
Introduction
Cancer formation and progression pass through a multistep process in which cancer cells acquire new properties and deregulate many homeostatic pathways. This, to enable them to sustain cell proliferation, escape cell death, disrupt the original niche of development by inducing neovascularization, increase the dimension of the tumoral mass and eventually metastasize. All these properties have been considered as hallmarks of cancer, as described by Hanahan and Weinberg in their seminal review in 2000 [1]. Nonetheless, years of advances in oncology have rendered that picture rather incomplete. As a result, researchers and oncologists have now turned their attention to characteristics which were previously under-valued, including the metabolic reprogramming of cancer cells and the role of tumour-invading inflammatory cells and inflammation [2].
In cancer cells, the tight balance between cell proliferation and cell dismissal is altered, the balancing point being tipped toward the former [3]. This uncontrolled cell proliferation needs energetic support, and is accompanied by changes in the metabolism, shifting from oxidative phosphorylation to glycolysis [4]. When cells are in a non-proliferative state and mitochondria are functional, pyruvate - the final product of glycolysis - is imported into the organelles and is completely oxidized in the TCA cycle to produce reducing equivalents (NADH); these molecules then fuel the mitochondria respiratory chain to produce ATP through oxidative phosphorylation [5]. Conversely, Otto Warburg observed that, even in the presence of oxygen, cancer cells rely instead on glycolysis for their metabolic needs, hence his coining the term “aerobic glycolysis” [6],[7]. However, for decades the “Warburg effect” was considered only as a metabolic signature of cancer, or an adaptation to an environment with low oxygen concentration within the tumoral mass. Moreover, the discovery of oncogenes and onco-suppressors shed light on the genetic basis of cancer, diverting attention away from cancer metabolism [8],[9],[10]. The discovery of the mitochondrial localization of the onco-protein Bcl-2 extended the role of these organelles from metabolism to the control of the apoptotic cascade, therefore presenting a novel scenario for mitochondria and metabolism in cancer development [11],[12],[13],[14]. Interestingly, the metabolic switch towards glycolysis was shown to be not limited to cancer cells, but shared with many other highly proliferative cell types: cells during embryogenesis, stem cells upon growth factors stimulation, T cells after antigen activation are a few examples [15],[16],[17]. Moreover, a connection between cancer and altered metabolism was clearly established when mutations in genes encoding for the two TCA cycle enzymes succinate dehydrogenase (SDH) [18] and fumarate hydratase (FH) [19] were identified in human tumours.
Despite these observations, a paradox emerges: why would active proliferating cells use a less efficient method to produce ATP, compared to oxidative phosphorylation? One explanation could be that, when resources are scarce, cells turn to an inefficient method to produce at least a minimal amount of ATP; however, this is not the case for cancer cells that are supplied with glucose and nutrients by the neo-angiogenic blood vessels. A more convincing explanation suggests that cancer cells have metabolic requirements that go beyond ATP production: the intermediates derived from glycolysis and TCA cycle could be diverted towards biosynthetic pathways or could activate signalling cascades, thus enabling the characteristic hallmarks of cancer to occur. As a consequence, changes in metabolism can sustain cell proliferation by feeding anabolic pathways; moreover, they can also modify the tumour microenvironment by altering the complex interaction and crosstalk between cancer cells and normal cells surrounding, or infiltrating, the tumoral mass [4],[20]. Indeed, we can speculate that the newly depicted hallmarks of cancer, glycolytic metabolism and pro-inflammatory microenvironment, are mutually regulated and influence each other so as to determine a tumour’s overall capacity for development.
Aerobic glycolysis can be triggered by mutations in oncogenes, whose broad effect on cellular reprogramming involves changes in the regulation of metabolism. Alternatively, and even more interestingly, it can arise directly from mutations in genes involved in glycolysis or TCA cycle [21]. In the latter case, a direct link between altered metabolism and cancer is provided (Figure 1). In a second step, the oncogenic properties acquired by metabolic intermediates, accumulating as a consequence of the Warburg effect, will influence the tumour growth. Consequently, by this mean they will amplify pro-oncogenic signals, while -in some cases, they will exert a paracrine effect on cancer-infiltrating inflammatory cells.
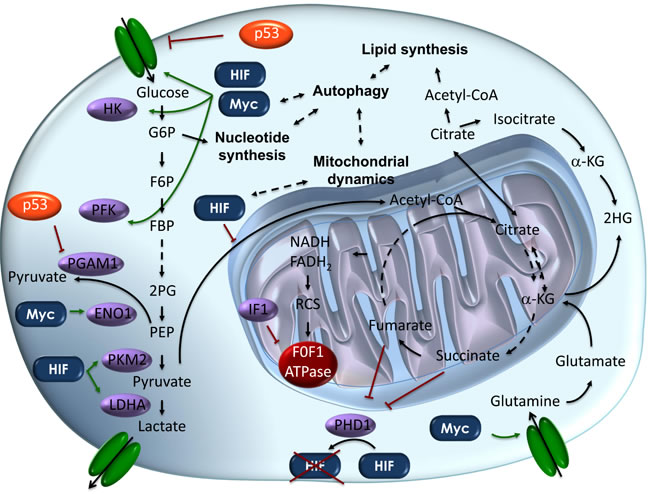
Figure 1: Schematic representation of the metabolic pathways altered in cancer cells. Metabolic pathways in cancer cells are directly controlled by the main oncogenes and oncosuppressors. This schematic picture depicts our current knowledge about how glycolysis and oxidative phosphorylation are inter-regulated through the synthesis of nucleotides and lipids. Key steps promoting the “Warburg effect” in cancer cells are shown. In orange onco-suppressor proteins, while in blue onco-proteins are shown. In violet, onco-proteins’ direct targets in the metabolic cascade are depicted. Dashed lines represent biunique interactions. Abbreviations: α-KG, α-Ketoglutarate; ENO1, Enolase1; FBP, Fructose-1,6-biphosphate; F6P, Fructose-6-phosphate; G6P, Glucose-6-phosphate, HK, hexokinase; HIF, hypoxia inducible factor; IF1, ATPase inhibitory factor 1; LDHA, Lactate dehydrogenase A; PGAM1, phosphoglycerate mutase 1; PFK, phosphofructokinase; PEP, Phosphoenolpyruvate; PHD1, prolyl hydroxylase domain 1; PKM2, pyruvate kinase isoform 2; 2HG, 2-hydroxyglutarate; 2PG, 2-phosphoglycerate; RCS, Respiratory Chain Supercomplexes.
Classic oncogenic signals divert metabolism from oxidative phosphorylation to glycolysis
Many tumours are driven by mutations in oncogenes or oncosuppressor genes such as c-Myc, RAS and p53. Interestingly, it has been shown that alterations in their function are responsible for the metabolic reprogramming observed in many cancer cells (Figure 1).
The multifaceted oncogene c-Myc is a master regulator of cellular growth and metabolism in cancer cells [22]. Although in some cancer types c-Myc has a primary oncogenic role, its DNA sequence being translocated downstream of promoters of either the light or the heavy immunoglobulin chain [23], its activity is usually up-regulated post-transcriptionally by other oncogenic signals [24]. The first hint that c-Myc has a direct role in up-regulating glycolysis in cancer came from the observation that LDH-A, the enzyme converting pyruvate to lactate, is a putative target of c-Myc [25]. Since then, different enzymes involved in glycolysis have been found over-expressed in a c-Myc-dependent manner (glucose transporter - GLUT1, hexokinase 2 - HK2, phosphofructokinase - PFKM and Enolase 1 - ENO1) [26],[27],[28]. Interestingly, not only glucose metabolism, but also glutaminolysis is increased in a c-Myc dependent manner, thus causing cancer cells to become addicted to glutamine and sensitive to its withdrawal [29].
