domain of the E-cadherin cytoplasmic tail contributes to its interaction with Myosin VI. Bioarchitecture. 2012; 1:185-188.
48. Chibalina MV, Puri C, Kendrick-Jones J, Buss F. Potential roles of myosin VI in cell motility. Biochem Soc Trans 2009; 37:966-970.
49. Warner CL, Stewart A, Luzio JP, Steel KP, Libby RT, Kendrick-Jones J, Buss F. Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell’s waltzer mice. EMBO J. 2003; 22:569-579.
50. Kussel-Andermann P, El-Amraoui A, Safieddine S, Nouaille S, Perfettini I, Lecuit M, Cossart P, Wolfrum U, Petit C. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J. 2000; 19:6020-6029.
51. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420:629-635.
52. Omelchenko T, Hall A. Myosin-IXA regulates collective epithelial cell migration by targeting rhogap activity to cell-cell junctions. Curr Biol. 2012; 22:278-288.
53. Berg JS, Cheney RE. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 2002; 4:246-250.
54. Bohil AB, Robertson BW, Cheney RE. Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. USA. 2006; 103:12411-12416.
55. Arjonen A, Kaukonen R, Mattila E, Rouhi P, Hognas G, Sihto H, Miller BW, Morton JP, Bucher E, Taimen P, Virtakoivu R, Cao YO, Sansom J, Joensuu H, Ivaska J. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J. Clin. Investig. 2014; 124:1069-1082.
56. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics (2002). CA Cancer J Clin. 2005; 55:74-108.
57. Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell. 1992; 71:543-546.
58. Demichelis F, Setlur SR, Beroukhim R, Perner S, Korbel JO, Lafargue CJ, Pflueger D, Pina C, Hofer MD, Sboner A, Svensson MA, Rickman DS, Urban A, et al. Distinct genomic aberrations associated with ERG rearranged prostate cancer. Genes Chromosomes Cancer. 2009; 48:366-380.
59. Wu Q, Sahasrabudhe RM, Luo LZ, Lewis DW, Gollin SM, Saunders WS. Deficiency in myosin lightchain phosphorylation causes cytokinesis failure and multipolarity in cancer cells. Oncogene. 2010; 29:4183-4193.
60. Jeong S, Lee J, Kim D, Seol MY, Lee WK, Jeong JJ, Nam KH, Jung SG, Shin DY, Lee EJ, Chung WY, Jo YS. Relationship of Focally Amplified Long Noncoding on Chromosome 1 (FAL1) lncRNA with E2F Transcription Factors in Thyroid Cancer. Medicine (Baltimore). 2016; 95:e2592.
61. Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006; 119:2717-2723.
62. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997; 386:623-627.
63. Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’.Trends Genet. 2008; 24:64-69.
64. Park SH, Maeda T, Mohapatra G, Waldman FM, Davis RL, Feuerstein BG. Heterogeneity, polyploidy, aneusomy, and 9p deletion in human glioblastoma multiforme.Cancer Genet Cytogenet. 1995; 83:127-135.
65. Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, Eastmond DA. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. 2006; 27:337-343.
66. Takanishi DM, Hart J, Covarelli P, Chappell R, Michelassi F. Ploidy as a prognostic feature in colonic adenocarcinoma. Arch Surg. 1996; 131:587-592.
67. Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005; 437:1038-1042.
68. King RW. When 2+2 = 5:the origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008; 1786:4-14.
69. Lothschütz D, Jennewein M, Pahl S, Lausberg HF, Eichler A, Mutschler W, Hanselmann RG, Oberringer M. Polyploidization and centrosome hyperamplification in inflammatory bronchi. Inflamm Res. 2002; 51:416-422.
70. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0:genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007; 9:66-80.
71. Schenker T, Trueb B. Down-regulated proteins of mesenchymal tumor cells. Exp Cell Res. 1998; 239:161-168.
72. Van Eldik LJ, Watterson DM, Burgess WH. Immunoreactive levels of myosin light-chain kinase in normal and virus-transformed chicken embryo fibroblasts. Mol Cell Biol. 1984; 4:2224-2226.
