
Introduction
Adaptive immune responses characteristically involve CD4+ T cells, CD8+ T cells and B cells, all of which may participate in spontaneous and induced responses to cancer in patients [1, 2]. Since cognate interactions between helper CD4+ T cells and B cells are required to produce the tumor-specific isotype switched B cells and antibodies detected in many cancer patients [3], the consequences of such interactions are of relevance to our rapidly growing understanding of the anti-tumor immune response.
The use of T cell receptor (TCR) transgenic mice has aided in understanding many of the mechanisms involved in anti-tumor immune responses in preclinical models. Early studies generally focused on direct tumor recognition by CD8+ T cells [4-7], while more recently developed models have employed CD4+ T cells [8, 9]. One of the best characterized is the Trp-1 model, in which CD4+ T cells expressing a transgenic MHC class II (MHCII)-restricted TCR specific for the tyrosinase related protein expressed by B16 melanoma were able to eradicate established tumors in lymphopenic hosts, via a direct cytotoxic attack against the B16 melanoma cells, with no requirement for CD8+ T cells or B cells [10].
We have developed an alternative preclinical model based on the response of MHCII-restricted TCR transgenic cells to tumor antigen [11]. In contrast to the Trp-1 model, the mechanism of tumor eradication in this model is an IFN-γ-dependent response that requires indirect recognition of tumor antigen presented by host cells. Thus our model mimics a common situation in which tumor antigen-specific CD4+ T cells are unable to directly recognize an MHCII-negative tumor. Once again tumor eradiation in immunodeficient hosts requires neither CD8+ T cells or B cells [11].
Here we have adapted our transgenic model to the study of B cells in tumor immunity. Despite a substantial body of work, there is as yet no consensus as to whether B cells have a positive or negative effect on tumor clearance [12]. Recent reports showing that immunotherapy with checkpoint inhibitors such as Ipilimumab can activate pre-existing and de novo B cell responses [1], in addition to de novo CD4+ T cell responses [13], have served to underline the ongoing clinical relevance of achieving a broader understanding of the role of T-B collaboration in anti-tumor immunity.
Several large-scale clinical studies have suggested that B cells are protective, since B cell infiltration into tumors has been correlated with increased survival of patients with a range of cancers [14-16]. In contrast, the presence of spontaneous serum antibody to tumor-associated antigens (TAAs) is usually either of no prognostic significance or shows a negative association with survival [17, 18]. However generation of antibody responses to TAAs in response to specific immunotherapy can be a positive prognostic indicator [1].
Positive and negative roles of B cells have also been explored in animal models of tumor immunity. T cell priming to tumor antigen is generally enhanced in the absence of B cell antigen presentation [19, 20], and B cells can acquire regulatory functions that negatively influence T cell-dependent anti-tumor immunity [21]. In contrast, pro-inflammatory antibody isotypes have been shown to mediate protection in metastatic disease models [22] but have also been implicated in driving chronic inflammation, which in turn may predispose to malignancy [23].
To examine how collaboration between tumor-specific CD4+ T cells and B cells, and the production of isotype switched antibodies to tumor antigens affect tumor growth, we made use of antigen receptor transgenic B cells and CD4+ T cells specific for a neo-antigen expressed by the B16 mouse melanoma. By co-transferring CD4+ T cells and B cells into tumor-bearing immunodeficient hosts, we determined the effects of B cell antigen presentation and antibody production on tumor protection and the anti-tumor CD4+ T cell response. Tumor-specific B cells reduced the number of tumor-reactive CD4+ T cells in secondary lymphoid tissues and the tumor itself, but had surprisingly little effect on the CD4+ T cell-derived cytokine profile. The absolute number of induced FoxP3+ regulatory T cells (iTregs) within the tumor-specific CD4+ T cell compartment was unaffected by the presence of B cells, although the B cell-dependent reduction in absolute numbers of CD4+ T cells caused iTregs to represent a higher proportion of CD4+ T cells. B cells responding to tumor antigen in the presence of CD4+ T cell help proliferated, differentiated into germinal center cells and secreted isotype switched anti-tumor antibodies detectable in the serum. In the absence of T cells, B cells activated by anti-CD40 mAb also produced tumor-specific isotype-switched antibodies, which had no effect on the growth of subcutaneous tumors but provided protection in a B16 lung metastasis model.
Results
To investigate the effects of tumor-specific B cells on tumor eradication by CD4+ T cells, we set up a mouse model combining B cell receptor transgenic hen egg lysozyme (HEL)-specific B cells capable of antibody isotype switching (SWHEL mouse model [24]) and TCR transgenic moth cytochrome c (MCC)-specific CD4+ T cells (5C.C7 TCR model [25]). Both transgenic mouse lines were bred on a Rag2-/- background to ensure that they generated only monospecific B and T cells, respectively, and contained no other T or B cells. The B16.F10 melanoma cell line was retrovirally transduced to express a fusion protein containing the relevant HEL and MCC epitopes (designated B16.mHELMCC) [11].
Immunodeficient Rag-/- mice were chosen as tumor hosts because of their capacity to support T cell-dependent tumor clearance after adoptive transfer of naive tumor-specific CD4+ T cells [11, 26]. This model is the first to combine BCR and TCR transgenic cells for the concurrent assessment of T cell, B cell and antibody responses during the course of a well-defined in vivo anti-tumor immune response.
Effect of B cells on the CD4+ T cell response to subcutaneous tumor
In a preliminary experiment, adoptive transfer of naive B cells from SWHEL Rag2-/- donors had no effect on the growth of subcutaneous B16 tumors, whereas survival was improved in mice adoptively transferred with naive CD4+ T cells (100% FoxP3-negative) harvested from 5C.C7 TCR Rag2-/- mice (Figure 1A). We next compared tumor growth in mice adoptively transferred with naive 5C.C7 TCR Rag2-/- T cells alone vs 5C.C7 T cells plus SWHEL B cells. All mice adoptively transferred with CD4+ T cells showed highly significant control of tumor growth compared with controls that received no cells (Figure 1B). Mice that received T plus B cells compared with T cells alone showed a trend towards reduced ability to control tumor growth after day 50, although the effect failed to reach statistical significance (p = 0.0516) (Figure 1B). Individual tumor growth curves in this experiment showed three patterns: exponential tumor growth in untreated controls, complete tumor clearance with long-term tumor-free survival in a proportion of adoptive hosts of T plus B cells or T cells alone (termed ‘responders’), and early tumor control followed by progressive outgrowth in the remainder of adoptive hosts (termed ‘failed responders’) (Figure 1C and 1D). The latter patterns correspond to those we have previously described in tumor-bearing Rag-/- recipients of 5C.C7 TCR Rag2-/- T cells [11]. In our previous experiments, we have never observed tumor recurrence in mice that are tumor-free at 120 days, even after regular observation for more than one year, and therefore in the present study, mice that were tumor-free at 120 days were deemed to have mounted a long-term response and were euthanized. Mean tumor growth in responders within the T plus B cell versus T cell group was indistinguishable, as was growth in failed responders from the two groups (Figure 1D).
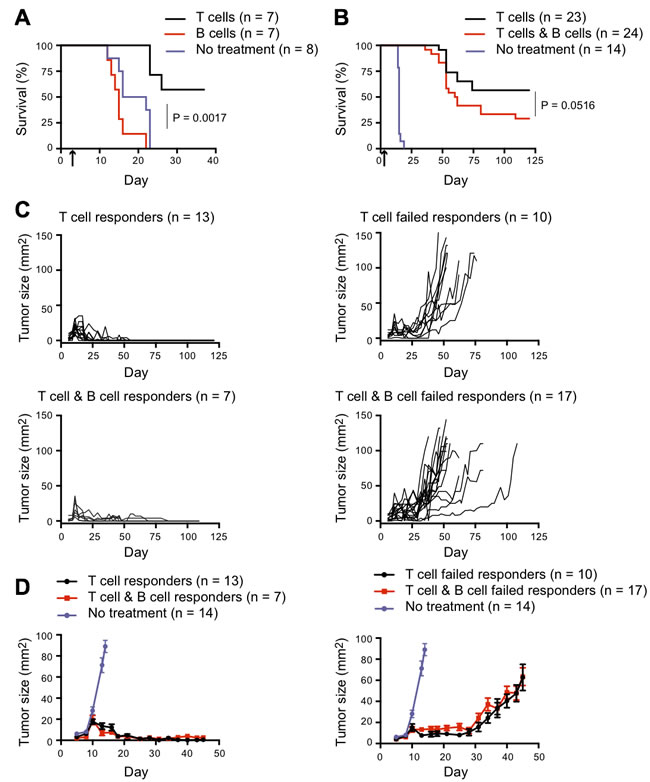
Figure 1: Effect of B cell co-transfer on CD4+ T cell-dependent tumor control. A. Rag2-/- mice were injected s.c. in the flank with 0.5x106 B16.mHELMCC tumor cells and adoptively transferred with 0.2x106 naive 5C.C7 T cells or 1x106 naive SWHEL B cells 5 days later (indicated by arrow on graph x-axis). Mice were euthanized when tumors reached 100mm2. Kaplan-Meier survival analysis of the three groups; black line: 5C.C7 T cells (n = 7), red line: SWHEL B cells (n = 7) and purple line: no treatment (n = 8). P = 0.0017 for comparison between no treatment and T cell groups, while difference between no treatment and B cell groups was not significant as calculated using Log-rank test with Gehan-Breslow-Wilcoxon post-test. (B-D) Rag2-/- mice were injected s.c. in the flank with 0.5x106 B16.mHELMCC tumor cells and 3 days later adoptively transferred with 0.2x106 naive 5C.C7 T cells or a combination of 0.2x106 naive 5C.C7 T cells and naïve 1x106 SWHEL B cells (indicated by arrow on graph x-axis). Data are representative of two independent experiments. Mice were euthanized when tumors reached 100mm2, with Kaplan-Meier survival analysis shown in B. Black line: 5C.C7 T cells (n = 23), red line: 5C.C7 T cells plus SWHEL B cells (n = 24), purple line: no treatment (n = 14). P = 0.0516 for comparison between T cell and B cell groups, by Log-rank test with Gehan-Breslow-Wilcoxon post-test. C. Individual tumor growth curves separated on the basis of tumor control into long-term responders (tumor-free at 120 days) and failed responders. Top left: 5C.C7 T cell group, responders (n = 13). Top right: 5C.C7 T cell group, failed responders (n = 10). Bottom left: 5C.C7 T cells plus SWHEL B cell group, responders (n = 7). Bottom right: 5C.C7 T cells plus SWHEL B cell group, failed responders (n = 17). D. Tumor growth (mean±SEM) of responders (left) and failed responders (right) overlaid with control group that received no treatment (purple dot). Note the change in scale, to highlight the early effects on tumor growth.