Other oncogenes also play a role in regulating metabolism. The MAP kinase pathway (RAS-RAF-MEK-ERK pathway) is often altered in cancer. In particular, single-point mutations in RAS lead to its constitutively active signalling in many solid cancers such as pancreas, lung and colorectal cancer [30]. Recently, an unexpected link between the RAS pathway and mitochondria has been described. Indeed, activated RAS (H-RAS) mediates the translocation of the signal transducer and activator of transcription 3 (STAT3) to mitochondria, where it regulates mtDNA transcription, so altering electron transport and increasing lactate production [31],[32]. Also RAF, which acts downstream RAS, is able to inhibit oxidative phosphorylation and down-regulate the master regulator of mitochondrial biogenesis PGC1α [33]. These observations show that, despite its complex role during cancer formation and progression, the MAP kinase pathway also impinges on cancer metabolism.
To note, c-Myc and RAS have also a prominent role in regulating the catabolic process of autophagy [34],[35], of which the metabolic regulator mTOR is the major inhibitor, most likely responding to the new metabolic demands during cancer progression [36]. To give some insights, c-Myc shows an intricate relationship with the autophagic signalling machinery component AMBRA1. This factor, indeed, is able to facilitate PP2A-dependent dephosphorylation and degradation of c-Myc, in conditions in which autophagy is active whilst mTOR and cell proliferation, hence cancer development, are inhibited [37].
More in general, the role of autophagy and organelle quality control (especially of mitochondria) during cancer development has long been debated. Briefly, the common believe today is that autophagy plays different roles in different stages of cancer development. In healthy tissues, or in early stages of cancer, autophagy represents a pivotal anti-tumoral defence. Instead, when cancer is established by autophagy-unrelated mutations, up-regulation of autophagy facilitates cell survival and metabolic adaptation of cancer cells. Indeed, the intriguing crosstalk between autophagy and cancer goes beyond the goal of this review and for this reason we direct the interested reader to other outstanding reviews [36],[38],[39].
Regarding onco-suppressors, the observations that the mutated forms of p53 have oncogenic properties led to the initial misclassification of p53 as an oncogene. Further studies showed that p53 mutations are gain-of-function mutations and that the wild-type protein is instead an onco-suppressor [40]. The p53 onco-suppressor activity is manifested when it is stabilized and its transcriptional activity is up-regulated in response to a variety of stress stimuli. This is in order to mediate apoptosis, DNA repair, cell cycle arrest or senescence, to maintain genome integrity and finally to limit cancer development [41]. Interestingly, recent studies also revealed a role for p53 in the regulation of metabolism [42]. In particular, wild-type p53 negatively regulates glycolysis, through negative regulation or mis-localization of glucose transporters (GLUT1 and GLUT4) [43] and other glycolytic enzymes (phosphoglycerate mutase - PGM; pyruvate dehydrogenase kinase 2 - PDK2) [44],[45]. Moreover, p53 also shunts glucose to the pentose phosphate pathway and to NADPH production [46]. NADPH production has a double role both in anabolic pathways and in restoring the reduced form of the anti-oxidant glutathione (GSH) [47], a molecule in the first line of defence versus the oxidative stress responsible for increased mutational rate. Hence, in the case of mutated p53, the electron transport chain (ETC) is compromised and cells switch to glycolysis to overcome the block in ATP production. In addition to directly regulating the expression of glycolytic enzymes and components of the ETC, p53 can repress glycolysis by inhibiting the AKT/mTOR and NF-kB signalling pathways whose activity is strongly up-regulated in cancer cells [48],[49]. Thus, the different “negative” gain-of-function properties of p53 include promotion of cell proliferation, angiogenesis, migration, invasion, metastatization, chemoresistence [40] and, importantly for the purposes of this review, the capacity to switch metabolism towards glycolysis [50],[51] (Figure 1).
When mitochondria are upstream of cancer development
Although of great interest, only recently the metabolic effects of tumour-associated mutations have been investigated; as such, a complete picture of the role of mutations in oncogenes and oncosuppressors in cancer metabolism is still lacking. Besides the mutations so far described, alterations in mitochondrial metabolism have also been directly linked to cancer development. Interestingly, inhibition of oxidative phosphorylation leads to a loss or inactivation of p53 due to the generation of reactive oxygen species [52]. Moreover, a number of mutations in genes directly implicated in glycolysis or TCA cycle have been shown to promote tumour formation [53].
Indeed, mutations in either fumarate hydratase (FH) or succinate dehydrogenase (SDH B, C and D), both enzymes of the TCA cycle, are known to promote different cancer types, from leiomyoma, leiomyosarcoma and renal cell carcinoma (FH mutations) to paraganglioma and pheochromocytoma (SDH mutations) [19],[18]. Interestingly, these mutations give rise to pseudo-hypoxia, a condition in which the complex metabolic and transcriptomic changes, usually taking place under low oxygen concentration, occur under normoxic conditions. Pseudo-hypoxia enhances cancer formation and aggressiveness [54],[55]. Indeed, all solid cancers experience cycling changes in oxygen pressure, varying from normal oxygen concentration to hypoxia or even anoxia in the innermost part of the tumoral mass [56]. During acute hypoxia, hypoxia inducible factor (HIF)1α - otherwise degraded - is stabilized and induces the transcription of its target genes including specific angiogenic and pro-metastatic factors [57]. The oncometabolites succinate and fumarate, which both accumulate due to mutations inactivating SDH or FH, inhibit HIF1α degradation, so causing pseudo-hypoxia and favouring tumour progression [58],[59],[60],[61]. Moreover, fumarate can affect and alter many different pathways, thus contributing to complex metabolic dysfunctions involved in tumour formation. For example, it can alter metabolism of urea [62] or the anti-oxidant response [63],[64],[65],[66].
Of note, SDH is the Complex II of the mitochondrial respiratory chain, feeding the OXPHOS with FADH2 reduced equivalents. In fact, in addition to mutations in the nuclear DNA-encoded SDH, mutations in mtDNA-encoded subunits of the Complex I, III and IV have been associated with tumour pre-conditioning or development [67],[68].
Further, inhibition of OXPHOS can also be achieved in cancer cells by direct inhibition of F1F0-ATPase, mediated by over-expression of its specific inhibitor IF1 [69]. The latter controls mitochondrial function and cell survival [70],[71] by directly acting on the mitochondrial structure and on F(1)F(o)-ATP synthase activity [69], thus representing a bad-prognosis predictor in a plethora of cancer types (ranging from liver to bladder and gastric cancer) [72],[73],[74].
In addition to mutations, alterations of the catalytic activity of metabolic enzymes can also be a secondary effect of microenvironmental signals. Hypoxia itself reduces the activity of SDH and components of the mitochondrial respiratory chain, so contributing to the metabolic shift toward glycolysis [75],[76],[77].
In addiction to hypoxic and pesudo-hypoxic signalling, also metabolic intermediates can “sensitize” back to the nucleus and control gene expression, thus unveiling an unexpected connection between oncometabolites and epigenetics. For instance, 2-hydroxyglutarate, whose accumulation is driven by mutations in the enzyme isocitrate dehydrogenase (IDH), was described to varying DNA methylation [78]. In similar studies, it was shown that increased cytoplasmic levels of acetyl-CoA and acidic pH, both due to the inhibition of TCA cycle and to the accumulation of lactic acid in the cytoplasm, modulate DNA acetylation [79],[80]. It would be of great interest to analyse whether these epigenetic alterations are stochastically distributed on the DNA or, still more intriguingly, if they are targeted to specific transcription factors or possible hot spots in promoters of cancer- or metabolism- related genes.
