
INTRODUCTION
Glioblastomas, also known as glioblastoma multiforme (GBM), are tumors usually found in the cerebral hemispheres of the brain. GBM is composed of poorly differentiated neoplastic astrocytes that exhibit aggressive proliferation and invasive properties [1, 2]. It is generally difficult to identify prognostic factors associated with tumors, and 95% of patients die within 3 months after diagnosis without therapy [3]. Although there has been considerable progress in the treatment of GBM over the past 20 years, the median survival of glioblastoma patients is still less than 15 months [4, 5]. The blood-brain barrier (BBB) and the blood-cerebrospinal fluid barrier impede the effects of both chemotherapy and target therapies. Therefore, the development of suitable drugs that can cross the BBB and act as adjuvant drugs to improve the efficacy of surgery and chemotherapy is important for the treatment of GBM.
Honokiol (Hono) and magnolol (Mag) are extracted from the root and stem bark of Magnolia officinalis. These two compounds are often used in traditional herbal medicine in East Asia for the treatment of gastrointestinal complaints, anxiety, thrombotic stroke, and nervous system dysfunction [6]. Hono or Mag treatments not only induce apoptosis but also suppress cell proliferation through cell cycle arrest and up-regulating the expression of pro-apoptosis molecules in human glioma cells [7–12]. Treatment with Mag promotes p21/Cip1 and p27/ Kip1 expression and inhibits proliferation in the human U373 cell line in vitro and in vivo [8]. Mag also induces autophagy and inhibits cell migration and invasion in human PC3 cells [13]. In recent studies, Hono treatment was found to significantly enhance reactive oxygen species (ROS) production, decrease mitochondrial membrane potential, release cytochrome c, and activate caspase-9 and caspase-3, which leads to apoptosis in human glioblastoma cells [11, 12]. Hono treatment blocks the phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway-mediated immunoresistance of glioma without inhibiting critical proinflammatory T cell functions [14]. In addition, Hono effectively inhibits U87MG invasion and tumor growth in a human U251 xenograft glioma model [15, 16]. In prostate cancer, robust autophagy is triggered by Hono or Mag treatment, which inhibits angiogenesis and causes cell death in human umbilical cord vein endothelial cells and PC-3 cells [13, 17]. Recently, both Hono and Mag were reported being able to cross the BBB and inhibit cancer cell progression; however, the synergistic effect and mechanisms of Hono and Mag in the inhibition of proliferation and induction of cell death in glioma cancer cells have not been elucidated.
To investigate the synergistic effect of these two compounds in GBM treatment, we examined anti-tumor effects both in vitro and in vivo. The combination treatment of Hono with Mag (Hono-Mag) greatly limited the proliferation and survival of human GBM cells. Tumor size was also substantially decreased after Hono-Mag treatment in an orthotopic xenograft animal model. The synergistic effect of Hono-Mag treatment strongly inhibited glioma tumor growth through the autophagy formation and apoptosis pathways. In summary, the Hono-Mag combination treatment is more effective and has more potential in glioma treatment than treatment with Hono or Mag alone.
RESULTS
Hono and mag reduced the survival rate of human glioblastoma cells in vitro
To determine the effect of Hono and Mag on the proliferation of human glioblastoma cells, the survival rates of LN229, U87MG and GBM8401 were assessed using MTT assays. The glioma chemotherapy drug TMZ was applied as a positive control. The survival rates of LN229, U87MG and GBM8401 were decreased in the concentration of 50 μM and above by TMZ treatment (Figure 1A, 1B, 1C). In the presence of 40 μM Hono for 24 hours, the survival rates of Hono-treated U87MG and LN229 cells were significantly reduced by more than 30% over control cells (Figure 1A, 1B). The survival rate of GBM8401 was also reduced by 10% after treatment with 40 μM Hono treatment for 24 hours (Figure 1C). In contrast, GBM cells treatment with Mag showed less decreased on the survival rate. However, when Hono was combined with Mag treatment at 40 μM for 24 hours, the survival rate remarkably decreased by over 50% in LN229 cells and 70% in U87MG cells compared with control. In addition, the survival rate of GBM8401 cells decreased by over 30% under the same combined treatment compared with control. The combination treatment of Hono and Mag at 60 μM and 80 μM substantially decreased the proliferation and survival rates of LN229, U87MG and GBM8401 cells as compared to the control group, Hono-alone group, or Mag-alone group.