To explore the mechanisms underlying the loss of CD4+ T cell-dependent tumor control in non-responder mice, we examined several aspects of the CD4+ T cell response. Mice in the experiment shown in Figure 1B were bled on day 40 for quantitation of T cell number and detection of FoxP3 as an indicator of conventional CD4+ T cell conversion to an iTreg phenotype (Figure 2A). B cells were also quantitated in the T plus B cell group. At day 40, the percentage of CD4+ T cells within peripheral blood mononuclear cells correlated with survival (Figure 2B). Only 1 of 26 mice with fewer than 6% circulating CD4+ T cells remained tumor free until sacrifice at day 120, compared with all 19 mice with more than 6% CD4+ T cells (Figure 2C). The ratio of B cells to CD4+ T cells was also predictive of survival, with a higher ratio significantly associated with a worse prognosis (Figure 2D). A significant reduction in the percentage of CD4+ T cells was seen in the T plus B cell group when compared with T cells alone (Figure 2E). Although FoxP3+ cells expressed as a percentage of CD4+ T cells also correlated with survival (Figure 2F), this effect appeared to be secondary to changes in the number of CD4+ T cells, as there was no correlation between survival and FoxP3+ cells when expressed as a percentage of total (non-B cell) circulating peripheral blood mononuclear cells (Figure 2G, 2H).
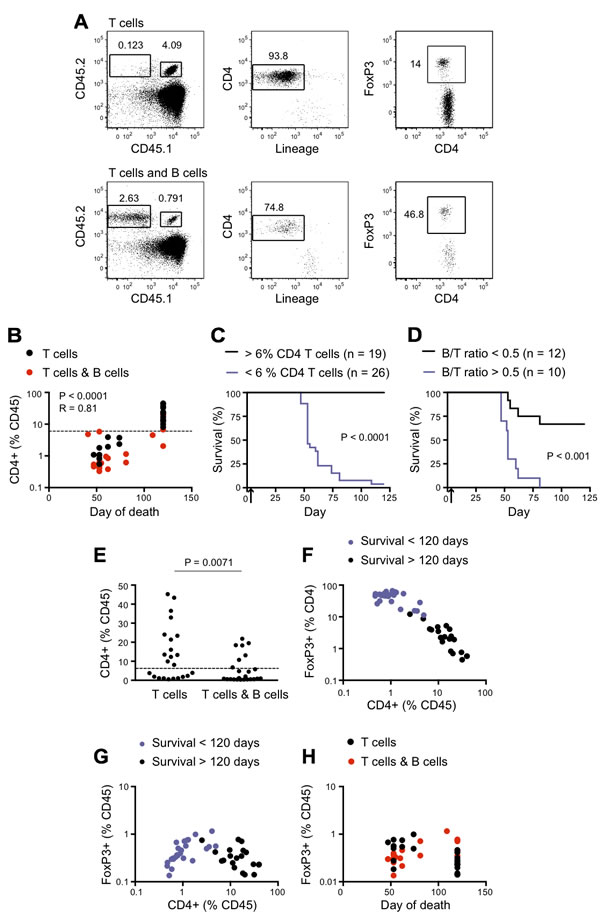
Figure 2: Immune determinants of long-term tumor control. For the experiment described in Figure 1B, donor T cell frequencies in the blood were analyzed on day 40. A. 5C.C7 T cells were gated as singlet+, CD45.1+ CD45.2+, CD4+ and lineage- (lineage cocktail included mAbs against CD11b, Gr1, CD8, NK1.1 and Ter119). iTreg cells were further identified by intracellular FoxP3 expression; the percentage positive is indicated. CD45.2 homozygous SWHEL B cells were identified as CD45.2+CD45.1- (tumor bearing hosts were CD45.1 homozygous). Top panel: Recipients of 5C.C7 CD4+ T cells alone. Bottom panel: Recipients of 5C.C7 CD4+ T cells plus SWHEL B cells. B. Frequency of CD4+ (% CD45) in the blood on day 40 vs day of death. The frequency of CD4+ was calculated as a percentage of total CD45.1+ cells (excluding B cells), to correct for the presence of B cells in the T plus B cell group. With one exception, all mice tumor free at the completion of the experiment (day 120) had more than 6% CD4+ (% CD45) (dashed line) on day 40. Statistical significance was calculated by Spearman correlation. C. Kaplan-Meier survival analysis after stratification for > 6% CD4+ T cells as a percentage of CD45.1+ cells in PBMC on day 40. Black line: > 6% CD4+ T cells (n = 19). Purple line: < 6% CD4+ T cells (n = 26). **** P < 0.0001 by Log-rank test with Gehan-Breslow-Wilcoxon post-test. D. Kaplan-Meier survival analysis after stratification for B cell/CD4+ T cell ratio > 0.5 in blood on day 40. Black line: B cells/CD4+ T cells < 0.5 (n = 12). Purple line: B cells/CD4+ T cells > 0.5 (n = 10). ). *** P < 0.001 by Log-rank test with Gehan-Breslow-Wilcoxon post-test. E. Frequency of CD4+ T cells as %CD45.1 in the blood on day 40 in mice that received either T cells alone or T cells plus B cells. Dashed line represents 6%. Data are representative of two independent experiments. (F, G) Frequency of FoxP3+ T cells as percent of CD4+ T cells F. or percent of CD45.1+ cells G. in the blood on day 40, plotted relative to frequency of CD4+ T cells in the blood on day 40. Black dots: > 120 days survival (n = 19). Purple dots: < 120 days survival (n = 26). Data are representative of two independent experiments. H. Frequency of FoxP3+ Tregs of total CD45.1+ in blood on day 40, plotted relative to the day of death. Black dots: T cell group. Red dots: T cell plus B cell group. No statistical significance was reached for either group by Spearman correlation. Data are representative of two independent experiments.
The percentages of circulating CD4+ T cells on day 40 correlated with the percentages in peripheral blood at death (Figure 3A, left panel). Subsequent analysis showed that at the time of euthanasia (when tumor size reached > 100mm2 or at day 120), the percentage of peripheral blood CD4+ T cells correlated with the CD4+ T cell proportions in the spleen and tumor-draining LN, and within the tumor itself in tumor-bearing mice (Figure 3A, right panels). This suggested that the adaptive anti-tumor response had essentially reached equilibrium by day 40. Similar correlations between circulating cell percentages at day 40 and at the time of euthanasia were seen for B cells (Figure 3B) and FoxP3+ T cells expressed as a percentage of CD4+ T cells (Figure 3C). Taken together, these data suggested that the number of tumor-reactive CD4+ T cells at equilibrium was a major determinant of tumor control. While early cognate interactions with tumor-specific B cells reduced the number of CD4+ T cells, the total number of iTreg cells was independent of the number of CD4+ T cells and B cells and was not predictive of survival.