As a final remark of this paragraph, we would like to mention that dysfunctional mitochondria are able to signal back to the autophagy machinery to selectively remove them, through a quality control process called mitophagy. Failure on modulating mitophagy upon mitochondrial oncogenic stimuli is an intriguing cancer-related phenotype, representing a field of research that still remains to be comprehensively approached [81],[82].
Cancer-related inflammation is driven by oncometabolites
Pathologic analysis of human cancers’ biopsies often shows an inflammatory microenvironment with recruitment to the tumoral mass of cells from both the innate and adaptive branch of the immune response [83]. Inflammation’s role in the context of cancer development has long been debated. Historically, according to the immune surveillance model, inflammation was thought to be part of the attempt of the immune system to eradicate cancer cells [84]. To support this idea, there are indeed several pieces of evidence of increased tumour formation rates in mouse models where components of the immune system are genetically ablated [85],[86]. Moreover, human immunocompromised individuals show a higher probability of developing virus-derived cancer types [87]. In particular, the roster of anti-tumoral immune cells includes CD8+ cytotoxic T (CTLs), CD4+ Th1 helper lymphocytes and natural killer (NK) cells. However, more recent evidence has challenged the classic immune surveillance model by showing that some specific immune cells have, instead, the role of promoting cancer [88],[89],[90] (Figure 2). Indeed, in particular macrophages (polarized to an M2 subtype), mast cells, neutrophils and some subtype of T and B lymphocytes are able to favour cancer. To this end, these cells supply growth factors to sustain cell proliferation, survival factors to escape apoptosis, molecules or enzymes to modify extracellular matrix and facilitate invasion and metastatization, or to inhibit the anti-tumoral counterparts of the immune response [91],[92],[93]. Noteworthy, apart from its role in the cellular quality control, autophagy has been related to an unconventional secretion pathway of inflammatory mediators, such as Interleukin-1β and Interleukin-18 [94],[95].
Of great interest, some publications have started to link glycolytic metabolism of cancer cells to the inflammatory state of the tumour microenvironment. Both processes would be engaged in a positive feedback in which oncometabolites act as paracrine molecules that modulate the tumour-infiltrating cell polarity and activity (Figure 2). For example, succinate accumulation has been shown to mediate HIF1α-dependent expression of the pro-inflammatory chemokine IL-1-β by bone-marrow-derived macrophages (BMDMs) [96]. Although this has been described as the result of lipopolysaccharide-mediated glycolytic shift in the context of microbial infection, it is reasonable to speculate that a similar mechanism could be involved in cancer. Two observations support this hypothesis. First, the concentration of circulating succinate (and HIF1α activity) in patients with tumours harbouring mutations in SDH is higher than in the healthy population [54],[97]; second, succinate can signal through its receptor GPR91 on dendritic cells in order to modulate the immune response [98].
Thus, macrophages seem to have a pivotal role in the crosstalk between cancer signalling and the inflammatory response. This is supported by the role of chemokines produced by the majority of the solid tumours, such as the colony-stimulating factor 1 (CSF1), which is a potent chemoattractant for macrophages [99]. Two main subpopulations of these immune cells are reported to invade the tumour: the classically activated macrophages (M1 population) and the alternatively activated ones (M2). M1 macrophages usually promote antigen presentation and immune activation, while M2 show pro-tumoral properties [100]. Remarkably, tumour-derived TGF-β1 shifts the population of macrophages towards a M2 phenotype [101]. On a feedback loop, squamous cell carcinoma (SCC) cells are differently sensitive to monocyte-derived TGF-β [102]. TGF-β responding SCC-stem cells cycle more slowly than TGF-β insensitive counterparts, responsible for tumour growth. TGF-β responding cells show increased invasiveness and resistance to anti-cancer therapies due to an enhanced antioxidant power mediated by a TGF-β-dependent change in metabolism, which ensures an adequate NADPH pool to reduce the oxidized form of glutathione [102]. The increase in the invasive properties associated with metabolic changes is further supported by the metabolic alterations during epithelial to mesenchymal transition (EMT) and metastasis formation. During EMT, it has been shown that TGF-β signaling mediates a transient increase in mitochondrial respiration to promote migration [103]. Moreover, super invasive properties of cancer cells have been linked to increased mitochondrial activity [104], together with an increased fragmentation of the organelles facilitating cell movement [105],[106],[107]. This property correlates with the observation that mitochondria fragment and localize at the posterior area of T lymphocytes during chemotaxis, where they provide the energy required for the myosin machineries to drive migration [108],[109].
The functional versatility of mitochondria is indeed paralleled by the dynamic nature of the organelles, which can fuse and divide through the action of the so-called mitochondria-shaping proteins (OPA1, MFN1, MFN2, DRP1, FIS1, among others) [110]. The mitochondrial shape is involved in many mechanisms contributing to cancer development and progression. For instance, mitochondria fragment during cell division in a CDK1/DRP1-dependent manner to achieve their stochastic distribution between daughter cells in highly proliferative tissues [111]. In addition, highly glycolytic cells show fragmented mitochondria when compared to cells relying on OXPHOS [112]. Moreover, DRP1 and OPA1 are both involved in the mitochondrial shape changes occurring upon apoptotic stimulation [113]. In particular, OPA1 is a multifaceted protein: it is able of keeping the mitochondria cristae shape under control, so confining the pro-apoptotic cytochrome c inside the cristae (and thus controlling apoptosis) [114],[115]; however, it also organizes the quaternary structure of the respiratory chain complexes in super-complexes boosting OXPHOS [116]. Future studies will be necessary to dissect the anti-apoptotic versus the pro-OXPHOS roles of this protein in cancer development. In addition to metabolism and apoptosis, as discussed above, the shape of mitochondria also controls cell migration during metastatization. Indeed, fragmented mitochondria re-localize within specific areas of migrating cells to generate the ATP necessary to “fuel” the cell motor myosin, so promoting migration and invasion [108],[105],[106],[107].
A complete analysis of the role of different mitochondria-shaping proteins in cancer development is still missing, although it represents a promising area for new ground-breaking discoveries in cancer biology.
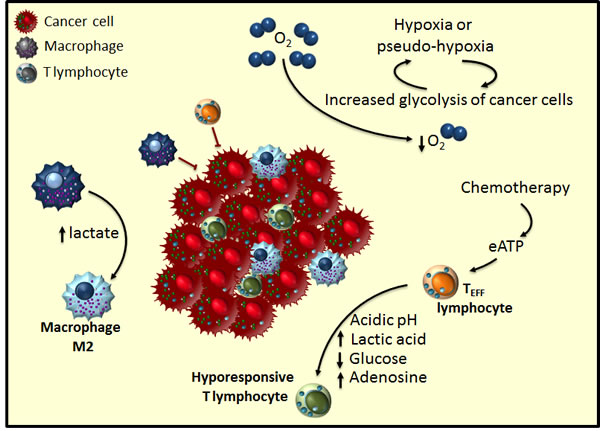
Figure 2: Tumour microenvironment hijacks anti-tumoral immune response. Cancer cells alter the immune response through metabolic competition establishing an inflammatory microenvironment. Increased glycolysis in tumoral cells depletes the tumour microenvironment of glucose and amino acids, so making TEFF cells hyporesponsive to tumour antigens. Moreover, high lactic acid levels generated by tumoral cells engage macrophages in a differentiation pathway towards a pro-inflammatory and tumour-promoting M2 subtype. Chemotherapy induces immunogenic cancer cell death, which through increased extracellular ATP (eATP) is able to partially re-activate TEFF cells.