73. Ponti G, Manfredini M, Pellacani G, Tomasi A. Role of microsatellite instability, immunohistochemistry and mismatch repair germline aberrations in immunosuppressed transplant patients: a phenocopy dilemma in Muir-Torre syndrome. Clin Chem Lab Med. 2016; [Epub ahead of print]
74. Sammalkorpi H, Alhopuro P, Lehtonen R, Tuimala J, Mecklin JP, Järvinen HJ, Jiricny J, Karhu A, Aaltonen LA. Background mutation frequency in microsatelliteunstable colorectal cancer. Cancer Res. 2007; 67:5691-5698.
75. van de Luijtgaarden KM, Heijsman D, Maugeri A, Weiss MM, Verhagen HJ, IJpma A, Brüggenwirth HT, Majoor-Krakauer D. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum Genet. 2015; 134:881-893.
76. Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH, Liu L, Tranquilli M, Bale AE, Elefteriades JA. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. 2015; 100:1604-1611.
77. Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol. Life Sci. 2004; 61:822-842.
78. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005; 437:1043-1047.
79. Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992; 70:923-935.
80. Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000; 406:641-645.
81. Buss F, Spudich G, Kendrick-Jones J. Myosin VI:cellular functions and motor properties. Annu. Rev. Cell Dev. Biol. 2004; 20:649-676.
82. Jung EJ, Liu G, Zhou W, Chen X. Myosin VI is a mediator of the p53-dependent cell survival pathway. Mol Cell Biol. 2006; 26:2175-2186.
83. Caulin C, Nguyen T, Lang GA, Goepfert TM, Brinkley BR, Cai WW, Lozano G, Roop DR. An inducible mouse model for skin cancer reveals distinct roles for gain- and loss-of-function p53 mutations. J Clin Invest. 2007; 117:1893-1901.
84. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009; 139:1327-1341.
85. Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008; 22:1337-1344.
86. Bova GS, Isaacs WB. Review of allelic loss and gain in prostate cancer.World Journal of Urology. 1996; 14:338-346.
87. Guan XY, Sham JS, Tai LS, Fang Y, Li H, Liang Q. Evidence for another tumor suppressor gene at 17p13.3 distal to TP53 in hepatocellular carcinoma.Cancer genetics and cytogenetics. 2003; 140:45-48.
88. Oldfors HC, Dios DG, Linder A, Visuttijai K, Samuelson E, Karlsson S, Nilsson S, Behboudi A. Analysis of an independent tumor suppressor locus telomeric to Tp53 suggested Inpp5k and Myo1c as novel tumor suppressor gene candidates in this region. BMC Genet. 2015; 16:80.
89. Liotta LA, Schiffmann E. Tumor motility factors. Cancer Surv. 1988; 7:631-652.
90. Zhong J, Chen Y, Wang LJ. Emerging molecular basis of hematogenous metastasis in gastric cancer. World J Gastroenterol. 2016; 22:2434-2340.
91. Hart IR, Saini A. Biology of tumor metastasis. Lancet. 1992; 339:1453-1457.
92. Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998; 279:527-533.
93. Babbin BA, Koch S, Bachar M, Conti MA, Parkos CA, Adelstein RS, Nusrat A, Ivanov AI. Non-muscle myosin IIA differentially regulates intestinal epithelial cell restitution and matrix invasion. Am J Pathol. 2009; 174:436-448.
94. Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis:an imbalance of positive and negative regulation. Cell. 1991; 63:327-336.
95. Olson MF, Sahai E.The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis. 2008; 26:273-287.
96. Lauffenburger DA, Horwitz AF. Cell migration:a physically integrated molecular process, Cell. 1996; 84:359-369.
97. Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions, J. Cell Sci. 2008; 121:11-18.
98. Hosono Y, Usukura J, Yamaguchi T, Yanagisawa K, Suzuki M, Takahashi T. MYBPH inhibits NM IIA assembly via direct interaction with NMHC IIA and reduces cell motility. Biochem Biophys Res Commun. 2012; 428:173-178.