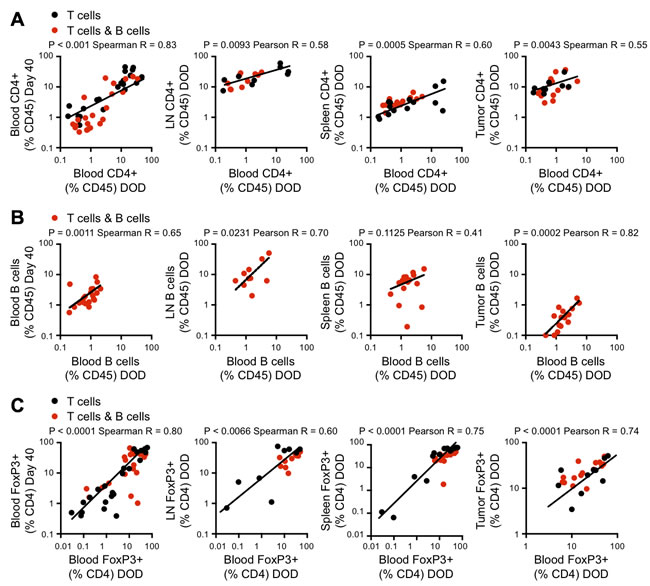
Figure 3: Analysis of immune cell populations in blood, lymph nodes, spleen and tumor. In the experiment described in Figure 1B, flow cytometric analysis of blood, lymph nodes, spleen and tumor was performed at the time of euthanasia (when tumors reached 100mm2). A. Correlation between CD4+ T cell frequency in the blood on the day of death (DOD) and on day 40 (as shown in Figure 2) (left panel) and CD4+ T cell frequencies in blood and lymph node, spleen and tumor on DOD (right panels). Black dots: 5C.C7 CD4+ T cell group. Red dots: 5C.C7 CD4+ T cell plus SWHEL B cell group. Log-transformed data. B. Correlation between B cell frequency in the blood on DOD and d40 (left panel) and between blood and lymph node, spleen and tumor on DOD (right panels). C. Correlation between Foxp3+ iTreg frequency as a percent of CD4+ T cells in the blood on DOD and d40 (left panel) and FoxP3+ (%CD4+) in blood and lymph node, spleen and tumor on the DOD (right panels). Black dots: 5C.C7 CD4+ T cell group. Red dots: 5C.C7 CD4+ T cell plus SWHEL B cell group. For each analysis, data from both groups was pooled, and a D’Agostino-Pearson omnibus normality test was performed prior to correlation analysis to determine the appropriate parametric or non-parametric test. Parametric Pearson correlation coefficients or nonparametric Spearman correlations were used as indicated on each panel.
To explore possible mechanisms of B cell modulation of the CD4+ T cell response, we tested cytokine production by splenic T cells in a cohort of T plus B cell versus T cell alone recipient mice from the experiment shown in Figure 1B, euthanized with > 100mm2 tumors on day 52. Intracellular production of IL-2, TNF and IFNγ by tumor-specific T cells was examined after overnight stimulation with MCC peptide (Figure 4A). Surprisingly, given previous studies suggesting that cognate T-B interactions cause deviation towards a Th2 cytokine profile [27], the frequencies of IL-2-, IFNγ- and TNF-producing cells were not decreased in the T plus B cell group. Indeed, the percentage of IL-2+ cells was significantly increased, and there was a trend towards an increase in TNF+ cells (Figure 4B). Secreted cytokine production was also analyzed after an overnight culture with a combination of MCC peptide and HEL protein. There was no significant difference between the groups in the levels of IFNγ, TNF, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, IL-13, IL-17, KC, MCP-1, MIG, MIP-1α and MIP-1β analyzed using a cytometric bead array (CBA) cytokine kit (not shown). The cytokine profile was strongly biased towards Th1, with levels of TNF and IFNγ at least 100-fold those of IL-4, IL-5 and IL-13.
Apart from their reported propensity to induce iTregs, B cells can also develop intrinsic regulatory function [28, 29], which might reduce T cell numbers in vivo. Several different phenotypes of regulatory B cells (Bregs) have been reported, including the B10 cell, which regulates anti-tumor responses via IL-10 production [21]. We used surface staining against the B10/Breg markers CD5, CD1d and Granzyme B in combination with CD19 and were able to identify cells with a Breg phenotype in wild-type controls but not in tumor-bearing recipients of T and B cells (data not shown). Although we measured IL-10 at levels of 30 pg/ml in culture supernatants (data not shown), there was no difference between cultures with and without B cells and ex vivo restimulation of B cells from tumor-bearing mice failed to detect the presence of IL-10 producing B cells (data not shown).
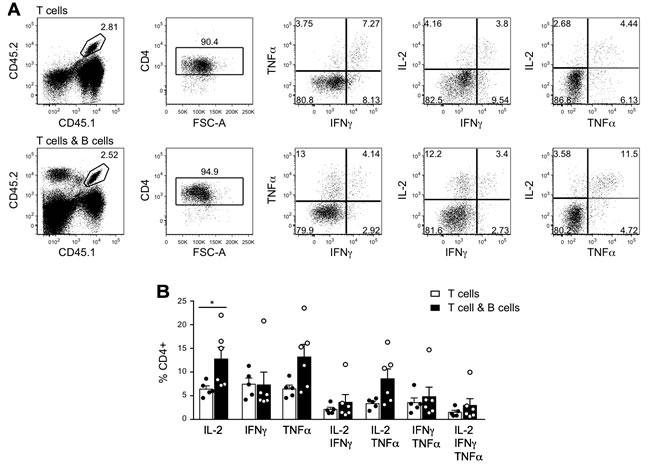
Figure 4: Cytokine production by CD4+ T cells in tumor-bearing mice. In the experiment described in Figure 1B, 6 mice that received T and B cells and 5 mice that received T cells alone were euthanized as tumors reached critical size on day 52 post tumor cell inoculation. Splenocytes were cultured overnight with MCC peptide and analyzed by flow cytometry for intracellular cytokine production. A. Representative dot plots for mice that received 5C.C7 CD4+ T cells alone (top panels) or 5C.C7 CD4+ T cells plus SWHEL+ B cells (bottom panels). T cells were gated as CD45.1+CD45.2+CD4+ and analyzed for IL-2, TNFα and IFNγ expression. Frequency of gated events is shown. B. Frequency of cytokine producing cells within CD4+ T cells. Individual measurements (dots) and mean±SEM of each group are shown. * = p < 0.05 by unpaired Student’s t test.
Effect of CD4+ T cells on the B cell response to subcutaneous tumor
To confirm that the HEL-specific B cells were mounting a CD4+ T cell dependent response to subcutaneous tumor, we inoculated mice s.c. with B16.mHELMCC cells, adoptively transferred 1x106 naïve SWHEL Rag2-/- B cells and 2x105 naive 5C.C7 Rag2-/- CD4+ T cells on day 3 and characterized splenic B cells by flow cytometry 13, 30 and 71 days later. Transferred SWHEL B cells were identified as HEL-binding B220+CD19+ cells, and CD95+GL7+ was used to identify the subpopulation of germinal center cells (Figure 5A). In the presence of cognate T cell help from CD4+ T cells, B cells proliferated extensively, with 106 present in the spleen on day 13 post cell transfer, compared with fewer than 2 x 104 in a control group 7 days after transfer of 106 T cells and 5x106 MHCII IE-negative B cells that could not present the MCC epitope in order to attract cognate T cell help. The number of splenic B cells in the T plus B cell group showed no significant decline over the next 2 months (Figure 5B). Germinal center B cells, which were absent from the original naive B cell inoculum, peaked at day 30 and then declined 100-fold by day 71 (Figure 5B). A strong isotype-switched anti-tumor antibody response was produced in response to cognate CD4+ T cell help, with large increases in the serum levels of IgG1, IgG2c (the functional equivalent of IgG2a, which is not present in C57BL/6 mice [30]) and IgG2b, and smaller increases in IgG3, IgA and IgE (Figure 5C and 5D). Note that most publications still refer to the C57BL/6 isotype as IgG2a rather than IgG2c, so we have termed the isotype we detected as IgG2a/c throughout.