A closer look at tumour-derived lactic acid: how tumour-associated macrophages are functionally polarized
To enhance this intricate relationship between cancer metabolism and tumour-invading immune cells a seminal paper from Medzhitov’s group shed light on a novel mechanism of communication between cancer cells and tumour-associated macrophages, based on tumour-derived lactic acid [20]. In this paper, the authors show that tumour-associated macrophages (TAM) - when compared to the peritoneal macrophages or to the tumoral cells - are characterized by a higher expression of the vascular endothelial growth factor (VEGF) and Arginase 1 (ARG1), both of which support tumour growth by neovascularization and by providing metabolic substrates respectively [20]. Remarkably, ARG1 can induce depletion of arginine from the microenvironment, thus leading to inhibition of cytotoxic CD8+ T lymphocyte function and their immune surveillance role [117]. Tumour conditioned medium shows the same capacity to induce VEGF and ARG1 expression in an HIF1α dependent manner [20], as indicated by the fact that their expression is prevented in an HIF1α null background. This suggests that some tumour-derived and secreted molecules can mediate VEGF and ARG1 expression in macrophages through a mechanism involving HIF1α stabilization under normoxic conditions. Strikingly, lactic acid - specifically produced by tumoral cells in many cancer types, in which mitochondrial respiration is inhibited and pyruvate is converted into lactic acid by the pyruvate kinase isoform M2 (PKM2) - mediates this communication loop between cancer and tumour invading cells [20]. Interestingly, the higher concentration of lactic acid, the larger and more aggressive are the tumoral masses observed. The uptake of lactic acid by TAM from the stroma is facilitated by the acidic pH also produced by the glycolytic metabolism of cancer cells. Eventually, in vivo co-injection of tumoral cells with lactate medium-conditioned macrophages results in tumours bigger than in the case of control-medium cultured macrophages, so confirming the pro-tumoral phenotype acquired by these immune cells [20]. In addition, the interaction between PKM2 and Transglutaminase type 2 (TG2), observed in a model of human fibrosarcoma, has been shown to significantly modulate autophagy, this facilitating the metabolic shift towards aerobic glycolysis [118],[119].
Tumour cell-derived lactic acid is also able to inhibit monocyte maturation to dendritic cells [120],[121]. This complex response to lactic acid could be an ancestral response reminiscent of the immune recognition of bacterial infection, in line with the observation that bacteria are able to inhibit oxidative phosphorylation and increase glycolysis in infected cells through LPS [96]. Above all, the discovery of this pro-tumoral paracrine effect of lactic acid could lead to the investigation of previously unexplored mechanisms endowing cancer cells with resistance to anti-cancer therapies. There is already evidence that accumulation of succinate and lactate facilitates epithelial to mesenchymal transition [122],[123]. One could also speculate, for example, that oncometabolites also exert a similar role in tropism of metastasis. Although current opinion favor the passive homing of metastasis (that is, metastasis forms in the first place where invasive or circulating cancer cells are entrapped), it is intriguing to speculate that homing of cancer cells could occur in eligible tissues according to the metabolic microenvironment, in a mechanism similar to that described for chemokines [124]. From this viewpoint, the tissue tropism of metastasis could be mediated by the favorable conditions in which to adapt and survive in the new micro-environmental niche. In line with this, as computational studies already suggested, normalizing the function of some metabolic targets could also inhibit cancer cell migration [125].
MetaboliC competition controls anti-tumoral function of T lymphocytes
Tumour microenvironment and imbalance of metabolites have also an important role in shaping and dampening the adaptive immune response (Figure 2) versus the tumoral cells, so resulting in a more aggressive tumour and poorer prognosis for the patients [126],[100]. Many publications in recent years showed that nutrient availability controls immune response by reducing the number and function of tumour-invading lymphocytes (TILs) while promoting an immunosuppressive environment [120],[20],[127],[128] (Figure 2).
T lymphocytes have characteristic metabolic features. In particular, naïve and memory T cells rely on mitochondrial respiration to fulfil their energetic demands [129],[130]. When engaged by an antigen and proper co-stimulation, T cells become effectors of the immune response, shifting their metabolism to a highly glycolytic one, this being more suitable for intense proliferation and production of cytokines [131],[132]. Importantly, cancer cells and the opponents fighting them share the same metabolic phenotype, this generating a strong competition for nutrients in the tumoral area. Pearce’s group showed that glucose consumption by the tumoral cells restricts T lymphocytes, reducing mTOR activity, glycolytic capacity and IFN-γ production, thus favouring tumour progression [127]. By decreasing the immune response in a coordinate fashion, lactate generated by the glycolytic tumoral mass is also able to dampen T effector lymphocytes glycolysis in a negative feedback loop through the inhibition of lactic acid release from T cells [120]. Moreover, although in a model of rheumatoid arthritis, lactate accumulation has been shown to inhibit motility of CD4 and CD8 T lymphocytes [133].
Regulatory T cells play an important role in shaping an immunosuppressive environment in the tumour [2] and they have been shown to metabolically rely on fatty acid oxidation [134],[135], as well as on glycolysis, in conditions different than cancer [136]. Nonetheless, the interplay between cancer cell metabolism and the actively immunosuppressive population of Treg cells has to be further investigated.
Tumour microenvironment is a complex milieu, where some molecules are depleted while others abound. In vivo studies showed that extracellular ATP levels are higher at tumour sites, when compared to tumour-free tissues [137]. Indeed, cancer cell demise is usually accompanied by release of ATP in an immunogenic fashion, especially in response to chemotherapeutic agents [138]. Extracellular ATP (eATP) is a potent alert signal for the immune system and differently targets various tumour-infiltrating cells. ATP acts, indeed, as an autocrine or paracrine co-stimulator for IL-2 production by activated T lymphocytes, thus regulating intracellular calcium waves and T cell activation and motility [139],[140],[141],[142]. Moreover, eATP attracts dendritic cell (DC) precursors into the tumour bed, so facilitating their stabilization in the proximal area of dying cells, and their development in mature cells with the capacity of presenting tumour-associated antigens [143].
To counteract the pro-immunogenic role of ATP, its levels can be reduced by the ecto-enzymes CD39 and CD73 (able to progressively hydrolyse ATP to adenosine), which are highly expressed on the immunosuppressive CD4+ FoxP3+ regulatory T cells [144],[145], on the intra-tumoral CD8+ FoxP3+ regulatory T cells [146] and on other immune cells subjected to TGFβ stimulation in the tumour microenvironment [147].
Reduced rates of glycolysis in T cells lead to the inhibition of T cell function by different molecular mechanisms. On the one hand, low levels of phosphoenolpyruvate (PEP, a glycolytic intermediate) result in unopposed activity of SERCA pumps, which reduce cytoplasmic calcium and, thus, the NFAT signalling necessary for translating a proper activation of the TCR [128]. On the other hand, low glycolytic activity leads to the interaction between GAPDH (not engaged in metabolic activity) and IFN-γ mRNA, suppressing its translation [148],[132].