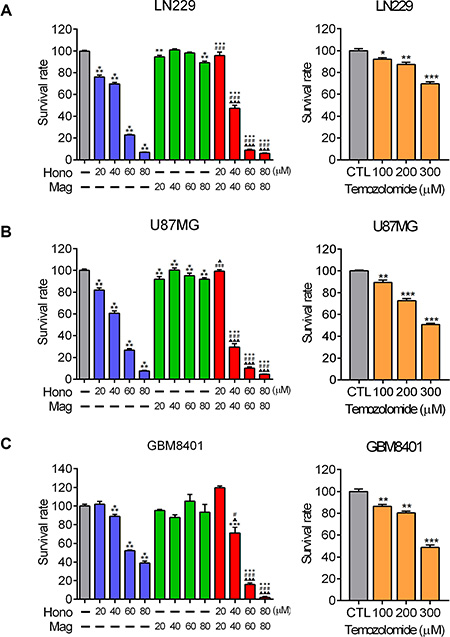
Figure 1: The effects of Hono, Mag and Hono-Mag combination treatment on cell viability. (A) LN229 glioma cells, (B) U87MG glioma cells, and (C) GBM8401 glioma cells were treated with DMSO or 20, 40, 60, or 80 μM of Hono, Mag or Hono-Mag combination for 24 hours. After treatment, the survival rate was analyzed using MTT tests. The right panels showed TMZ-treated glioma cells which were regarded as a positive control. *p < 0.05; **p < 0.01; ***p < 0.001 compared to the control group. #p < 0.05; ##p < 0.01; ###p < 0.001 compared to the same concentration of Hono-treated group. ▲p < 0.05; ▲▲p < 0.01; ▲▲▲p < 0.001 compared to the same concentration of Mag-treated group.
Hono and mag arrested the cell cycle and decreased the expression of cell cycle-related proteins
To investigate the cell cycle regulatory mechanisms of Hono and Mag in cell growth inhibition, LN229, U87MG and GBM8401 cells were cultured with or without 40 μM Hono (H40), 40 μM Mag (M40), or a 40 μM Hono-Mag combination (HM40). As shown in Figure 2A, 2B, the Hono treatment resulted in concentration-dependent cell cycle arrest at the G0/G1 phase in human LN229, U87MG and GBM8401 cells. However, Mag-stimulated cell cycle arrest at the G0/G1 phase was only observed in GBM8401 cells. HM40 treatment increased the sub-G1 phase cell population and caused apoptosis. Besides, the Hono-Mag treatment reduced the protein expression of Ki-67 (Figure 3A, 3C) and Ki-67-positive cells in LN229 and U87MG cells (Figure 3B, 3D and Supplementary Figure 1). In addition, the HM20 and HM40 treatments decreased the PI3K and Akt in LN229 and U87MG cells (Figure 3A, 3C). In LN229 and U87MG glioma cells, the protein expression of cyclin A was decreased in the H40, HM20 and HM40 groups. Moreover, the expression of cyclin D1 was decreased under the H40 and the HM40 treatment (Figure 4A, 4B). Furthermore, the protein levels of CDK2, CDK4, and CDK6 declined significantly after HM20 and HM40 treatment compared to other groups in both U87MG and LN229 cells.
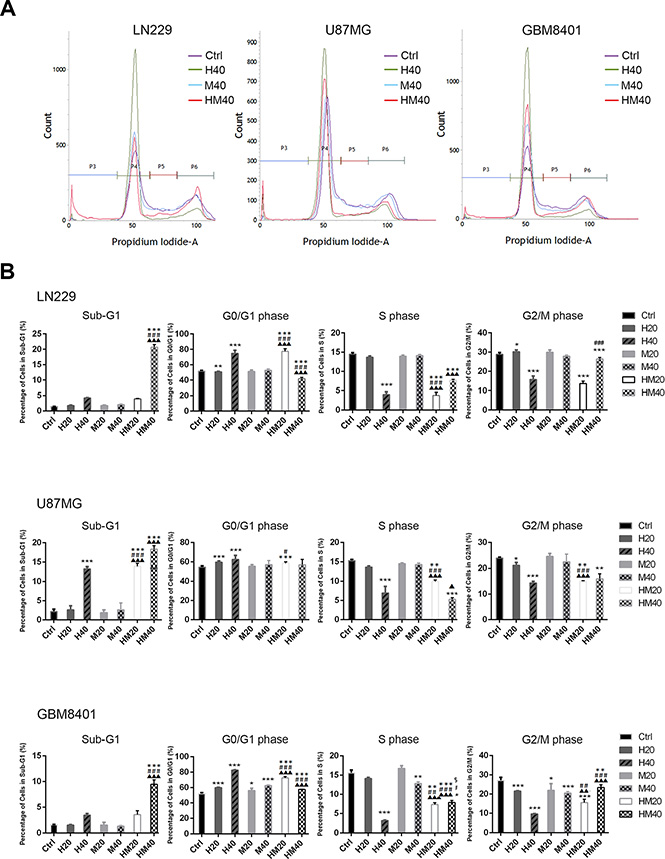
Figure 2: Cell cycle regulation by Hono, Mag and Hono-Mag combination treatment in human GBM cells. (A) LN229, U87MG and GBM8401 glioma cells were treated with Hono, Mag or Hono-Mag (20–40 μM) for 24 hours individually. The cell cycle expression was then analyzed by FACS. (B) Quantitative analyses of cell populations in the sub-G1, G0/G1, S, and G2/M phases of the cell cycle were conducted using BD FACSuite analysis software. *p < 0.05; **p < 0.01; ***p < 0.001 compared to the control group. #p < 0.05; ##p < 0.01; ###p < 0.001 compared to the same concentration of Hono-treated group. ▲p < 0.05; ▲▲p < 0.01; ▲▲▲p < 0.001 compared to the same concentration of Mag-treated group.