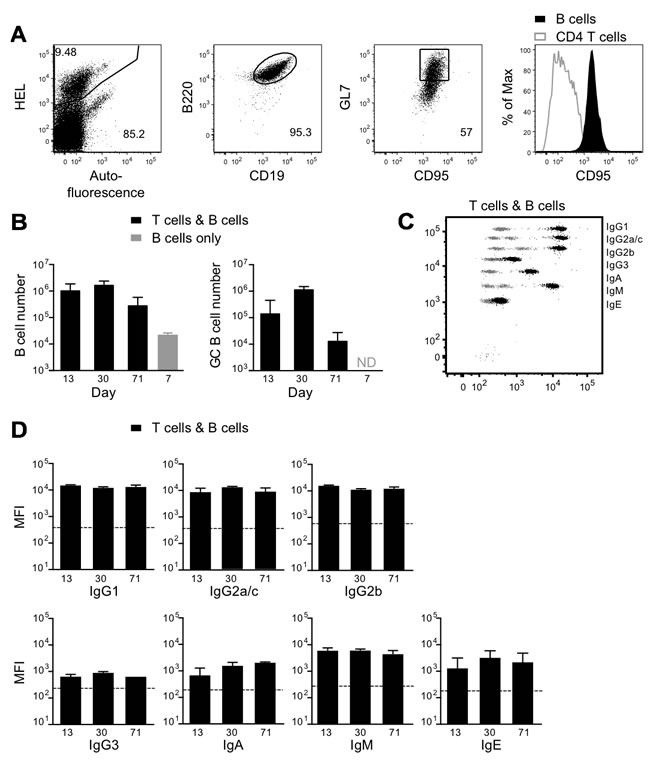
Figure 5: Effect of CD4+ T cells on activation, differentiation and antibody production by tumor-specific B cells. Rag2-/- mice were injected s.c. in the flank with 1x106 B16.mHELMCC tumor cells. On day 3, mice received 1x106 SWHEL B cells plus 0.2x106 5C.C7 T cells i.v. T cell and B cell numbers and serum antibodies were measured 13, 30 and 71 days later. A. Representative dot plots show gating strategy for donor B cells in the spleen. SWHEL B cells were identified as HEL+B220+CD19+ and germinal center SWHEL B cells as HEL+B220+CD19+CD95+GL7+. Expression of CD95 by germinal center B cells is compared with that of T cells (right panel, T cells gated as HEL-CD45.2+ within the HEL-CD45.2- host, as shown in Figure 4A lower panels). B. Black bars: mean±SEM of the number of splenic SWHEL B cells (left) and splenic germinal center (CD95+GL7+) SWHEL B cells (right) in recipients of B cells and 5C.C7 T cells, on days 13, 30 and 71 post T and B cell transfer (n = 4/group). Grey bar: absolute number of splenic SWHEL B cells in s.c. tumor-bearing mice 7 days after transfer of 1x106 5C.C7 T cells and 5x106 SWHEL+ B cells lacking expression of the MHCII allele IE required for presentation of the MCC epitope to T cells. Germinal center B cells were not detected (ND) in the B cell only control group. C. Representative dot plot depicting serum antibody isotype CBA data from recipients of SWHEL B cells plus T cells (black), overlaid on control CBA plot (grey). D. MFI of CBA signals for each antibody isotype on days 13, 30 and 71 post T and B cell transfer (mean±SEM, n≥4/group), with geometric mean of CBA bead only control for each isotype indicated by the dashed line. Data are representative of 2 independent experiments.
Effects of B cells on tumor growth in the absence of T cells
The experiment shown in Figure 1A indicated that adoptive transfer of naive SWHEL B cells had no effect on the growth of subcutaneous B16.mHELMCC tumors in Rag-/- hosts. Since tumor-specific antibody has been shown to protect from lung metastases after i.v. injection of B16 tumor [22], we tested whether SWHEL mice on a Rag2-/- background would be more resistant to i.v. challenge with 1x106 B16.mHELMCC tumor than their SWHEL-negative Rag2-/- littermates that lack all T and B cells. The number of lung metastases was not significantly different in the two groups of mice (Figure 6A). The predominant antibody isotype detected in the serum of naïve SWHEL mice was IgM (Figure 6B), with some spontaneous age-dependent switching to IgG2b and minor amounts of IgG1, IgG3 and IgA (Figure 6B and 6C). Serum antibodies in i.v. tumor-challenged SWHEL mice were similar prior to challenge (naïve state) and at the time of death (day 21), as expected given the absence of T cell help in the Rag2-/- mice (Figure 6B and 6C). Thus the presence of naïve tumor-specific B cells secreting IgM did not afford protection from tumor metastasis.
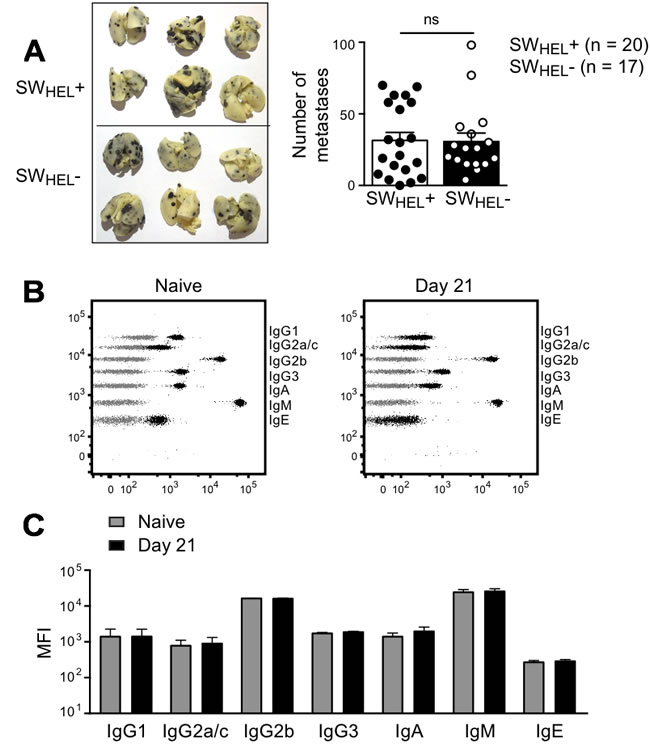
Figure 6: Effect of naive B cells in a lung metastasis model. A. Rag2-/- mice that did or did not express the SWHEL BCR (termed SWHEL+ and SWHEL-) were injected i.v. with 1x106 B16.mHELMCC tumor cells. Left: Representative photos of lungs from the two groups. Right: Number of metastases on day 21. Each dot represents an individual animal, with the bar representing mean±SEM (Rag2-/- SWHEL+: n = 20, Rag2-/- SWHEL-: n = 17). Data are pooled from 2 independent experiments. ns = not significant by unpaired Student’s t test B. Representative dot plots depicting serum antibody isotype CBA data from Rag2-/- SWHEL+ mice (black) overlaid on control CBA plot (grey). Sera were collected at the time of death. Left plot: naïve (prior to tumor challenge) SWHEL+ mouse. Right plot: SWHEL+ mouse after i.v. tumor challenge (time of death: Day 21). C. Serum antibody isotype analysis. MFI of CBA signals for each isotype before tumor challenge and on day of death, mean±SEM, n≥7/group. Data are from a single experiment. All isotypes not significantly different by unpaired Student’s t test.
To test the potency of isotype-switched tumor-specific antibody in the metastasis model without the confounding influence of tumor-specific CD4+ T cells, SWHEL Rag2-/- mice were challenged i.v. with tumor cells, followed by 2 i.p. injections of an agonistic anti-CD40 monoclonal antibody to provide helper signals to the B cells. In vivo B cell activation in response to anti-CD40 was confirmed by increased expression of MHCII and CD95 on HEL-binding B cells (Figure 7A). The number of lung metastases was significantly decreased in anti-CD40 treated SWHEL-positive Rag2-/- mice compared to SWHEL-negative Rag2-/- controls (Figure 7B). The combination of i.v. tumor plus anti-CD40 stimulated a large increase in HEL-specific serum IgG2a/c, with smaller increases in IgG1, IgG3, IgA and IgE and no change in IgG2b or IgM (Figure 7C and 7D). Our finding that IgG2a/c was most prominently associated with the control of B16 lung metastasis is consistent with previous work in which purified exogenous anti-tumor antibody was administered [22].
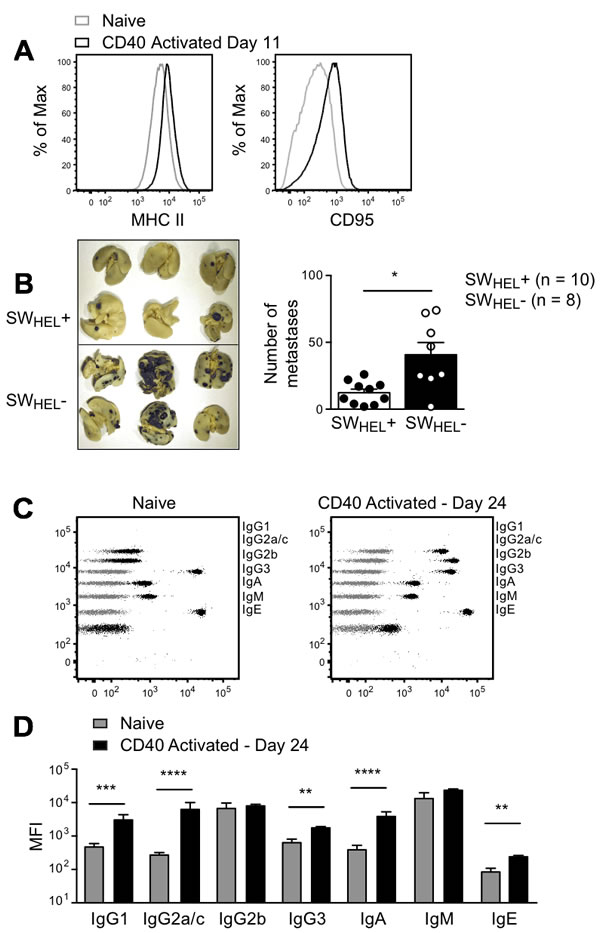
Figure 7: Effect of anti-CD40 activated B cells and isotype switched anti-tumor antibody in a lung metastasis model. Rag2-/- SWHEL+ and Rag2-/- SWHEL- mice were injected i.v. with 1x106 tumor cells followed by i.p. anti-CD40 on days 3 and 6. A. Splenocytes from two anti-CD40 treated Rag2-/- SWHEL+ mice 11 days after tumor challenge were pooled and stained with B cell activation markers, as indicated. Black histograms: anti-CD40 activated Rag2-/- SWHEL+ B cells; grey histograms: naive Rag2-/- SWHEL+ mice. Additional mice were euthanized on day 24, lungs were collected to count metastatic foci and serum was sampled to measure antibody concentration. B. Left: Representative photos of lungs from Rag2-/- SWHEL+ and Rag2-/- SWHEL- mice. Right: Number of metastases. Each dot represents an individual animal, with the bar representing mean±SEM (SWHEL+: n = 10, SWHEL-: n = 8). * = P < 0.05 by nonparametric Mann-Whitney test. C. Representative dot plots depicting antibody isotype CBA data from Rag2-/- SWHEL+ mice (black) overlaid on control CBA plot (grey). Left panel: naïve Rag2-/- SWHEL+ mouse. Right panel: anti-CD40 treated Rag2-/- SWHEL+ mouse after i.v. tumor challenge (time of death: Day 24). D. MFI of CBA signals for each isotype before tumor challenge and on day of death, mean±SEM, n≥9/group. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 by unpaired Student’s t test.