Given the promising results already observed in clinics, special focus has now being put on immune checkpoint blockade exploitation, so as to treat a range of tumours more efficiently [149],[150]. The expression of inhibitory checkpoint receptors PD-1, Lag3, CTLA-4 increases in tumour infiltrating T lymphocytes, contributing to the dampening of the immune response [151],[152]. Interestingly, it has been shown that these receptors also have a role in the metabolic modification of TILs. For example, PD-1-expressing CD8+ T cells fail to fully activate glycolysis upon TCR engagement [152]. Moreover, treatment with checkpoint blockade antibodies against CTLA-4, PD-1, PD-L1 restores glucose availability in tumour microenvironment, re-establishing the condition for proper T effector cell function [127]. In particular, the action of PD-L1 directly on tumours reduces glycolysis by impinging on mTOR activation and on the expression of glycolytic enzymes, this being sufficient to restore T cell activity [127].
Nevertheless, nutrient availability issues do not stop with glucose. Tumour microenvironment is depleted of amino acids as well. Some of these are necessary for immune function of T cells [153]. High expression of Arginase-1 in myeloid-derived suppressor cells generates low levels of arginine, which in turn are responsible for reduced expression of TCR components limiting its proper activation [154]. Depletion of tryptophan and accumulation of immunosuppressive tryptophan metabolites are instead mediated by Indoleamine 2,3-dioxygenase (IDO) and are associated with poor prognosis in different cancer types, including endometrial and ovarian cancer [155],[156]. A similar mechanism involves the depletion of cysteine from the tumour microenvironment mediated by myeloid cells [157].
Of note, culturing tumour-specific T cells in vitro for a short time (6-24hours), in nutrient deplete condition, allows the recovery of effector function [158]. It comes as no surprise that some frontier anti-cancer treatments are trying to use the modulation of metabolism of cancer cells and/or T cells to improve their anti-cancer properties. Whatever the result, certainly more attention should be given to the therapeutic use of drugs impinging on tumour cell glycolysis since the same therapies would also modulate the function of the anti-tumoral TILs [159]. Fascinatingly, a recent paper by Restifo’s group proposes a simple and clinically feasible method to identify - by cytofluorimetric analysis of mitochondrial membrane potential - the T cell population with the best metabolic fitness to survive and accomplish long-term effector functions [160]. The authors show that isolating T cells based only on their lower mitochondrial membrane potential - which likely parallels the fact that these cells have active OXPHOS - allows the selection of cells with superior antitumour activity [160]. This method may be applied in different clinical settings that rely on adoptive transfer strategies; coupled with approaches aiming at metabolic re-activation of T cells, it could provide a promising strategy for cancer treatment.
Conclusions
Alterations in metabolism have long been considered only as a consequence (rather than as a pivotal factor) of tumour formation and progression. Despite this oversight, in the recent decades mitochondria and metabolism have been returned to the scientific crime scene to be further investigated for their role in cancer. We know now that the metabolic shift from mitochondrial respiration to glycolysis is not a mere modification of cellular metabolism. The metabolic intermediates, which accumulate due to the Warburg effect, acquire new functions impinging on a plethora of mechanisms ranging from pro-angiogenic to epigenetic alterations, from pro-inflammatory to immune-evasive effects (Figure 3). Additionally, an outstanding crosstalk has been discovered between cancer cells and tumour-infiltrating immune cells where the oncometabolites modify, or inhibit, the function of the latter to favour cancer cells’ proliferation, tumour expansion, and metastasis formation (Figure 3). New therapeutic approaches could be drawn up to restore tumoral cells to mitochondrial respiration; this would limit the pro-tumoral advantages of glycolysis on tumour aggressiveness itself, and would boost the anti-tumoral immune response in order to synergize the efforts in the battle against cancer.
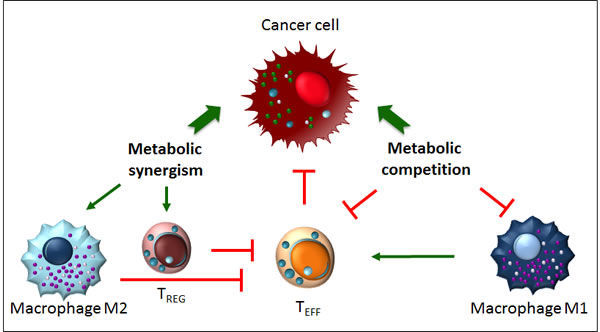
Figure 3: Metabolic competition and metabolic synergism in the tumour microenvironment. Metabolism influences the function of tumour-invading immune cells. Metabolic competition dampens the anti tumoral properties of M1 macrophages and TEFF cells. Metabolic synergism, instead, favors the development of pro-tumoral immune TREG cells and M2 macrophages.
Acknowledgments
We are extremely grateful to C Frezza for helpful suggestions and for critically reading the manuscript.
Funding
Our lab is supported by the Italian Ministry of Health (GR-2011-02351643).
Conflicts of Interest
The Authors have no conflicting interests to declare.
references
1. Hanahan D, Weinberg R A. The hallmarks of cancer. Cell. 2000; 100, 57-70.
2. Hanahan D, Weinberg R A. Hallmarks of cancer: the next generation. Cell. 2011; 144, 646-674.
3. Evan G I, Vousden K H. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001; 411, 342-348.
4. Vander Heiden M G, Cantley L C, Thompson C B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324, 1029-1033.
5. Slater E C. Mechanism of oxidative phosphorylation. Annu Rev Biochem. 1977; 46, 1015-1026.
6. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927; 8, 519-530.
7. Warburg O. On respiratory impairment in cancer cells. Science. 1956; 124, 269-270.
8. McAllister R M. Search for oncogenes in human rhabdomyosarcoma cells. Prog Immunobiol Stand. 1971; 5, 237-242.
9. Newmark P. Tyrosine phosphorylation and oncogenes. Nature. 1981; 292, 15-16.
10. Meier H, Huebner R J. Host-gene control of C-type tumor virus-expression and tumorigenesis: relevance of studies in inbred mice to cancer in man and other species. Proc Natl Acad Sci U S A. 1971; 68, 2664-2668.
11. McDonnell T J, Deane N, Platt F M, Nunez G, Jaeger U, McKearn J P, Korsmeyer S J. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989; 57, 79-88.
12. Vaux D L, Cory S, Adams J M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988; 335, 440-442.
13. Hockenbery D, Nunez G, Milliman C, Schreiber R D, Korsmeyer S J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990; 348, 334-336.
14. Liu X, Kim C N, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996; 86, 147-157.
15. Harvey A J, Kind K L, Thompson J G. REDOX regulation of early embryo development. Reproduction. 2002; 123, 479-486.
16. Rafalski V A, Mancini E, Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012; 125, 5597-5608.
17. Wang R, Green D R. Metabolic checkpoints in activated T cells. Nat Immunol. 2012; 13, 907-915.
18. Baysal B E, Ferrell R E, Willett-Brozick J E, Lawrence E C, Myssiorek D, Bosch A, van der Mey A, Taschner P E, Rubinstein W S, Myers E N, Richard C W r, Cornelisse C J, Devilee P et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000; 287, 848-851.
19. Tomlinson I P, Alam N A, Rowan A J, Barclay E, Jaeger E E, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance R R, Olpin S, Bevan S et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002; 30, 406-410.
20. Colegio O R, Chu N Q, Szabo A L, Chu T, Rhebergen A M, Jairam V, Cyrus N, Brokowski C E, Eisenbarth S C, Phillips G M, Cline G W, Phillips A J, Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014; 513, 559-563.