99. Takano S, Kamiyama H, Tsuboi K, Matsumura A. Angiogenesis and antiangiogenic therapy for malignant gliomas. Brain Tumor Pathol. 2004; 21:69-73.
100. Akiyama H, Yamamoto M, Sakashita C, Umezawa Y, Kurosu T, Murakami N, Miura O. Therapy-related leukemia with Inv(16)(p13.1q22) and type D CBFB/MYH11 developing after exposure to irinotecan-containing chemoradiotherapy. Intern Med. 2015; 54:651-655.
101. Eppinga RD, Peng IF, Lin JL, Wu CF, Lin JJ. Opposite effects of overexpressed myosin Va or heavy meromyosin Va on vesicle distribution, cytoskeleton organization, and cell motility in nonmuscle cells. Cell Motility Cytoskeleton. 2008; 65:197-215.
102. Du YC, Lewis BC, Hanahan D, Varmus H. Assessing tumor progression factors by somatic gene transfer into a mouse model:Bcl-xL promotes islet tumor cell invasion. PLoS Biol. 2007; 5:e276.
103. Provance DW, Wei M, Ipe V, Mercer JA. Cultured melanocytes from dilute mutant mice exhibit dendritic morphology and altered melanosome distribution. Proc. Natl. Acad. Sci. USA. 1996; 93: 14554-14558.
104. Catlett NL, Weisman LS. The terminal tail region of a yeast myosin-V mediates its attachment to vacuole membranes and sites of polarized growth. Proc Natl Acad Sci USA. 1998; 95:14799-14804.
105. Fehrenbacher N, Jäättelä M. Lysosomes as targets for cancer therapy. Cancer Res. 2005; 65: 2993-2995.
106. Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004; 23:2746-2756.
107. Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004; 23:2881-2890.
108. Aneta G, Tomáš V, Daniel R, Jan B. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget. 2016. doi: 10.18632/oncotarget.7214.
109. Royer C, Lu X. Epithelial cell polarity:a major gatekeeper against cancer? Cell Death Differ. 2011; 18:1470-1407.
110. Chen CP, Su YN, Chern SR, Wu PC, Wang W. Prenatal diagnosis of microvillus inclusion disease. Taiwan J Obstet Gynecol. 2011; 50:399-400.
111. Müller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz-Erian P, Ponstingl H, Partsch J, Röllinghoff B, Köhler H, Berger T, Lenhartz H, Schlenck B, Houwen RJ, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008; 40:1163-1165.
112. Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protusion, adhesion dynamics,and polarity by myosins IIA and IIB in migration cells. J Cell Biol. 2007; 176:573-580.
113. Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIB is involved in the guidance of fibroblast migration. Mol. Biol. Cell. 2004; 15:982-989.
114. Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009; 10:778-790.
115. Abercrombie M, Dunn GA, Heath JP. The shape and movement of fibroblasts in culture. Soc. Gen. Physiol. Ser. 1977; 32:57-70.Adams, J.C., 2001. Cell-matrix contact structures. Cell. Mol. Life Sci. 58, 371-392.
116. Gupton SL, Gertler FB. Filopodia:the fingers that do the walking. Sci. STKE. 2007; 400:re5.
117. Cramer LP. Organization and polarity of actin filament networks in cells:implications for the mechanism of myosin-based cell motility. Biochem. Soc. Symp. 1999; 65:173-205.
118. Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, Sixt M. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008; 453:51-55.
119. Hynes RO. Integrins:bidirectional, allosteric signaling machines. Cell. 2002; 110:673-687.
120. Katoh K, Kano Y, Amano M, Onishi H, Kaibuchi K, Fujiwara K. Rho-kinase-mediated contraction of isolated stress fibers. J. Cell Biol. 2001; 153:569-584.
212. Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009; 28:35-49.
122. Sameni M, Moin K, Sloane BF. Imaging proteolysis by living human breast cancer cells. Neoplasia. 2001; 2:496-504.
123. Kim EJ, Helfman DM. Characterization of the metastasis-associated protein, S100A4.Roles of calcium binding and dimerization in cellular localization and interaction with myosin. J Biol Chem. 2003; 278:30063-30073.