We also tested whether anti-CD40-activated SWHEL B cells could exert a protective effect against s.c. B16.mHELMCC tumors. SWHEL Rag2-/- mice and SWHEL-negative Rag2-/- littermates were inoculated with tumor cells in the flank, followed by anti-CD40 on days 3 and 6. No difference in tumor growth or survival was observed between SWHEL-positive and SWHEL-negative mice (Figure 8A and 8B). As a control for B cell independent effects of anti-CD40, a second group of SWHEL-negative Rag2-/- littermates was inoculated with tumor but did not receive anti-CD40. Median survival was reduced from 22 to 17 days, consistent with the previously reported effect of anti-CD40 on macrophages [31]. The antibody response of the anti-CD40-treated SWHEL-positive mice to s.c. tumor challenge (Figure 8C) was indistinguishable from that to i.v. challenge (Figure 7D).
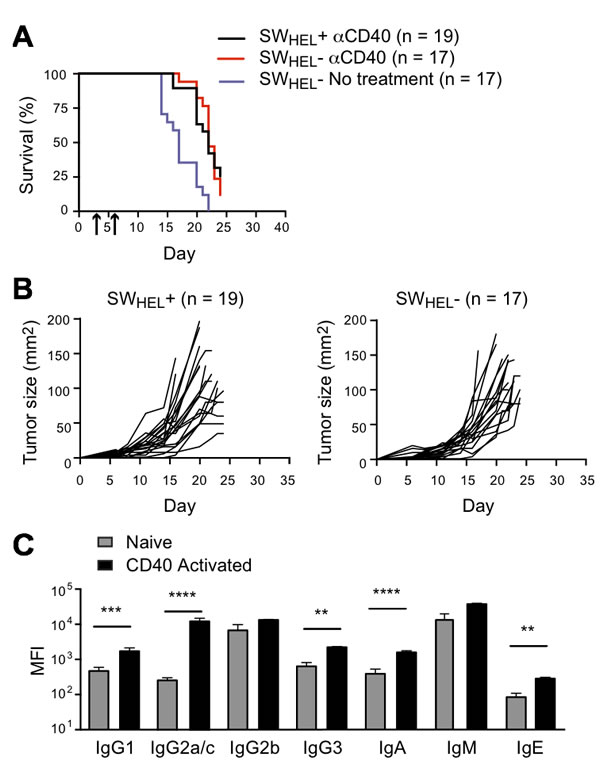
Figure 8: Effect of anti-CD40 activated B cells and isotype switched anti-tumor antibody on growth of subcutaneous tumor. Rag2-/- SWHEL+ and Rag2-/- SWHEL- mice were injected s.c. with 1x106 tumor cells followed by i.p. anti-CD40 on days 3 and 6. Tumor growth was measured, and serum was collected at the time of death to measure antibody isotypes. A. Kaplan-Meier survival analysis. Mice were euthanized when tumors reached 100mm2 or at the termination of the experiment on day 24 after tumor cell inoculation. Black line: anti-CD40 antibody treated Rag2-/- SWHEL+ mice (n = 19), red line: CD40 antibody treated Rag2-/- SWHEL- mice (n = 17), with arrows indicating days of anti-CD40 dosing. Purple line: Rag2-/- SWHEL- mice not treated with CD40 antibody. B. Individual tumor growth curves. Left: Rag2-/- SWHEL+ mice (n = 19). Right: Rag2-/- SWHEL- mice (n = 17). C. Serum antibody isotypes. MFI of CBA signals for each isotype before tumor challenge (naive) and on day of death (CD40 activated) (mean±SEM, n = 10/group). ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 by unpaired Student’s t test.
To rule out the possibility that the failure of antibody to protect against rapidly growing subcutaneous tumors was because the antibody response developed too slowly, we immunized prior to tumor challenge, to generate high levels of isotype switched antibodies. SWHEL-positive mice and SWHEL-negative Rag2-/- littermates were immunized s.c. with irradiated tumor cells emulsified in CFA, followed by anti-CD40 on days 3 and 6. Live tumor challenge was performed s.c. or i.v. on day 14. High levels of IgG2a/c were generated prior to challenge in the SWHEL mice, and these levels were maintained until the mice were sacrificed (Figure 9A). Once again, protection against i.v. but not s.c. challenge was seen (Figure 9B-9D). Taken together, these data suggest that isotype-switched antibodies produced by activated tumor-specific B cells can protect mice against B16 tumor metastases, but have no effect on s.c. tumor growth.
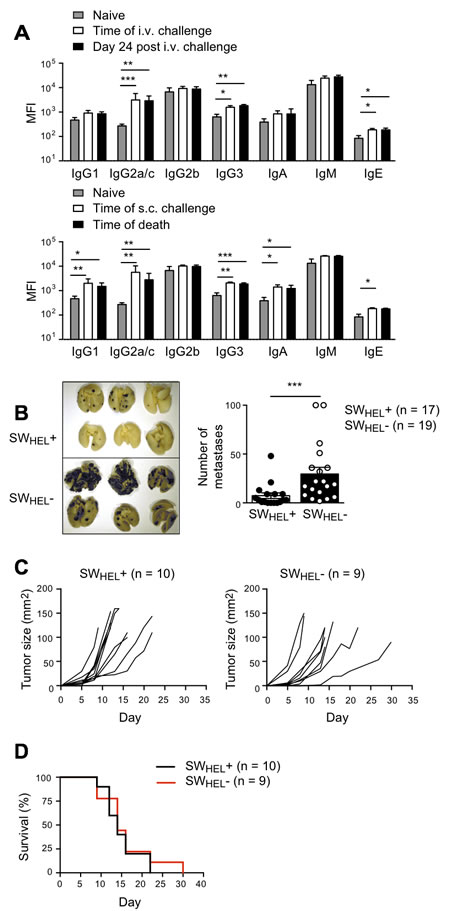
Figure 9: Effect of immunization on B cell control of tumor in subcutaneous and lung metastasis models. Rag2-/- SWHEL+ and Rag2-/- SWHEL- mice were immunized s.c. with 5x106 irradiated tumor cells emulsified in CFA, followed by i.p. anti-CD40 on days 3 and 6. Two weeks later mice were challenged with 1x106 live tumor cells i.v. or s.c. Mice were euthanized on day 24 after i.v. challenge, or when s.c. tumors reached 100mm2. A. Serum antibody isotypes. MFI of CBA signals for each isotype before and after immunization, and on day of death (mean±SEM, n≥7/group). Grey bar, naïve Rag2-/- SWHEL+ mice. White bar, Rag2-/- SWHEL+ mice at time of i.v. or s.c. challenge (day 14 after immunization). Black bar, Rag2-/- SWHEL+ mice on day of death). Top: i.v. challenge. Bottom: s.c. challenge. * = P < 0.05, ** = P < 0.01, *** = P < 0.001 by unpaired Student T test B. Left panel: Representative photos of lungs from Rag2-/- SWHEL+ and Rag2-/- SWHEL- mice after i.v. tumor challenge. Right panel: Number of metastases. Each dot represents an individual animal, with the bar representing mean±SEM (SWHEL+, n = 17, SWHEL-, n = 19). Data are pooled from 2 independent experiments. *** = P < 0.001 by nonparametric Mann-Whitney test. C. Individual tumor growth curves after s.c. tumor challenge. Left panel: Rag2-/- SWHEL+ mice. Right panel: Rag2-/- SWHEL- mice. D. Kaplan-Meier survival analysis after s.c. tumor challenge. Mice were euthanized when tumors reached 100mm2. Black line, Rag2-/- SWHEL+ mice (n = 10). Red line, Rag2-/- SWHEL- mice (n = 9).
Discussion
The role of T-B collaboration and production of tumor-specific antibodies in anti-tumor immunity remains controversial. Here we developed a novel pre-clinical model in which the effects of tumor-specific CD4+ T cells, B cells and antibodies could be studied under controlled conditions in a well-defined transgenic system. To the best of our knowledge, this is the first report to make use of tumor antigen-specific monoclonal B and T cells to define the effects of T-B collaboration on the anti-tumor response. Our study confirms that tumor-specific B cells can have both positive and negative effects on the growth of transplanted tumor cells in preclinical models, and further defines the immune mechanisms that drive those effects.