21. Frezza C. The role of mitochondria in the oncogenic signal transduction. Int J Biochem Cell Biol. 2014; 48, 11-17.
22. Miller D M, Thomas S D, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012; 18, 5546-5553.
23. Lombardi L, Newcomb E W, Dalla-Favera R. Pathogenesis of Burkitt lymphoma: expression of an activated c-myc oncogene causes the tumorigenic conversion of EBV-infected human B lymphoblasts. Cell. 1987; 49, 161-170.
24. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins J R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000; 14, 2501-2514.
25. Shim H, Dolde C, Lewis B C, Wu C S, Dang G, Jungmann R A, Dalla-Favera R, Dang C V. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997; 94, 6658-6663.
26. Dang C V, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009; 15, 6479-6483.
27. Kim J W, Gao P, Liu Y C, Semenza G L, Dang C V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007; 27, 7381-7393.
28. Osthus R C, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee L A, Dang C V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000; 275, 21797-21800.
29. Wise D R, DeBerardinis R J, Mancuso A, Sayed N, Zhang X Y, Pfeiffer H K, Nissim I, Daikhin E, Yudkoff M, McMahon S B, Thompson C B. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008; 105, 18782-18787.
30. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011; 11, 761-774.
31. Gough D J, Corlett A, Schlessinger K, Wegrzyn J, Larner A C, Levy D E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009; 324, 1713-1716.
32. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, Chiao P J, Keating M J, Garcia-Manero G et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012; 22, 399-412.
33. Lee M H, Lee S E, Kim D W, Ryu M J, Kim S J, Kim S J, Kim Y K, Park J H, Kweon G R, Kim J M, Lee J U, De Falco V, Jo Y S et al. Mitochondrial localization and regulation of BRAFV600E in thyroid cancer: a clinically used RAF inhibitor is unable to block the mitochondrial activities of BRAFV600E. J Clin Endocrinol Metab. 2011; 96, E19-30.
34. Guo J Y, Chen H Y, Mathew R, Fan J, Strohecker A M, Karsli-Uzunbas G, Kamphorst J J, Chen G, Lemons J M, Karantza V, Coller H A, Dipaola R S, Gelinas C et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011; 25, 460-470.
35. Tsuneoka M, Umata T, Kimura H, Koda Y, Nakajima M, Kosai K, Takahashi T, Takahashi Y, Yamamoto A. c-myc induces autophagy in rat 3Y1 fibroblast cells. Cell Struct Funct. 2003; 28, 195-204.
36. Galluzzi L, Pietrocola F, Bravo-San Pedro J M, Amaravadi R K, Baehrecke E H, Cecconi F, Codogno P, Debnath J, Gewirtz D A, Karantza V, Kimmelman A, Kumar S, Levine B et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015; 34, 856-880.
37. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini P F, De Zio D, Nazio F, Antonioli M, D’Orazio M, Skobo T, Bordi M, Rohde M et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015; 17, 706.
38. Goldsmith J, Levine B, Debnath J. Autophagy and cancer metabolism. Methods Enzymol. 2014; 542, 25-57.
39. White E, Mehnert J M, Chan C S. Autophagy, Metabolism, and Cancer. Clin Cancer Res. 2015; 21, 5037-5046.
40. Muller P A, Vousden K H. p53 mutations in cancer. Nat Cell Biol. 2013; 15, 2-8.
41. Vousden K H, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009; 137, 413-431.
42. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015; 356, 197-203.
43. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, Chan C S, Hu W, Feng Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014; 5, 5535-5546. doi: 10.18632/oncotarget.2137.
44. Kondoh H, Lleonart M E, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005; 65, 177-185.
45. Contractor T, Harris C R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012; 72, 560-567.
46. Bensaad K, Tsuruta A, Selak M A, Vidal M N, Nakano K, Bartrons R, Gottlieb E, Vousden K H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006; 126, 107-120.
47. Pai E F, Schulz G E. The catalytic mechanism of glutathione reductase as derived from x-ray diffraction analyses of reaction intermediates. J Biol Chem. 1983; 258, 1752-1757.
48. Feng Z, Levine A J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010; 20, 427-434.
49. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008; 10, 611-618.
50. Freed-Pastor W A, Mizuno H, Zhao X, Langerod A, Moon S H, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell M J, Osborne T F, Tian B et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012; 148, 244-258.
51. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine A J, Hu W, Feng Z. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013; 4, 2935.
52. Bartesaghi S, Graziano V, Galavotti S, Henriquez N V, Betts J, Saxena J, A D, Karlsson A, Martins L M, Capasso M, Nicotera P, Brandner S, De Laurenzi V et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc Natl Acad Sci U S A. 2015; 112, 1059-1064.
53. Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014; 2, 10.
54. Gimenez-Roqueplo A P, Favier J, Rustin P, Mourad J J, Plouin P F, Corvol P, Rotig A, Jeunemaitre X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001; 69, 1186-1197.
55. Pollard P J, Briere J J, Alam N A, Barwell J, Barclay E, Wortham N C, Hunt T, Mitchell M, Olpin S, Moat S J, Hargreaves I P, Heales S J, Chung Y L et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005; 14, 2231-2239.
56. Rofstad E K, Galappathi K, Mathiesen B, Ruud E B. Fluctuating and diffusion-limited hypoxia in hypoxia-induced metastasis. Clin Cancer Res. 2007; 13, 1971-1978.
57. Wang G L, Jiang B H, Rue E A, Semenza G L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92, 5510-5514.
58. Selak M A, Armour S M, MacKenzie E D, Boulahbel H, Watson D G, Mansfield K D, Pan Y, Simon M C, Thompson C B, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005; 7, 77-85.
59. Bruick R K, McKnight S L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001; 294, 1337-1340.
60. Hirsila M, Koivunen P, Gunzler V, Kivirikko K I, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003; 278, 30772-30780.
61. Isaacs J S, Jung Y J, Mole D R, Lee S, Torres-Cabala C, Chung Y L, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe P J, Linehan W M, Neckers L. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005; 8, 143-153.
62. Zheng L, Mackenzie E D, Karim S A, Hedley A, Blyth K, Kalna G, Watson D G, Szlosarek P, Frezza C, Gottlieb E. Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 2013; 1, 12.
63. Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp G W, Wolhuter K, Stevens M, Fischer R, Carmeliet P et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011; 20, 524-537.
64. Ooi A, Wong J C, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min B W, Tan M H, Zhang Z, Yang X J, Zhou M, Gardie B, Molinie V et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011; 20, 511-523.
65. Sullivan L B, Martinez-Garcia E, Nguyen H, Mullen A R, Dufour E, Sudarshan S, Licht J D, Deberardinis R J, Chandel N S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013; 51, 236-248.
66. Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, Berquand A, Kessler B M, Lappin T et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013; 3, 689-700.
67. Brandon M, Baldi P, Wallace D C. Mitochondrial mutations in cancer. Oncogene. 2006; 25, 4647-4662.
68. Lu J, Sharma L K, Bai Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009; 19, 802-815.
69. Campanella M, Casswell E, Chong S, Farah Z, Wieckowski M R, Abramov A Y, Tinker A, Duchen M R. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008; 8, 13-25.
70. Faccenda D, Tan C H, Seraphim A, Duchen M R, Campanella M. IF1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ. 2013; 20, 686-697.