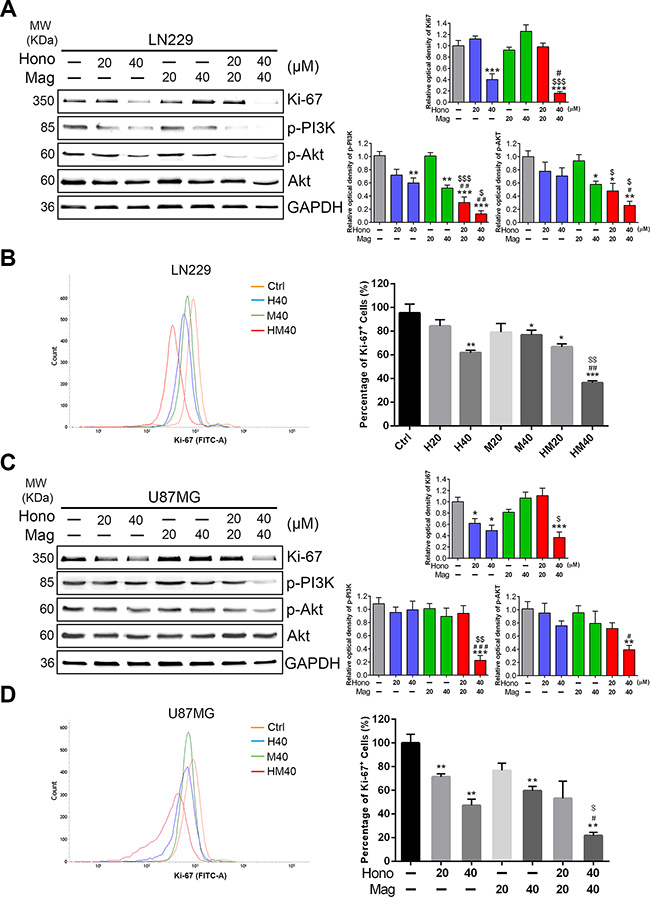
Figure 3: Hono-Mag combination treatment reduced the expression of proliferation-related protein in human GBM cells. LN229 and U87MG glioma cells were treated with Hono, Mag or Hono-Mag (20–40 μM) for 24 h. The cell lysates of (A) LN229 and (C) U87MG were then analyzed for Ki-67, p-PI3K and p-Akt using Western blotting. GAPDH was used as the loading control. The right panels show the quantitative analyses. The protein expression of Ki-67 in (B) LN229 and (D) U87MG cells was analyzed using FACS. The right panels show the quantitative analyses. **p < 0.01; ***p < 0.001 compared to the control group. #p < 0.05; ##p < 0.01 compared to the same concentration of Hono-treated group. $p < 0.05; $$p < 0.01;$$$p < 0.001 compared to the same concentration of Mag-treated group.