We have previously shown that CD4+ T cells are capable of eradicating established subcutaneous B16.mHELMCC tumors without any requirement for either CD8+ T cells or B cells [11]. In this model, the response of naive tumor-specific CD4+ T cells is initiated by tumor-derived antigen presented by host migratory dendritic cells in draining lymph nodes. The T cells then differentiate into Th1-type cells making high levels of IFNγ, which we identified as a major mediator of tumor control [11].
In the current study, the presence of B cells reduced the numbers of tumor-reactive CD4+ T cells, which in turn correlated with a reduction in the effectiveness of long-term tumor control, defined as > 120 days tumor-free survival. We found that CD4+ T cells in blood, lymph nodes, spleen and the tumors themselves were in equilibrium, and observed a highly significant correlation between the percentages of circulating tumor-specific CD4+ T cells at day 40 and at the time of euthanasia. A significant reduction in circulating tumor-specific CD4+ T cells was already apparent at day 40 in animals that failed to control tumor growth after an initial phase of stable disease between days 10 and 30. Thus our data indicate that the measurement of tumor-specific lymphocytes in blood can provide a highly relevant correlate of ongoing anti-tumor immunity.
A B cell-dependent reduction in the T cell response to tumor has previously been reported in B cell knockout models [19, 20], but the mechanism is not well understood. Our results indicate that tumor-specific B cells are capable of mediating this effect in the absence of non-specific B cells, suggesting involvement of B cell antigen presentation and/or B cell differentiation in response to specific antigen. Reduction in CD4+ T cell numbers and longevity in response to antigen presentation by a combination of B cells and dendritic cells, compared with dendritic cells alone, would be consistent with the findings of Candolfi et al., who used B cell-specific deletion of Prdm1 to show that B cell differentiation to antibody secretion was not required for their negative effect on T cell-dependent tumor control [32]. The ratio of circulating tumor-specific B to T cells at day 40 was highly predictive of survival in our model, with ratios greater than 0.5 associated with median survival of 53 versus > 120 days. Of 11 mice with a B:T ratio of < 0.5, 7 cleared their tumors and remained tumor-free until day 120, while none of those with a ratio > 0.5 survived more than 81 days. These findings raise the possibility that competition between antigen-primed T cells and B cells for access to survival factors within the lymphopenic environment may be involved in the negative effect of B cells on T cell numbers. The major factors required for CD4+ T cell proliferation and survival are IL-2 and IL-7. Exogenous IL-2 does not enhance proliferation of CD4+ T cells in immunodeficient mice, suggesting that IL-2 is unlikely to be limiting for CD4+ T cells under lymphopenic conditions [33]. While IL-7 is important for murine B cell development, very little IL-7 receptor is expressed by primed murine B cells. Thus the identity of factors that might underpin competition between CD4+ T and B cells remains unclear.
In addition to measuring the relative numbers of B and T cells, we analyzed cytokine production in tumor-bearing mice that received T and B cells versus T cells alone. CD4+ T cell priming in the presence of B cells has been reported to drive the response towards a Th2 phenotype [34], which may in turn reduce the effectiveness of T cell-dependent tumor control. However intracellular cytokine staining of splenic CD4+ T cells from mice in which tumor escape had occurred indicated an increased rather than decreased proportion of IL-2+ T cells in the presence of tumor-specific B cells, with the majority of T cells co-expressing TNF. Production of IFNγ was comparable in the two groups. This bias towards Th1 differentiation is likely due to the high affinity of the 5C.C7 T cells [35], which make IFNγ even under Th2 priming conditions such as use of alum as adjuvant (unpublished observations).
Measurement of cytokine levels in supernatants from overnight culture showed no significant differences in either Th1 or Th2 cytokines between the T plus B versus T cell groups, and confirmed the strong Th1 bias. Thus deviation of CD4+ T cells away from a type 1 response could not account for tumor escape in our model. We found no evidence of IL-10 production by HEL-specific B cells themselves, and could not identify B cells expressing typical Breg phenotypic markers such as CD5, CD1d and Granzyme B. This is consistent with published results indicating that monoclonal B cells of pre-defined specificity are not recruited into the Breg compartment in vivo [36].
Since B cells can induce conversion of CD4+ T cells to Foxp3+ iTregs [37, 38], we analyzed CD4+ T cells for Foxp3 expression at day 40 and the time of euthanasia. We detected iTregs in all recipients of naive FoxP3- 5C.C7 T cells, and their numbers were unaffected by the presence of B cells. In addition, iTregs did not appear to control the effectiveness of CD4+ T cell tumor clearance, which was independent of iTreg number. This conclusion is consistent with recent studies reporting that interfering with regulatory T cell function by means of anti-CTLA-4 antibody therapy recruits new T cells into the anti-tumor response, rather than “releasing” already primed cells from Treg-dependent control [39, 40].
We detected very few B cells in the subcutaneous tumors in this model. The percentage of B cells in tumors was approximately 10-fold lower than in blood, while that of CD4+ T cells was more than 10-fold higher. Taken together with the linear correlation between B cell numbers in blood and tumor, this suggests that those within the tumor sample likely represent blood contamination. A recent report indicated that B cell infiltration into non-lymphoid tissues may be CD8+ T cell dependent, explaining the lack of infiltration in our model [41]. Supporting this interpretation, infiltrating B cells in human tumors are generally associated with a prominent CD8+ T cell infiltrate, and the co-location of the 2 cell types correlates with an improved prognosis in many, but not all, tumor types. In contrast, prominent B cell infiltration in the pre-neoplastic phase is highly correlated with progression to frank malignancy in mouse models [23].
In addition to their effects on CD4+ T cells, B cells responding to tumor in the presence of T cell help secreted tumor-specific anti-HEL antibody, which we detected in the serum. In tumor-bearing hosts adoptively transferred with both T and B cells, T cell production of IFNγ supported switching to the IFNγ-dependent IgG2a/c isotype. However we also saw large increases in IgG1 and IgG2b, plus lower amounts of IgG3, IgA and IgE. In the absence of T cells, SWHEL B cells could also be activated to secreted isotype switched antibody by administration of anti-CD40 antibody. Compared with CD4+ T cell help, anti-CD40 induced an even stronger bias towards IgG2a/c, and protected against lung metastases after i.v. administration of B16.mHELMCC tumor cells. We showed that isotype-switched antibody generated either before or after i.v. B16 tumor administration was protective, indicating that antibody could reduce the growth of already seeded lung metastases. In contrast, we detected no effect of antibody or B cells against subcutaneous tumors in the absence of CD4+ T cells, even when the mice had generated optimal isotype-switched antibody responses prior to live tumor cell inoculation. Multiple factors could account for this difference, including enhanced penetration of antibodies into lung metastases compared with skin tumors, and differential presence of FcγR-expressing innate cells at the different sites. In a previous study in which administration of purified anti-tumor antibody synergized with peptide vaccination in a subcutaneous B16 model, FcγR-expressing innate cells in the tumor microenvironment were implicated in early tumor control [42]. Administration of anti-CD40 antibody reduced subcutaneous tumor growth rates in a B cell-independent manner, consistent with the effect on macrophages reported by Beatty et al. [31]. It remains to be determined whether the positive effects of anti-CD40 administration on tumor control in B cell sufficient animals [43] are dependent on macrophages, B cells or both, and whether anti-CD40-mediated B cell activation would alter the outcome of their cognate interactions with T cells in response to specific tumor antigen.
Clinical studies indicate that the presence of spontaneous anti-tumor antibodies is associated with a worse prognosis in some settings [17, 18]. In contrast, de novo antibody responses to immunological interventions such as vaccination with tumor antigen may correlate with an improved prognosis [1]. It is likely that the generation of a potent CD4+ T cell-dependent anti-tumor response is the immunological driver behind beneficial clinical responses to cancer, and that the provision of CD4+ T cell help to CD8+ T cells and B cells generates further positive and negative amplification of anti-tumor immunity, respectively, resulting in eventual control of tumor growth by a combination of CD4+ and CD8+ T cells, despite the negative effects of B cells.
Our studies suggest that B cell depletion may have beneficial effects when incorporated into a cancer immunotherapy regimen that generates de novo CD4+ T cell responses. In the limited number of reports of B cell depletion in patients with non-B cell tumors, it was not administered in conjunction with an immune checkpoint inhibitor, and no major clinical benefit was seen [44, 45]. However an increase in secondary solid tumors was reported after effective anti-CD20 mAb therapy of B cell malignancies [46]. Thus further studies will be needed to test whether B cell depletion has any future role in clinical tumor immunotherapy. In B cell replete settings, de novo appearance of anti-tumor antibodies could usefully serve as a surrogate marker for the generation of a potentially beneficial tumor-specific response to immunotherapy. In addition, it is possible that protective antibody responses may provide a means of decreasing the long-term burden of metastatic tumors, although there is as yet little clinical evidence to support this claim [47]. To date, no clinical protocols have been shown to enhance protective antibody responses while inhibiting B cell-dependent effects on CD4+ T cells. Our model provides a basis for the development of such protocols in a controlled setting that yields mechanistic insights.