71. Santacatterina F, Sanchez-Cenizo L, Formentini L, Mobasher M A, Casas E, Rueda C B, Martinez-Reyes I, Nunez de Arenas C, Garcia-Bermudez J, Zapata J M, Sanchez-Arago M, Satrustegui J, Valverde A M et al. Down-regulation of oxidative phosphorylation in the liver by expression of the ATPase inhibitory factor 1 induces a tumor-promoter metabolic state. Oncotarget. 2016; 7, 490-508. doi: 10.18632/oncotarget.6357.
72. Faccenda D, Campanella M. Molecular Regulation of the Mitochondrial F(1)F(o)-ATPsynthase: Physiological and Pathological Significance of the Inhibitory Factor 1 (IF(1)). Int J Cell Biol. 2012; 2012, 367934.
73. Yin T, Lu L, Xiong Z, Wei S, Cui D. ATPase inhibitory factor 1 is a prognostic marker and contributes to proliferation and invasion of human gastric cancer cells. Biomed Pharmacother. 2015; 70, 90-96.
74. Wu J, Shan Q, Li P, Wu Y, Xie J, Wang X. ATPase inhibitory factor 1 is a potential prognostic marker for the migration and invasion of glioma. Oncol Lett. 2015; 10, 2075-2080.
75. Frezza C, Zheng L, Tennant D A, Papkovsky D B, Hedley B A, Kalna G, Watson D G, Gottlieb E. Metabolic profiling of hypoxic cells revealed a catabolic signature required for cell survival. PLoS One. 2011; 6, e24411.
76. Merlo A, de Quiros S B, Secades P, Zambrano I, Balbin M, Astudillo A, Scola B, Aristegui M, Suarez C, Chiara M D. Identification of a signaling axis HIF-1alpha/microRNA-210/ISCU independent of SDH mutation that defines a subgroup of head and neck paragangliomas. J Clin Endocrinol Metab. 2012; 97, E2194-200.
77. Puissegur M P, Mazure N M, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, Maurin T, Lebrigand K, Cardinaud B, Hofman V, Fourre S, Magnone V, Ricci J E et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011; 18, 465-478.
78. Figueroa M E, Abdel-Wahab O, Lu C, Ward P S, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez H F, Tallman M S, Sun Z, Wolniak K et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010; 18, 553-567.
79. Cai L, Sutter B M, Li B, Tu B P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011; 42, 426-437.
80. McBrian M A, Behbahan I S, Ferrari R, Su T, Huang T W, Li K, Hong C S, Christofk H R, Vogelauer M, Seligson D B, Kurdistani S K. Histone acetylation regulates intracellular pH. Mol Cell. 2013; 49, 310-321.
81. Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy. 2013; 9, 1720-1736.
82. Chourasia A H, Boland M L, Macleod K F. Mitophagy and cancer. Cancer Metab. 2015; 3, 4.
83. Dvorak H F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986; 315, 1650-1659.
84. Swann J B, Smyth M J. Immune surveillance of tumors. J Clin Invest. 2007; 117, 1137-1146.
85. Teng M W, Swann J B, Koebel C M, Schreiber R D, Smyth M J. Immune-mediated dormancy: an equilibrium with cancer. J Leukoc Biol. 2008; 84, 988-993.
86. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007; 121, 1-14.
87. Vajdic C M, van Leeuwen M T. Cancer incidence and risk factors after solid organ transplantation. Int J Cancer. 2009; 125, 1747-1754.
88. Coffelt S B, Lewis C E, Naldini L, Brown J M, Ferrara N, De Palma M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am J Pathol. 2010; 176, 1564-1576.
89. DeNardo D G, Andreu P, Coussens L M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010; 29, 309-316.
90. Johansson M, Denardo D G, Coussens L M. Polarized immune responses differentially regulate cancer development. Immunol Rev. 2008; 222, 145-154.
91. Murdoch C, Muthana M, Coffelt S B, Lewis C E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008; 8, 618-631.
92. De Palma M, Murdoch C, Venneri M A, Naldini L, Lewis C E. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007; 28, 519-524.
93. Biswas S K, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010; 11, 889-896.
94. Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990; 9, 1503-1510.
95. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011; 30, 4701-4711.
96. Tannahill G M, Curtis A M, Adamik J, Palsson-McDermott E M, McGettrick A F, Goel G, Frezza C, Bernard N J, Kelly B, Foley N H, Zheng L, Gardet A, Tong Z et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013; 496, 238-242.
97. Dahia P L, Ross K N, Wright M E, Hayashida C Y, Santagata S, Barontini M, Kung A L, Sanso G, Powers J F, Tischler A S, Hodin R, Heitritter S, Moore F et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005; 1, 72-80.
98. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwarzler C, Junt T, Voshol H, Meingassner J G, Mao X, Werner G, Rot A et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008; 9, 1261-1269.
99. Wyckoff J, Wang W, Lin E Y, Wang Y, Pixley F, Stanley E R, Graf T, Pollard J W, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004; 64, 7022-7029.
100. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454, 436-444.
101. Martinez F O, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008; 13, 453-461.
102. Oshimori N, Oristian D, Fuchs E. TGF-beta Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell. 2015; 160, 963-976.
103. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-O M, Deberardinis R J, Boothman D A. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene. 2014; 0,
104. Porporato P E, Payen V L, Perez-Escuredo J, De Saedeleer C J, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014; 8, 754-766.
105. Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, Scorrano L, Maximo V, Campello S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One. 2015; 10, e0122308.
106. Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff D W, Abel P W, Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013; 32, 4814-4824.
107. Wan Y Y, Zhang J F, Yang Z J, Jiang L P, Wei Y F, Lai Q N, Wang J B, Xin H B, Han X J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol Rep. 2014; 32, 619-626.
108. Campello S, Lacalle R A, Bettella M, Manes S, Scorrano L, Viola A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med. 2006; 203, 2879-2886.
109. da Silva A F, Mariotti F R, Maximo V, Campello S. Mitochondria dynamism: of shape, transport and cell migration. Cell Mol Life Sci. 2014; 71, 2313-2324.
110. Corrado M, Scorrano L, Campello S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int J Cell Biol. 2012; 2012, 729290.
111. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007; 282, 11521-11529.
112. Hackenbrock C R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 1966; 30, 269-297.
113. Suen D F, Norris K L, Youle R J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008; 22, 1577-1590.
114. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko G V, Rudka T, Bartoli D, Polishuck R S, Danial N N, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006; 126, 177-189.
115. Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D’Adamio L, Derks C, Dejaegere T, Pellegrini L et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006; 126, 163-175.
116. Cogliati S, Frezza C, Soriano M E, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes L C, Perales-Clemente E, Salviati L, Fernandez-Silva P et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013; 155, 160-171.
117. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005; 5, 641-654.
118. Altuntas S, Rossin F, Marsella C, D’Eletto M, Diaz-Hidalgo L, Farrace M G, Campanella M, Antonioli M, Fimia G M, Piacentini M. The transglutaminase type 2 and pyruvate kinase isoenzyme M2 interplay in autophagy regulation. Oncotarget. 2015; 6, 44941-44954. doi: 10.18632/oncotarget.6759
119. Rossin F, D’Eletto M, Falasca L, Sepe S, Cocco S, Fimia G M, Campanella M, Mastroberardino P G, Farrace M G, Piacentini M. Transglutaminase 2 ablation leads to mitophagy impairment associated with a metabolic shift towards aerobic glycolysis. Cell Death Differ. 2015; 22, 408-418.
120. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007; 109, 3812-3819.
121. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, Hellerbrand C, Kastenberger M, Kunz-Schughart L A, Oefner P J, Andreesen R, Gottfried E, Kreutz M P. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. 2010; 184, 1200-1209.