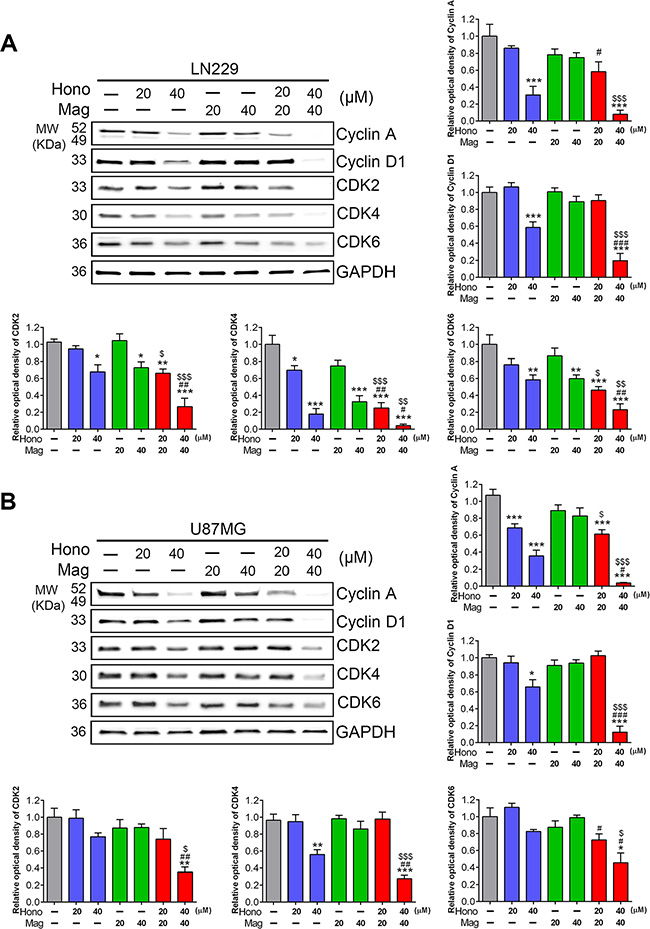
Figure 4: Hono-Mag treatment altered the expression of cell cycle-related proteins in human GBM cells. LN229 and U87MG cells were treated with different concentrations of Hono, Mag or Hono-Mag for 24 hours. Protein expression levels of (A) LN229 and (B) U87MG were analyzed for cyclin A, cyclin D1, CDK2, CDK4 and CDK6 by Western blotting. GAPDH was used as an internal control. The panels show the quantitative analyses. *p < 0.05; **p < 0.01; ***p < 0.001 compared to the control group. #p < 0.05; ##p < 0.01; ###p < 0.001 compared to the same concentration of Hono-treated group. ▲p < 0.05; ▲▲p < 0.01; ▲▲▲p < 0.001 compared to the same concentration of Mag-treated group.
The effect of hono and mag on apoptosis, autophage-associated proteins and MAPKS in human glioblastoma cells
Annexin V and PI staining showed that HM40 treatment promoted apoptosis in U87MG and LN229 cells (Figure 5A, 5C). To further reveal the underlying mechanism of HM40-induced apoptosis, apoptotic-related proteins were examined. The cleaved caspase-8, caspase-9 (Supplementary Figure 2), caspase-3 and PARP (Figure 5B, 5D) were increased after HM40 treatment in LN229 and U87MG cells. In addition, the anti-apoptosis protein Bcl-2 was decreased after HM40 treatment (Figure 5B, 5D). Moreover, the expression of LC3-βII, an autophagy marker, was observed after HM40 treatment. As shown in Figure 6A, the HM40 treatment decreased the protein expression of p-p38 and p-JNK but increased p-ERK in LN229 cells (Figure 6A). A similar effect was observed in U87MG cells (Figure 6B). To investigate the roles of p-ERK and autophagy induced by HM40 treatment, the HM40 treatment groups were pretreated with ERK inhibitor (PD98059) or autophagy inhibitor (3-MA). Both PD98059 and 3-MA increased the HM40-induced cell population at the sub-G1 phase (Figure 7A). Furthermore, both PD98059 and 3-MA reduced the Hono-Mag-increased p-ERK, LC3βII, and Bcl-2, but enhanced the expression of cleaved caspase-3 and cleaved PARP in LN229 cells (Figure 7B). The HM40 treatments with PD98059 or 3-MA significantly enhanced Hono-Mag-induced cell apoptosis, suggesting that ERK activation and autophagy played rescue roles.