Materials and Methods
Mice
All mice were bred and housed under SPF conditions in the Centenary Institute Animal Facility. SWHEL mice expressing the HyHEL10 B cell receptor (BCR) specific for Hen Egg Lysozyme (HEL) were a gift from Robert Brink [24] and were maintained on a C57BL/6 Rag2-/- background [48]. 5C.C7 T cell receptor (TCR) transgenic mice specific for Moth Cytochrome C (MCC) [25] were maintained on a B10.BR Rag2-/- background (backcrossed to B10.BR for > 10 generations). To ensure histocompatibility between B cells (maintained on C57BL/6) and T cells (maintained on B10.BR), cell donor mice were bred on a [C57BL/6 x B10.BR]F1 background. Intercrosses expressing CD45.2 or CD45.1/CD45.2 were bred from four colonies of Rag2-/- mice on C57BL/6 CD45.1, C57BL/6 CD45.2, B10.BR CD45.1 and B10.BR CD45.2 backgrounds (the latter 2 backcrossed onto B10.BR for > 10 generations), allowing us to unequivocally identify each population of adoptively transferred cells. In some experiments, Rag1-/- [49] mice on a [C57BL/6 x B10.BR]F1 background (bred in house from C57BL/6 Rag1-/- and B10.BR Rag1-/- mice (backcrossed for > 10 generations)) were used as tumor hosts instead of [C57BL/6 x B10.BR]F1 Rag2-/- mice. We have shown that Rag1-/- and Rag2-/- F1 immunodeficient host mice behave in an indistinguishable manner in our experimental model. All experiments were performed with the consent of the University of Sydney Animal Ethics Committee, or Sydney Local Health District Animal Welfare Committee.
Tumor models
The B16.F10 melanoma cell line originally obtained from ATCC was kindly provided by Nikolas Haass. B16.F10 cells were retrovirally transduced to express membrane HELMCC, which consists of HEL protein with residues 64-76 replaced with residues 87-103 of MCC, fused at the C-terminus to the transmembrane and cytoplasmic domains of H-2Kb [50]. Transduced tumor cells were screened by flow cytometry and cloned to select a stable line designated B16.mHELMCC, which was used for all experiments [11].
Viable B16.mHELMCC tumor cells were injected either intravenously (i.v.) via the tail vein or subcutaneously (s.c.) in the flank. For i.v. tumor cell inoculation, additional test mice were inoculated and euthanized between days 20 and 24 to assess the size of lung metastases and determine the optimal timepoint to euthanize the experimental groups. After euthanasia, mice were perfused with saline solution, and lungs collected in Fekete’s solution. The number of metastases was counted visually in a blinded fashion [51]. When the number of metastases was > 100, a score of 100 was given. Subcutaneous tumors were measured in a blinded fashion 2-3 times per week with digital calipers. Mice were euthanized when the tumor area reached 100mm2.
For immunization of SWHEL mice, 5x106 irradiated B16.mHELMCC cells (3500cGy) emulsified in complete Freund’s adjuvant (CFA) were injected s.c. in one flank. Two intraperitoneal (i.p.) injections of anti-CD40 (FGK45, 25µg/injection) were given on days 3 and 6, and mice were challenged i.v. or s.c. with live tumor cells on day 14. The site of s.c. challenge was the flank opposite to the immunization site.
Adoptive cell transfer and flow cytometry analysis
For adoptive transfer, splenic SWHEL B cells and TCR transgenic T cells from pooled lymph nodes were co-transferred at a 5:1 ratio (1x106 B cells, 0.2x106 T cells). For flow cytometric analysis of blood, lymph node, spleen and tumor, samples were prepared as described previously [11]. The following monoclonal Abs were used to stain cells: anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-CD11b (M1/70), anti-NK1.1 (PK136), anti-CD45 (30-F11), anti-CD95 (Jo2), anti-Ly-77 (GL7), anti-MHCII (M5/114.15.2), anti-Ter119 (TER 119) and anti-B220 (RA3-6B2) obtained from BD Biosciences (Franklin Lakes, NJ, USA); anti-CD1d (1B1), anti-CD5 (53-7.3), anti-CD19 (6D5), anti-CD45.2 (104), anti-CD45.1 (A20), anti-Gr1 (RB6-8C5) and anti-Granzyme B (GB11) obtained from BioLegend (San Diego, CA, USA). All antibodies were directly conjugated with FITC, PE, allophycocyanin, Pacific Blue or cyanin conjugates PE-Cy7, PerCP-Cy5.5 or allophycocyanin-Cy7. Non-specific binding to Fc receptors blocked using anti-CD16/32 purified in house from the 2.4G2-hybdridoma. Intracellular staining of FoxP3 was performed using a murine FoxP3 staining kit and anti-Foxp3 mAb (FJK-16s) from eBioscience. SWHEL B cells were detected with HEL protein conjugated to Alexa647, kindly provided by Chris Jolly. Samples were analyzed on LSR-II, Fortessa and FACSCanto BD flow cytometers.
Cytokine measurements
For intracellular cytokine detection, 2 x107 splenocytes were plated at 1 x107 cells/mL in 12 well plates in tissue culture medium containing 10µM MCC peptide and 500ng/mL HEL protein. After 2 hours incubation at 37°C, Brefeldin A (Sigma) was added to a final concentration of 5ng/mL and cells were further cultured for 16 hours. Cells were then washed and stained for cell surface CD45.1, CD45.2 and CD4, followed by intracellular staining with mAbs against IL-2 (JES6-5H4) and IFNγ (XMG1.2) from BD Biosciences, and anti-TNFα (MP6-XT33) from BioLegend. For measurement of cytokines in culture supernatant, splenocytes were plated at 1 x106 cells/well in 96 well plates in tissue culture medium containing 10µM MCC peptide and 500ng/mL HEL protein. Supernatants were collected after 24 hrs incubation at 37°C and cytokines measured using a CBA cytokine kit (BD, 562246)
Mouse immunoglobulin isotyping kit
For relative measurement of serum antibody isotypes, a CBA Mouse Immunoglobulin Isotyping Kit (BD, 550026) was used according to manufacturer’s instructions. 1µL of serum was used in most assays except when samples were titered to generate dose-response curves for each isotype, allowing estimation of fold concentration changes based on the mean fluorescence intensity (MFI) values. The isotyping kit included monoclonal antibody R19-15, which recognizes both the IgG2a isotype present in most mouse strains and the functionally equivalent IgG2c isotype present in C57BL/6 mice [30], and therefore expressed by SWHEL positive mice. Since most publications still refer to the C57BL/6 isotype as IgG2a rather than IgG2c, we have termed the isotype we detected as IgG2a/c throughout.
Statistics
All analyses were performed using GraphPad Prism Software. For experiments with 2 groups, Mann-Whitney (nonparametric) or unpaired Student’s t (parametric) tests were used. Data are shown as mean +/- SEM. Kaplan-Meier survival curves were analyzed using Log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests. Pearson correlation coefficients were used for parametric data and Spearman’s rank correlation coefficient was used for non-parametric data. A D’Agostino-Pearson omnibus normality test was performed prior to correlation analysis to determine the appropriate parametric or non-parametric test.
Acknowledgments
We thank C. Jolly for providing HEL-Alexa647 and scientific discussion; B. Roediger for scientific discussion; A. Buckley and J. Holst for generating B16.mHELMCC; L. Beebe for technical assistance; the staff of the Centenary Institute Flow Cytometry and Animal Facilities for technical support, and members of the T cell biology and Immune Imaging Labs for scientific discussion.
Conflicts of interest
The authors have no conflicts of interest.
Funding
This work was supported by Australian National Health and Medical Research Council grants 1001020, 1012930 and 1051843 and a New South Wales Cancer Council grant RG 13-13. TVG and DGH were supported by scholarships from the Cancer Institute New South Wales, and HMM and BFdeStG by fellowships from the National Health and Medical Research Council of Australia.
REFERENCES
1. Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL, Kuk D, Panageas KS, Ritter G, Sznol M, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A. 2011; 108:16723-16728. doi: 10.1073/pnas.1110814108.
2. Sasada T, Suekane S. Variation of tumor-infiltrating lymphocytes in human cancers: controversy on clinical significance. Immunotherapy. 2011; 3:1235-1251. doi: 10.2217/imt.11.106
3. Reuschenbach M, von Knebel Doeberitz M, Wentzensen N. A systematic review of humoral immune responses against tumor antigens. Cancer Immunol Immunother. 2009; 58:1535-1544. doi: 10.1007/s00262-009-0733-4.
4. Hermans IF, Daish A, Moroni-Rawson P, Ronchese F. Tumor-peptide-pulsed dendritic cells isolated from spleen or cultured in vitro from bone marrow precursors can provide protection against tumor challenge. Cancer Immunol Immunother. 1997; 44:341-347. doi:
5. Dobrzanski MJ, Reome JB, Dutton RW. Therapeutic effects of tumor-reactive type 1 and type 2 CD8+ T cell subpopulations in established pulmonary metastases. J Immunol. 1999; 162:6671-6680.
6. Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, Shiku H, Schreiber RD, Allen PM. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000; 13:265-276.
7. Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003; 198:569-580. doi: 10.1084/jem.20030590.
8. Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007; 109:5346-5354. doi: 10.1182/blood-2006-10-051318.
9. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008; 112:362-373. doi: blood-2007-11-120998 [pii]10.1182/blood-2007-11-120998.
10. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010; 207:637-650. doi: jem.20091918 [pii]10.1084/jem.20091918.
11. Shklovskaya E, Terry AM, Guy TV, Buckley A, Bolton HA, Zhu E, Holst J, Fazekas de St Groth B. Tumour-specific CD4 T cells eradicate melanoma via indirect recognition of tumour-derived antigen. Immunol Cell Biol. 2016. doi: 10.1038/icb.2016.14.
12. Fremd C, Schuetz F, Sohn C, Beckhove P, Domschke C. B cell-regulated immune responses in tumor models and cancer patients. Oncoimmunology. 2013; 2:e25443. doi: 10.4161/onci.25443.
13. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, Visser M, Stratton MR, Haanen JB, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015; 21:81-85. doi: 10.1038/nm.3773.
14. Ladanyi A, Kiss J, Mohos A, Somlai B, Liszkay G, Gilde K, Fejos Z, Gaudi I, Dobos J, Timar J. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol Immunother. 2011; 60:1729-1738. doi: 10.1007/s00262-011-1071-x.
15. Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, Nelson BH. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012; 18:3281-3292. doi: 10.1158/1078-0432.CCR-12-0234.
16. Schmidt M, Hellwig B, Hammad S, Othman A, Lohr M, Chen Z, Boehm D, Gebhard S, Petry I, Lebrecht A, Cadenas C, Marchan R, Stewart JD, et al. A comprehensive analysis of human gene expression profiles identifies stromal immunoglobulin kappa C as a compatible prognostic marker in human solid tumors. Clin Cancer Res. 2012; 18:2695-2703. doi: 10.1158/1078-0432.CCR-11-2210.
17. Zornig I, Halama N, Lorenzo Bermejo J, Ziegelmeier C, Dickes E, Migdoll A, Kaiser I, Waterboer T, Pawlita M, Grabe N, Ugurel S, Schadendorf D, Falk C, et al. Prognostic significance of spontaneous antibody responses against tumor-associated antigens in malignant melanoma patients. Int J Cancer. 2015; 136:138-151. doi: 10.1002/ijc.28980.
18. Yamaguchi T, Takii Y, Maruyama S. Usefulness of serum p53 antibody measurement in colorectal cancer: an examination of 1384 primary colorectal cancer patients. Surg Today. 2014; 44:1529-1535. doi: 10.1007/s00595-013-0703-5.
19. Qin Z, Richter G, Schuler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat Med. 1998; 4:627-630.
20. Shah S, Divekar AA, Hilchey SP, Cho HM, Newman CL, Shin SU, Nechustan H, Challita-Eid PM, Segal BM, Yi KH, Rosenblatt JD. Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int J Cancer. 2005; 117:574-586. doi: 10.1002/ijc.21177.
21. Horikawa M, Minard-Colin V, Matsushita T, Tedder TF. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J Clin Invest. 2011; 121:4268-4280. doi: 10.1172/JCI59266.
22. Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005; 310:1510-1512. doi: 10.1126/science.1118948.
23. de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005; 7:411-423. doi: 10.1016/j.ccr.2005.04.014.
24. Phan TG, Amesbury M, Gardam S, Crosbie J, Hasbold J, Hodgkin PD, Basten A, Brink R. B cell receptor-independent stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. J Exp Med. 2003; 197(7):845-860.
25. Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med. 1992; 176:1091-1098.
26. Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010; 207:651-667. doi: jem.20091921 [pii]10.1084/jem.20091921.
27. Macaulay AE, DeKruyff RH, Goodnow CC, Umetsu DT. Antigen-specific B cells preferentially induce CD4+ T cells to produce IL-4. J Immunol. 1997; 158:4171-4179.
28. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, Beyer T, Reister F, Fabricius D, Lotfi R, Lunov O, Nienhaus GU, Simmet T, et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013; 73:2468-2479. doi: 10.1158/0008-5472.CAN-12-3450.
29. Balkwill F, Montfort A, Capasso M. B regulatory cells in cancer. Trends in immunology. 2013; 34:169-173. doi: 10.1016/j.it.2012.10.007.
30. Martin RM, Brady JL, Lew AM. The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods. 1998; 212:187-192.
31. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011; 331:1612-1616. doi: 10.1126/science.1198443.
32. Candolfi M, Curtin JF, Yagiz K, Assi H, Wibowo MK, Alzadeh GE, Foulad D, Muhammad AK, Salehi S, Keech N, Puntel M, Liu C, Sanderson NR, et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia. 2011; 13:947-960.
33. Bolton HA, Zhu E, Terry AM, Guy TV, Koh W-P, Tan S-Y, Power CA, Bertolino P, Lahl K, Sparwasser T, Shklovskaya E, Fazekas de St Groth B. Selective Treg reconstitution during lymphopenia normalizes DC costimulation and prevents graft-versus-host disease. J Clin Invest. 2015; 125:3627-3641. doi: 10.1172/JCI76031.
34. Linton PJ, Bautista B, Biederman E, Bradley ES, Harbertson J, Kondrack RM, Padrick RC, Bradley LM. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J Exp Med. 2003; 197:875-883. doi: 10.1084/jem.20021290.
35. Berg LJ, Pullen AM, Fazekas de St Groth B, Mathis D, Benoist C, Davis MM. Antigen/MHC-specific T cells are preferentially exported from the thymus in the presence of their MHC ligand. Cell. 1989; 58:1035-1046.
36. Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol. 2009; 182:7459-7472. doi: 10.4049/jimmunol.0900270.
37. Han Y, Wu J, Bi L, Xiong S, Gao S, Yin L, Jiang L, Chen C, Yu K, Zhang S. Malignant B cells induce the conversion of CD4+CD25- T cells to regulatory T cells in B-cell non-Hodgkin lymphoma. PloS one. 2011; 6:e28649. doi: 10.1371/journal.pone.0028649.
38. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res. 2011; 71:3505-3515. doi: 10.1158/0008-5472.CAN-10-4316.
39. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189-2199. doi: 10.1056/NEJMoa1406498.
40. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJ, van der Burg S, Kapiteijn E, Michielin O, Romano E, Linnemann C, Speiser D, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014; 6:254ra128. doi: 10.1126/scitranslmed.3008918.
41. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014; 346:98-101. doi: 10.1126/science.1254536.
42. Ly LV, Sluijter M, van der Burg SH, Jager MJ, van Hall T. Effective cooperation of monoclonal antibody and peptide vaccine for the treatment of mouse melanoma. J Immunol. 2013; 190:489-496. doi: 10.4049/jimmunol.1200135.
43. Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, Lesokhin AM, Avogadri-Connors F, Yuan J, Li Y, Houghton AN, Merghoub T, Wolchok JD. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med. 2012; 209:2113-2126. doi: 10.1084/jem.20120532.
44. Aklilu M, Stadler WM, Markiewicz M, Vogelzang NJ, Mahowald M, Johnson M, Gajewski TF. Depletion of normal B cells with rituximab as an adjunct to IL-2 therapy for renal cell carcinoma and melanoma. Ann Oncol. 2004; 15:1109-1114. doi: 10.1093/annonc/mdh280.
45. Barbera-Guillem E, Nelson MB, Barr B, Nyhus JK, May KF, Jr., Feng L, Sampsel JW. B lymphocyte pathology in human colorectal cancer. Experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunol Immunother. 2000; 48:541-549.
46. Tarella C, Passera R, Magni M, Benedetti F, Rossi A, Gueli A, Patti C, Parvis G, Ciceri F, Gallamini A, Cortelazzo S, Zoli V, Corradini P, et al. Risk factors for the development of secondary malignancy after high-dose chemotherapy and autograft, with or without rituximab: a 20-year retrospective follow-up study in patients with lymphoma. J Clin Oncol. 2011; 29:814-824. doi: 10.1200/JCO.2010.28.9777.
47. Nelson BH. CD20+ B cells: the other tumor-infiltrating lymphocytes. J Immunol. 2010; 185(9):4977-4982. doi: 10.4049/jimmunol.1001323.
48. Shinkai Y, Rathbun G, Lam K-P, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992; 68:855-867.
49. Spanopoulou E, Roman CAJ, Corcoran LM, Schlissel MS, Silver DP, Nemazee D, Nussenzweig MC, Shinton SA, Hardy RR, Baltimore D. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. Genes Dev. 1994; 8:1030-1042.
50. Shklovskaya E, Roediger B, Fazekas de St Groth B. Epidermal and dermal dendritic cells display differential activation and migratory behavior while sharing the ability to stimulate CD4+ T cell proliferation in vivo. J Immunol. 2008; 181:418-430.
51. Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol. 2001; Chapter 20:Unit 20 21. doi: 10.1002/0471142735.im2001s39.