124. Wigton EJ, Thompson SB, Long RA, Jacobelli J. Myosin-IIA regulates leukemia engraftment and brain infiltration in a mouse model of acute lymphoblastic leukemia. J Leukoc Biol. 2016 [Epub ahead of print].
125. Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR. Actin and -actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nature Cell Biol. 2008; 10:1039-1050.
126. Kim KY, Kovacs M, Kawamoto S, Sellers JR, Adelstein RS. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J. Biol. Chem. 2005; 280:22769-22775.
127. Cai Y, Biais N, Giannone G, Tanase M, Jiang G, Hofman JM, Wiggins CH, Silberzan P, Buguin A, Ladoux B, Sheetz MP. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys. J. 2006; 91:3907-3920.
128. Wei Q, Adelstein RS. Conditional expression of a truncated fragment of nonmuscle myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol. Biol. Cell. 2000; 11:3617-3627.
129. Bao J, Jana SS, Adelstein RS. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J. Biol. Chem. 2005; 280:19594-19599.
130. Ouderkirk JL, Krendel M. Myosin 1e is a component of the invadosome core that contributes to regulation of invadosome dynamics. Exp Cell Res. 2014; 322:265-276.
131. Watanabe TM, Tokuo H, Gonda K, Higuchi H, Ikebe M. Myosin-X induces filopodia by multiple elongation mechanism. J. Biol. Chem. 2010; 285:19605-19614.
132. Cao R, Chen J, Zhang X, Zhai Y, Qing X, Xing W, Zhang L, Malik YS, Yu H, Zhu X. Elevated expression of myosinX in tumours contributes to breast cancer aggressiveness and metastasis. Br J Cancer.2014; 111:539-550.
133. Mischel PS, Shai R, Shi T, Horvath S, Lu KV, Choe G, Seligson D, Kremen TJ, Palotie A, Liau LM, Cloughesy TF, Nelson SF. Identification of molecular subtypes of glioblastoma by gene expression profiling. Oncogene. 2003; 22:2361-2373.
134. Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, Liu HC, Mahfouz RS, Raimondi C, Lenny N, Patel A, Downing JR. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003; 102:2951-2959.
135. Montell DJ. Command and control: regulatory pathways controlling invasive behavior of the border cells. Mech. Dev. 2011; 105:19-25.
136. Geisbrecht ER, Montell DJ. Myosin VI is required for E-cadherin-mediated border cell migration. Nat Cell Biol. 2002; 4:616-620.
137. Naora H, Montell DJ. Ovarian cancer metastasis:integrating insights from disparate model organisms. Nat Rev Cancer. 2005; 5:355-366.
138. Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008; 121:727-735.
139. Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005; 6:622-634.
140. Batlle E, Sancho E, Franci C, Dom´nguez D, onfar M, Baulida J, Garc´a De, Herreros A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000; 2:84-89.
141. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH in tumour progression:an alliance against the epithelial phenotype? Nat Rev Cancer. 2007; 7:415-428.
142. Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000; 148:779-790.
143. Derycke LD, Bracke ME. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 2004; 48:463-476.
144. Mege RM, Gavard J, Lambert M. Regulation of cell-cell junctions by the cytoskeleton.Curr.Opin. Cell Biol. 2006; 18:541-548.
145. Maddugoda MP, Crampton MS, Shewan AM, Yap AS. Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell cell contacts in mammalian epithelial cells. J Cell Biol. 2007; 178:529-540.
146. Shewan AM, Maddugoda M, Kraemer A, Stehbens SJ, Verma S, Kovacs EM, Yap AS. Myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell-cell contacts. Mol. Biol. Cell. 2005; 16:4531-4532.
147. Cano A, Pe´rez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor Snail controls epithelialmesenchymal transitions by repressing E-cadherin expression. Nat Cell Bio. 2000; 2:76-83.
148. Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001; 106:489-498.
149. Hsu MY, Meier FE, Nesbit M, Hsu JY, Van BP, Elder DE, Herlyn M. E-Cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000; 156:1515-1525.