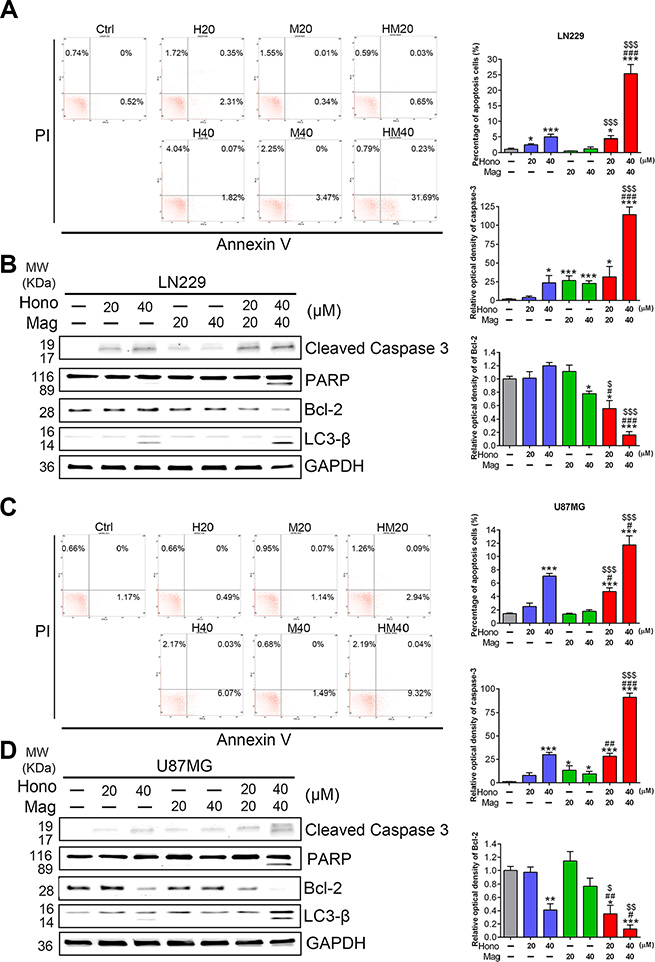
Figure 5: Hono-Mag promoted apoptosis and autophagy-associated proteins in GBM cells. LN229 and U87MG cells were treated with Hono, Mag or Hono-Mag (20–40 μM) for 24 hours. The apoptotic cells of (A) LN229 and (C) U87MG were determined using Annexin V staining and detected by flow cytometry analysis. The right panels show the percentage of the apoptotic cells. The cell lysates of (B) LN229 and (D) U87MG cells were then analyzed for cleaved caspase 3, cleaved PARP, Bcl-2 and LC3β by Western blotting. GAPDH was used as the loading control. The right panels show the quantitative analyses of the proteins. *p < 0.05; ***p < 0.001 compared to the control group. #p < 0.05; ###p < 0.001 compared to the same concentration of Hono-treated group. ▲p < 0.05; ▲▲▲p < 0.001 compared to the same concentration of Mag-treated group.
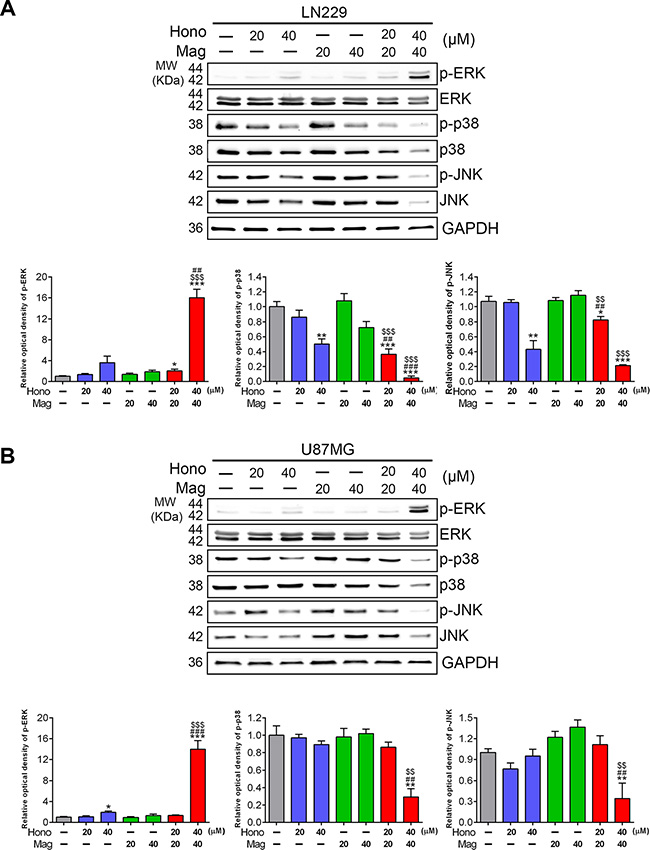
Figure 6: Hono-Mag down-regulated the activation of MAPKs in human GBM cells. LN229 and U87MG were treated with Hono, Mag or Hono-Mag (20–40 μM) for 24 hours to confirm the activation of MAPKs. The expression levels of p-ERK, ERK, p-p38, p38, p-JNK, and JNK in (A) LN229 and (B) U87MG cells were detected using Western blotting. GAPDH was used as the loading control. The lower panels show the quantitative analyses of the proteins. *p < 0.05; **p < 0.01; ***p < 0.001 compared to the control group. ##p < 0.01; ###p < 0.001 compared to the same concentration of Hono-treated group. ▲▲p < 0.01; ▲▲▲p < 0.001 compared to the same concentration of Mag-treated group.