122. Aspuria P J, Lunt S Y, Varemo L, Vergnes L, Gozo M, Beach J A, Salumbides B, Reue K, Wiedemeyer W R, Nielsen J, Karlan B Y, Orsulic S. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014; 2, 21.
123. Baumann F, Leukel P, Doerfelt A, Beier C P, Dettmer K, Oefner P J, Kastenberger M, Kreutz M, Nickl-Jockschat T, Bogdahn U, Bosserhoff A K, Hau P. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009; 11, 368-380.
124. Joyce J A, Pollard J W. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009; 9, 239-252.
125. Yizhak K, Le Devedec S E, Rogkoti V M, Baenke F, de Boer V C, Frezza C, Schulze A, van de Water B, Ruppin E. A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol Syst Biol. 2014; 10, 744.
126. Mockler M B, Conroy M J, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front Oncol. 2014; 4, 107.
127. Chang C H, Qiu J, O’Sullivan D, Buck M D, Noguchi T, Curtis J D, Chen Q, Gindin M, Gubin M M, van der Windt G J, Tonc E, Schreiber R D, Pearce E J et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015; 162, 1229-1241.
128. Ho P C, Bihuniak J D, Macintyre A N, Staron M, Liu X, Amezquita R, Tsui Y C, Cui G, Micevic G, Perales J C, Kleinstein S H, Abel E D, Insogna K L et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015; 162, 1217-1228.
129. van der Windt G J, O’Sullivan D, Everts B, Huang S C, Buck M D, Curtis J D, Chang C H, Smith A M, Ai T, Faubert B, Jones R G, Pearce E J, Pearce E L. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A. 2013; 110, 14336-14341.
130. van der Windt G J, Everts B, Chang C H, Curtis J D, Freitas T C, Amiel E, Pearce E J, Pearce E L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012; 36, 68-78.
131. Verbist K C, Wang R, Green D R. T cell metabolism and the immune response. Semin Immunol. 2012; 24, 399-404.
132. Gubser P M, Bantug G R, Razik L, Fischer M, Dimeloe S, Hoenger G, Durovic B, Jauch A, Hess C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013; 14, 1064-1072.
133. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland E J, Bombardieri M, Pitzalis C, Perretti M, Marelli-Berg F M, Mauro C. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015; 13, e1002202.
134. Michalek R D, Gerriets V A, Jacobs S R, Macintyre A N, MacIver N J, Mason E F, Sullivan S A, Nichols A G, Rathmell J C. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011; 186, 3299-3303.
135. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015; 36, 81-91.
136. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, Romano A, De Simone S, Procaccini C, La Rocca C, Carrieri P B, Maniscalco G T, Salvetti M et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. 2015; 16, 1174-1184.
137. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008; 3, e2599.
138. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala A Q, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini J L, Zitvogel L, Kroemer G. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014; 21, 79-91.
139. Schenk U, Westendorf A M, Radaelli E, Casati A, Ferro M, Fumagalli M, Verderio C, Buer J, Scanziani E, Grassi F. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal. 2008; 1, ra6.
140. Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, Ferrari V, Insel P A, Junger W G. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J. 2009; 23, 1685-1693.
141. Wang C M, Ploia C, Anselmi F, Sarukhan A, Viola A. Adenosine triphosphate acts as a paracrine signaling molecule to reduce the motility of T cells. EMBO J. 2014; 33, 1354-1364.
142. Goere D, Flament C, Rusakiewicz S, Poirier-Colame V, Kepp O, Martins I, Pesquet J, Eggermont A, Elias D, Chaput N, Zitvogel L. Potent immunomodulatory effects of the trifunctional antibody catumaxomab. Cancer Res. 2013; 73, 4663-4673.
143. Ma Y, Adjemian S, Yang H, Catani J P, Hannani D, Martins I, Michaud M, Kepp O, Sukkurwala A Q, Vacchelli E, Galluzzi L, Zitvogel L, Kroemer G. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology. 2013; 2, e24568.
144. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell’Acqua M L, Rossini P M, Battistini L, Rotzschke O et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007; 110, 1225-1232.
145. Deaglio S, Dwyer K M, Gao W, Friedman D, Usheva A, Erat A, Chen J F, Enjyoji K, Linden J, Oukka M, Kuchroo V K, Strom T B, Robson S C. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007; 204, 1257-1265.
146. Parodi A, Battaglia F, Kalli F, Ferrera F, Conteduca G, Tardito S, Stringara S, Ivaldi F, Negrini S, Borgonovo G, Simonato A, Traverso P, Carmignani G et al. CD39 is highly involved in mediating the suppression activity of tumor-infiltrating CD8+ T regulatory lymphocytes. Cancer Immunol Immunother. 2013; 62, 851-862.
147. Regateiro F S, Howie D, Nolan K F, Agorogiannis E I, Greaves D R, Cobbold S P, Waldmann H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-beta. Eur J Immunol. 2011; 41, 2955-2965.
148. Chang C H, Curtis J D, Maggi L B J, Faubert B, Villarino A V, O’Sullivan D, Huang S C, van der Windt G J, Blagih J, Qiu J, Weber J D, Pearce E J, Jones R G et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013; 153, 1239-1251.
149. Gubin M M, Zhang X, Schuster H, Caron E, Ward J P, Noguchi T, Ivanova Y, Hundal J, Arthur C D, Krebber W J, Mulder G E, Toebes M, Vesely M D et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014; 515, 577-581.
150. Kreamer K M. Immune Checkpoint Blockade: A New Paradigm in Treating Advanced Cancer. J Adv Pract Oncol. 2014; 5, 418-431.
151. Baitsch L, Legat A, Barba L, Fuertes Marraco S A, Rivals J P, Baumgaertner P, Christiansen-Jucht C, Bouzourene H, Rimoldi D, Pircher H, Rufer N, Matter M, Michielin O et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One. 2012; 7, e30852.
152. Parry R V, Chemnitz J M, Frauwirth K A, Lanfranco A R, Braunstein I, Kobayashi S V, Linsley P S, Thompson C B, Riley J L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005; 25, 9543-9553.
153. Mockler M B, Conroy M J, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front Oncol. 2014; 4, 107.
154. Rodriguez P C, Quiceno D G, Zabaleta J, Ortiz B, Zea A H, Piazuelo M B, Delgado A, Correa P, Brayer J, Sotomayor E M, Antonia S, Ochoa J B, Ochoa A C. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004; 64, 5839-5849.
155. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde B J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003; 9, 1269-1274.
156. Ino K, Yoshida N, Kajiyama H, Shibata K, Yamamoto E, Kidokoro K, Takahashi N, Terauchi M, Nawa A, Nomura S, Nagasaka T, Takikawa O, Kikkawa F. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer. 2006; 95, 1555-1561.
157. Srivastava M K, Sinha P, Clements V K, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010; 70, 68-77.
158. Wang S F, Fouquet S, Chapon M, Salmon H, Regnier F, Labroquere K, Badoual C, Damotte D, Validire P, Maubec E, Delongchamps N B, Cazes A, Gibault L et al. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. PLoS One. 2011; 6, e17621.
159. Munn D H, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. 2015; 39, 1-6.
160. Sukumar M, Liu J, Mehta G U, Patel S J, Roychoudhuri R, Crompton J G, Klebanoff C A, Ji Y, Li P, Yu Z, Whitehill G D, Clever D, Eil R L et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016; 23:63-76.