150. Korzeniewska-Dyl I. Caspases—structure and function. Pol Merkur Lekarski. 2007; 23:403-407.
151. Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003; 3:51-62.
152. Fazal F, Gu L, Ihnatovych I, Han Y, Hu W, Antic N, Carreira F, Blomquist JF, Hope TJ, Ucker DS, de Lanerolle P. Inhibiting myosin light chain kinase induces apoptosis in vitro and in vivo. Mol. Cell. Biol. 2005; 25:6259-6266.
153. Rivinoja A, Kokkonen N, Kellokumpu I, Kellokumpu S. Elevated Golgi pH in breast and colorectal cancer cells correlates with the expression of oncofetal carbohydrate T-antigen. J Cell Physiol. 2006; 208:167-174.
154. Sütterlin C, Hsu P, Mallabiabarrena A, Malhotra V. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell. 2002; 109:359-369.
155. Liu Z, Vong QP, Zheng Y. CLASPing microtubules at the trans-Golgi network. Dev Cell. 2007; 12:839-840.
156. Govindan B, Bowser R, Novick P. The role of Myo2, a yeast class V myosin, in vesicular transport. J Cell Biol. 1995; 128: 1055-1068.
157. Buss F, Kendrick-Jones J. How are the cellular functions of myosin VI regulated within the cell? Biochem Biophys Res Commun. 2008; 369:165-175.
158. Warner CL, Stewart A, Luzio JP, Steel KP, Libby RT, Kendrick-Jones J, Buss F. Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell’s waltzer mice. Embo J. 2003; 22:569-579.
159. Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, Kendrick-Jones J, Buss F. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol. 2005; 169:285-295.
160. Aschenbrenner L, Lee T, Hasson T. Myo6 facilitates the translocation of endocytic vesicles from cell peripheries. Mol Biol Cell. 2003; 14:2728-2743.
161. Block TM, Comunale MA, Lowman M, Steel LF, Romano PR, Fimmel C, Tennant BC, London WT, Evans AA, Blumberg BS, Dwek RA, Mattu TS, Mehta AS. Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc Natl Acad Sci USA. 2005; 102:779-784.
162. Karolczak J, Pavlyk I, Majewski , Sobczak M, Niewiadomski P, Rzhepetskyy Y, Sikorska A, Nowak N, Pomorski P, Prószyski T, Ehler E, Rdowicz MJ. Involvement of unconventional myosin VI in myoblast function and myotube formation. Histochem Cell Biol. 2015; 144:21-38.
163. Spudich G, Chibalina MV, Au JS, Arden SD, Buss F, Kendrick-Jones J. Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat Cell Biol. 2007; 9:176-183.
164. Wei S, Dunn TA, Isaacs WB, De Marzo AM, Luo J. GOLPH2 and MYO6:putative prostate cancer markers localized to the Golgi apparatus. Prostate. 2008; 68:1397-1395.
165. Barrett K, Leptin M, Settleman J. The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell. 1997; 91:905-915.
166. Khoo P, Allan K, Willoughby L, Brumby AM, Richardson HE. In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signaling. Dis Model Mech. 2013; 6:661-78.
167. Jiang Y, Scott KL, Kwak SJ, Chen R, Mardon G. Sds22/PP1 links epithelial integrity and tumor suppression via regulation of myosin II and JNK signaling. Oncogene. 2011; 30:3248-3260.
168. Ohkura H, Yanagida MS. Pombe gene sds22+ essential for a midmitotic transition encodes a leucinerich repeat protein that positively modulates protein phosphatase-1. Cell. 1991; 6:149-157.
169. Grusche FA, Hidalgo C, Fletcher G, Sung HH, Sahai E, Thompson BJ. Sds22, a PP1 phosphatase regulatory subunit, regulates epithelial cell polarity and shape. BMC Dev Biol. 2009; 9:14.
170. Suganuma M, Fujiki H, Suguri H, Yoshizawa S, Hirota M, Nakayasu M, Ojika M, Wakamatsu K, Yamada K, Sugimura T. Okadaic acid:an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc Natl Acad Sci USA. 1988; 85:1768-1771.