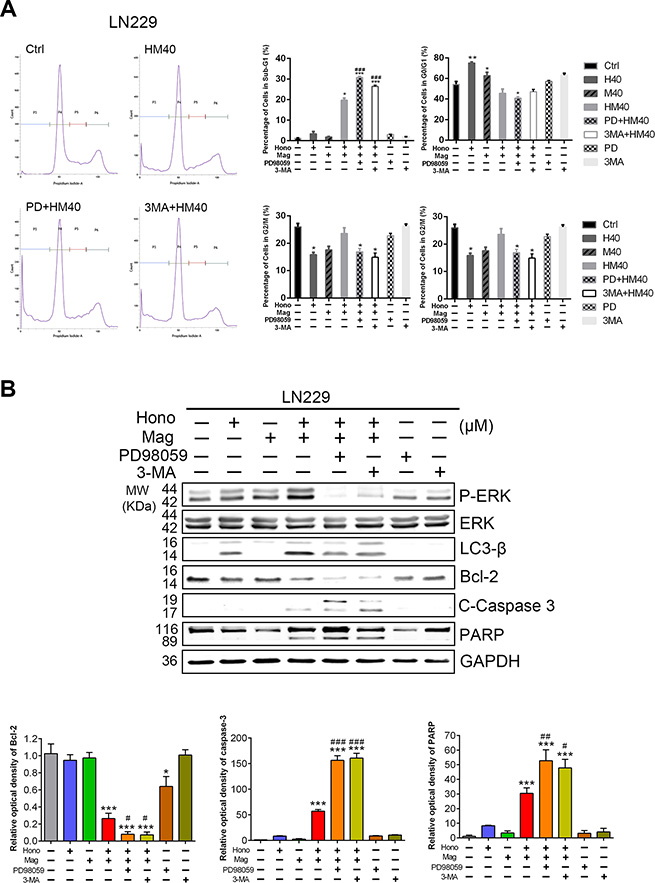
Figure 7: Suppression of p-ERK or autophagy enhanced Hono-Mag-stimulated apoptosis in human GBM Cells. The 40 μM of Hono, Mag, or Hono-Mag groups were combined with PD98059 or 3-MA in LN229 cells. After 24 hours of treatment, these cells were analyzed for populations in different stages of the cell cycle and protein expression. (A) The percentage of the cell population in the sub-G1, G0/G1, S, and G2/M phases of the cell cycle was determined using BD FACSuite analysis software. The right panels show quantitative analyses. (B) The protein expression levels of p-ERK, ERK, LC3β, Bcl-2, cleaved caspase 3 and PARP in LN229 cells were detected by Western blotting. GAPDH was used as the loading control. The lower panels show the quantitative analyses of the proteins. *p < 0.05; ***p < 0.001 compared to the control group. #p < 0.05; ##p < 0.01; ###p < 0.001 compared to the same concentration of Hono-Mag treatment group.
The effect of hono-mag combination treatment on orthotropic human glioblastoma xenograft mouse models
LN229 human glioblastoma cells, labeled with luciferase, were implanted intracranially. After treatment with Hono-Mag, tumor growth was significantly decreased compared to the Hono, Mag, TMZ and vehicle control groups (Figure 8A). The TMZ treatment only partially restricted tumor growth. Besides, the body weight in Hono and Hono-Mag groups was higher than in the TMZ and PBS groups (Figure 8B). HE and IHC staining revealed that Hono-Mag combination treatment not only inhibited Ki-67 expression and tumor growth but also activated the caspase-3 to enhance cells apoptosis in the tumors of nude mice (Figure 8C). Thus, the Hono-Mag combination treatment inhibited the progression and survival of glioblastoma cells both in vitro and in vivo.