171. Jasper H, Benes V, Schwager C, Sauer S, Clauder-Munster S, Ansorge W, Bohmann D. The genomic response of the Drosophila embryo to JNK signaling. Dev Cell. 2011; 1:579-586.
172. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006; 25:9-34.
173. Beaucher M, Hersperger E, Page-McCaw A, Shearn A. Metastatic ability of Drosophila tumors depends on MMP activity. Dev Biol. 2007; 303:625-634.
174. Suzuki T, Hara H. Phytate hydrolysate induces circumferential F-actin ring formation at cell-cell contacts by a Rho-associated kinase-dependent mechanism in colorectal cancer HT-29 cells. Mol Nutr Food Res. 2010; 54:1807-1818.
175. Vucenik I, Shamsuddin AM. Cancer inhibition by inositol hexaphosphate (IP6) and inositol:from laboratory to clinic. J. Nutr. 2003; 133:3778-3784.
176. Owaribe K, Kodama R, Eguchi G. Demonstration of contractility of circumferential actin bundles and its morphogenetic significance in pigmented epithelium in vitro and in vivo. J. Cell Biol. 1981; 90:507-514.
177. Woods A, Couchman JR. Syndecan 4 heparan sulfate proteoglycan is a selectively enriched and widespread focal adhesion component. Mol Biol Cell. 1994; 5:183-192.
178. Carey DJ. Syndecans:Multifunctional cell-surface co-receptors. Biochem J. 1997; 327:1-16.
179. Yip GW, Smollich M, Gotte M. Therapeutic value of glycosaminoglycans in cancer.Mol Cancer Ther. 2006; 5:2139-2148.
180. Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol. 2007; 8:957-969.
181. Matesic DF, Ali A, Sidorova TS, Burns TJ. A cell-cell communication marker for identifying targeted tumor therapies. Curr Bioact Compd. 2013; 9:255-262.
182. Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of rock (rhokinase) and mlck in spatial regulation of mlc phosphorylation for assembly of stress fibers and focal adhesions in 3t3 fibroblasts. J Cell Biol. 2000; 150:797-806.
183. Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, Zhou Q, Majumder S, Bi E, Liu DX, Huang S, Lu Z. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun. 2014; 5:5566.
184. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, Wei C, Guo F, Chen Y, Lu Z. PKM2 Regulates Chromosome Segregation and Mitosis Progression of Tumor Cells. Molecular cell. 2013; 53:75-87.
185. Scruggs SB, Solaro RJ. The significance of regulatory light chain phosphorylation in cardiac physiology. Archives of biochemistry and biophysics. 2011; 510:129-134.
186. Euteneuer U, Schliwa M. Mechanism of centrosome positioning during the wound response in BSC-1 cells. J. Cell Biol. 1992; 116:1157-1166.
187. Gomes ER, Jani S, Gundersen GG. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell. 2005; 121:451-463.
188. Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999; 144:1235-1244.
189. Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr.Opin. Cell Biol. 2001; 13:584-592.
190. Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardinrelated transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis, Nat. Cell. Biol. 2009; 11:257-268.
191. Sahai E, Marshall CJ. RHO-GTPases and cancer. Nature Rev. Cancer. 2002; 2:133-142.
192. Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ, Miano JM. Defining the mammalian CArGome. Genome Res. 2006; 16:197-207.
193. Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000; 406:532-535.
194. Evans DG, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the NorthWest of England over a 10-year period:higher incidence than previously thought. Otol Neurotol. 2005; 26:93-97.
195. De Vries M, van der Mey AG, Hogendoorn PC. Tumor biology of vestibular schwannoma:A review of experimental data on the determinants of tumor genesis and growth characteristics. Otol. Neurotol. 2015; 36:1128-1136.
196. Arpin M, Chirivino D, Naba A, Zwaenepoel I. Emerging role for ERM proteins in cell adhesion and migration. Cell Adh Migr. 2011; 5:199-206.
197. Okada T, Lopez-Lago M, Giancotti FG. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J Cell Biol. 2005; 171:361-371.
198. Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002; 277:883-886.
199. Sherman L, Sleeman J, Herrlich P, Ponta H. Hyaluronate receptors:key players in growth, differentiation, migration and tumor progression. Curr Opin Cell Biol. 1994; 6:726-733.
200. Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006; 442:576-579.
201. Maeda M, Johnson E, Mandal SH, Lawson KR, Keim SA, Svoboda RA, Caplan S, Wahl JK, Wheelock MJ, Johnson KR. Expression of inappropriate cadherins by epithelial tumor cells promotes endocytosis and degradation of E-cadherin via competition for p120(ctn). Oncogene, 2006; 25:4595-4604.
202. Kowalczyk AP, Nanes BA. Adherens junction turnover:regulating adhesion through cadherin endocytosis, degradation and recycling. Subcell Biochem, 2012; 60:197-222.
203. Doherty P, Williams G, Williams EJ. CAMs and axonal growth:a critical evaluation of the role of calcium and the MAPK cascade. Mol. Cell. Neurosci. 2000; 16:283-295.
204. Kim JB, Islam S, Kim YJ, Prudoff RS, Sass KM, Wheelock MJ, Johnson KR. N-Cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J. Cell Biol. 2000; 151:1193-1206.
205. Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. Ecadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004; 23:1739-1748.
206. Theisen CS, Wahl JK, Johnson KR, Wheelock MJ. NHERF links the N-cadherin/catenin complex to the platelet-derived growth factor receptor to modulate the actin cytoskeleton and regulate cell motility. Mol. Biol. Cell. 2007; 18:1220-1232.
207. Ayollo DV, Zhitnyak IY, Vasiliev JM, Gloushankova NA. Rearrangements of the actin cytoskeleton and E-cadherin-based adherens junctions caused by neoplasic transformation change cell-cell interactions. PLoS One. 2009; 4:e8027.
208. Gillett CE, Miles DW, Ryder K, Skilton D, Liebman RD, Springall RJ, Barnes DM, Hanby AM. Retention of the expression of E-cadherin and catenins is associated with shorter survival in grade III ductal carcinoma of the breast. J Pathol. 2001; 193:33-41.
209. Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008; 36:149-155.
210. Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol Biol Cell. 2005; 16:2636-2650.
211. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003; 112:453-465.
212. Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999; 145:1009-1026.
213. Small J, Rinnerthaler G, Hinssen H. Organization of actin meshworks in cultured cells:the leading edge. Cold Spring Harb.Symp. Quant. Biol. 1982; 46:599-611.
214. Wang W, Wyckoff JB, Frohlich VC, Oleynikov Y, Huttelmaier S, Zavadil J, Cermak L, Bottinger EP, Singer RH, White JG, Segall JE, Condeelis JS. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002; 62:6278-6288.
215. Carlier MF, Pantaloni D. Control of actin assembly dynamics in cell motility. J Biol Chem. 2007; 282:23005-23009.
216. Baarlink C, Wang H, Grosse R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science. 2013; 340:864-867.
217. Du J, Fan YL, Chen TL, Feng XQ. Lifeact and Utr230 induce distinct actin assemblies in cell nuclei. Cytoskeleton. 2015; 72:570-575.
218. Wu G, Chen PL. Structural requirements of chromokinesin Kif4A for its proper function in mitosis. Biochem Biophys Res Commun. 2008; 372:454-458.
219. Woolner SO, Brien LL, Wiese C, Bement WN. Myosin-10 and actin filaments are essential for mitotic spindle function. J. Cell Biol. 2008; 182:77-88.
220. Rosenblatt J, Cramer LP. Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly. Cell vol. 2004; 117:361-372.
221. Weber KL, Sokac AM, Berg JS, Cheney RE, Bement WM. A microtubulebindin myosin required for nuclear anchoring and spindle assembly. Nature. 2004; 431:325-330.