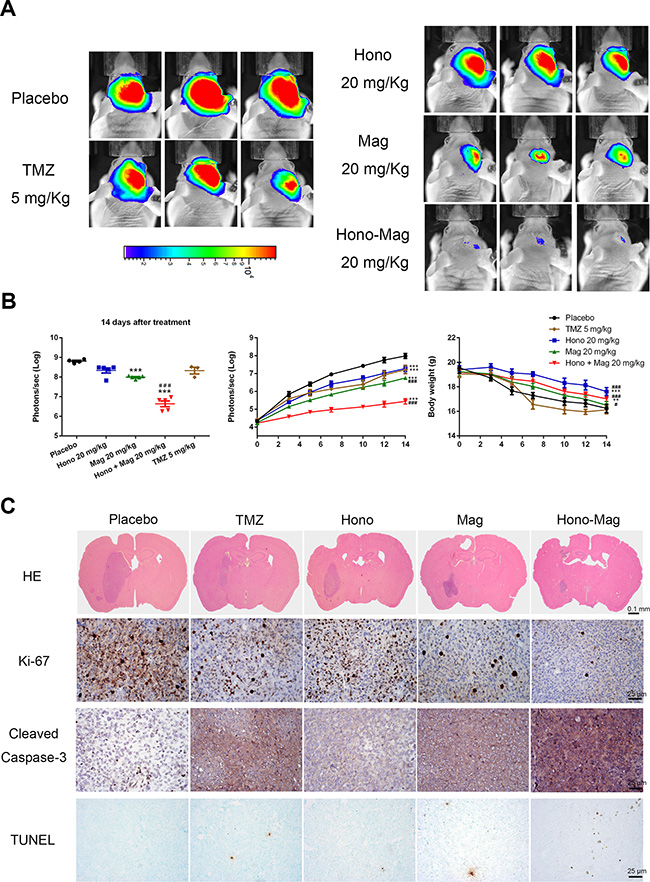
Figure 8: Hono-Mag treatment reduced tumor progression in orthotropic human GBM xenograft mouse models. (A) The in vivo bioluminescent imaging data were analyzed in different groups via the IVIS system. The lower panels show the quantitative analyses of the tumor progression. ***p < 0.001 compared to the placebo group. ###p < 0.001 compared to the TMZ-treated group. (B) The body weight was measured during drug treatment. **p < 0.05; ***p < 0.001 compared to the placebo group. #p < 0.05; ###p < 0.001 compared to the TMZ-treated group. (C) Representative images of histological staining and immunohistochemical stainings in xenograft orthotropic brain tumors. Protein levels of Ki-67, cleaved caspase-3 and TUNEL assay were performed. Bar, 25 μm.
DISCUSSION
Both Mag and Hono have been reported to exert anti-cancer effects via inhibiting proliferation, inducing apoptosis, countering metastasis, suppressing angiogenesis, and reversing multidrug resistance [18–20]. Until now, no studies had explored the effect of Mag-Hono combination treatment on glioblastoma; however, the present study found that both in vitro and in vivo, Mag-Hono combination treatment showed more effective in anti-proliferation and apoptosis than single treatment in GBM cells.
In glioma therapy, Mag treatment inhibits human U373 cell proliferation and induces apoptosis through p21/Cip1 and p27/Kip1 induction in vitro [10]. Hono also caused cell cycle arrest by decreasing the expression of cell cycle-related proteins, such as cyclin D1, CDK4 and CDK6 in PC-3 prostate cancer cells [21]. However, no studies affirmatively supported the combination of these two compounds or explored the underlying mechanism in glioma therapy. Hono treatment decreases the phosphorylation of Akt, signal transducer and activator of transcription 3 and ERK2 in colon cancer, malignant melanoma, and SVR endothelial tumors [21–23]. In addition, Mag inhibits EGFR, PI3K, and Akt activation in human prostate cancer [24]. The activation of PI3K and Akt plays a critical role in the survival and progression of GBM cells [25, 26]. Hyperactivation of PI3K and Akt may cause poor prognosis and chemotherapeutic/radiotherapeutic resistance [27]. The phosphorylation of Akt inhibits GSK3, contributing to the stabilization of cyclin D1 [28]. In LN229 and U87MG cells, the HM20 and HM40 treatments decreased the activation of PI3K, Akt and Akt-associated cell cycle-related proteins, such as cyclin D1 and cyclin A. In addition, Hono-Mag combination treatment inactivated CDK4, CDK6, Akt and decreased the proliferation through cell cycle arresting at the G0/G1 phase in GBM cells.
Apoptosis and autophagy were induced by 40 μM Hono-Mag combination treatment. Hono-Mag-induced Akt dephosphorylation may activate the pro-apoptotic Bcl-2 family member Bad and decrease the expression of the anti-apoptotic proteins Bcl-XL and Bcl-2, which prevent the release of cytochrome c from mitochondria [30, 31]. According to our results, the HM40 treatment decreased Bcl-2 expression and caused the cleavage of caspase-8, caspase-9, caspase-3 and PARP, which contributed to apoptosis through both intrinsic and extrinsic pathway in LN229 and U87MG cells. JNK and p38 have been reported to have either pro- or anti-apoptotic functions, depending on the cell types and microenvironments [32–34]. The inhibition of p38 triggers the TNF-mediated and caspase-dependent apoptotic pathway in human lymphoma U937 cells and increases sensitivity to the cytotoxic effect of TMZ in human glioma U87MG cells [35, 36]. In addition, the inhibition of JNK suppresses the activation of Akt and its downstream mediators, GSK-3β and Bad, but potentiates TMZ-induced cytotoxicity in U87 glioma cells [37]. In the present study, both phosphorylated p38 and JNK decreased after Hono-Mag combination treatment. These results revealed that apoptosis induced by Hono-Mag treatment may be caused by the attenuation of Akt, p38 MAPK and JNK in U87MG and LN229 cells.