222. Heald R. Motor function in the mitotic spindle. Cell. 2000; 102:399-402.
223. Bezanilla M, Wilson JM, Pollard TD. Fission yeast myosin-II isoforms assemble into contractile rings at distinct times during mitosis. Curr. Biol. 2000; 10:397-400.
224. Straight AF, Cheung A, Limouze L, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003; 299:1743-1747.
225. de Lanerolle P. Nuclear actin and myosins at a glance. J Cell Sci, 2012; 125: 4945-4949.
226. Zhang YS, Liu B, Luo XJ, Li TB, Zhang JJ, Peng JJ, Zhang XJ, Ma QL, Hu CP, Li YJ, Peng J, Li Q. A novel function of nuclear non muscle myosin regulatory light chain in promotion of xanthine oxidase transcription after myocardial ischemia/reperfusion. Free Radical Biology and Medicine. 2015; 85: 115-128.
227. Philimonenko VV, Janacek J, Harata M, Hozak P. Transcription-dependent rearrangements of actin and nuclear myosin I in the nucleolus. Histochem.Cell Biol. 2010; 134:243-249.
228. Rodgers BD. Insulin-like growth factor-I downregulates embryonic myosin heavy chain (eMyHC) in myoblast nuclei. Growth Horm IGF Res. 2005; 15:377-383.
229. Li Q, Sarna SK. Nuclear myosin II regulates the assembly of preinitiation complex for ICAM-1 gene transcription. Gastroenterology. 2009; 137:1051-1060.
230. Pranchevicius MC, Baqui MM, Ishikawa-Ankerhold HC, Lourenco EV, Leao RM, Banzi SR, dos Santos CT, Roque-Barreira MC, Espreafico EM, Larson RE. Myosin Va phosphorylated on Ser1650 is found in nuclear speckles and redistributes to nucleoli upon inhibition of transcription. Cell Motil. Cytoskeleton. 2008; 65:441-456.
231. Lindsay AJ, McCaffrey MW. Myosin Vb localises to nucleoli and associates with the RNA polymerase I transcription complex. Cell Motil. Cytoskeleton. 2009; 66:1057-1072.
232. Vreugde S, Ferrai C, Miluzio A, Hauben E, Marchisio PC, Crippa MP, Bussi M, Biffo S. Nuclear myosin VI enhances RNA polymerase II-dependent transcription. Mol. Cell. 2006; 23:749-755.
233. Reck-Peterson SL, Novick PJ, Mooseker MS. The tail of a yeast class V myosin, Myo2p, functions as a localization domain. Mol Biol Cell. 1999; 10:1001-10017.
234. Cameron RS, Liu C, Mixon AS, Pihkala JP, Rahn RJ, Cameron PL. Myosin16b: The COOH-tail region directs localization to the nucleus and overexpression delays S-phase progression. Cell Motil. Cytoskeleton. 2007; 64:19-48.
235. Salamon M, Millino C, Raffaello A, Mongillo M, Sandri C, Bean C, Negrisolo E, Pallavicini A, Valle G, Zaccolo M. Human MYO18B, a novel unconventional myosin heavy chain expressed in striated muscles moves into the myonuclei upon differentiation. J. Mol. Biol. 2003; 326:137-149.
236. Mehta IS, Amira M, Harvey AJ, Bridger JM. Rapid chromosome territory relocation by nuclear motor activity in response to serum removal in primary human fibroblasts. Genome Biol. 2010; 11:R5.
237. Hofmann WA, Johnson T, Klapczynski M, Fan JL, de Lanerolle P. From transcription to transport: emerging roles for nuclear myosin I. Biochem Cell Biol. 2006; 84:418-426.
238. Cameron RS, Liu C, Pihkala JP. Myosin 16 levels fluctuate during the cell cycle and are downregulated in response to DNA replication stress. Cytoskeleton (Hoboken). 2013; 70:328-348.
239. Wang SZ, Adler R. Chromokinesin: a DNA-binding, kinesin-like nuclear protein. J. Cell Biol. 1995; 128:761-768.
240. Mazumdar M, Misteli T. Chromokinesins: multitalented players in mitosis. Trends Cell Biol. 2005; 15:349-355.