Autophagy, a process conventionally considered as a survival mechanism, is increasingly being applied to facilitate cancer cell death by chemotherapy [38, 39]. Several studies indicate that both Mag and Hono are involved in autophagy induction [17, 40]. In human umbilical cord vein endothelial cells and prostate cancer cells, Mag-caused autophagy inhibited cell proliferation, migration, invasion and tube formation [13]. In human non-small-cell lung cancer cells, the combination of Hono and autophagy inhibitor inhibited the Hono-stimulated autophagy and induced apoptosis in a caspase-dependent manner [41]. Thus, an effective treatment may involve a combined and coherent action of apoptosis as well as non-apoptotic programs to minimize the chance of relapse [42]. Here, Hono-Mag-induced autophagy was blocked by both ERK and autophagy inhibitors. Recently, Hono was reported to induce autophagy and suppress cell migration through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK signaling pathways in neuroblastoma cells [43]. However, the attenuation of Hono-induced cell autophagy also down-regulates cell apoptosis in vitro [43]. According to our results, Hono-Mag treatment combined with PD98059 or 3-MA, significantly enhanced Hono-Mag-stimulated apoptosis in LN229 cells. Therefore, the induction of ERK phosphorylation resulting from Hono-Mag treatment played a critical role in rescuing apoptosis.
The first-line chemotherapeutic drug for GBM is the alkylating agent TMZ. Through oral absorption, TMZ is converted to an alkylating methyldiazonium cation, leading to cell death by breaking the DNA double strand [44, 45]. Human glioblastoma cell line LN229 has been reported to be temozolomide resistant [46, 47]. In the orthotopic xenograft mouse model, compared with the TMZ, Hono, and Mag treatment groups, the Hono-Mag combination treatment induced apoptosis and reduced the growth and progression of GBM tumors. These results indicated that the Hono-Mag combination has additional potential as an effective anti-tumor agent to target TMZ-resistant cells. Moreover, Hono-Mag combination treatment significantly maintained the body weight of mice comparing with the TMZ and PBS groups. Taking into account the ability to cross the BBB and the synergistic effect of Hono and Mag on glioma cell growth and progression inhibition, the Hono-Mag combination treatment may be applied as an adjuvant therapy to improve the therapeutic efficacy of GBM treatment.
MATERIALS AND METHODS
Cell culture
Human U87MG and LN229 glioblastoma cell lines were purchased from the American Type Culture Collection (Rockville, MD). LN229-Luc2 cells were derived via the stable transfection of pLuc2-iRFP and selected with a FACS Aria Fusion Sorter. The human glioblastoma multiforme (GBM8401) cell line was provided by Dr. Hueng from the National Defense Medical Center, Taiwan. The cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum and 100 IU/ml penicillin and streptomycin (pH 7.4) (all obtained from Gibco) in a humidified atmosphere of 5% CO2 at 37°C.
Drugs
The 2.3.3-[4, 5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) used in this study was obtained from Sigma-Aldrich (St. Louis, MO). Propidium iodide (PI), honokiol, magnolol, PD98059 and 3-methyladenine (3-MA) were purchased from Sigma-Aldrich (Sigma). Temozolomide (TMZ) was purchased from MedChem Express (MCE).
Cell survival assays
LN229, U87MG and GBM8401 glioblastoma cells were plated at 2 × 104 cells per well in a 24-well plate. Different concentrations of drugs or vehicle control were then added to the culture medium for 24 hours. After the cells were washed with phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8 mM Na2HPO4, and pH 7.4), 0.5 mg/ml of MTT was added and incubation continued for another 4 hours. The cells were then lysed with DMSO (Sigma). Absorbance at 590 nm was measured.
Flow cytometric analysis
The cell cycle, apoptosis assays and expression of Ki-67 were prepared by seeding 2 × 105 LN229, U87MG or GBM8401 glioma cells in 6-well plates. After cell attachment, growth medium either with or without different concentrations of Hono and Mag was added for 24 hours. To examine the expression of Ki-67, the cells were fixed and permeabilized at room temperature. Ki-67 primary antibody (Genetex) was incubated for 40 minutes. After PBS wash, the secondary antibodies conjugated to goat anti-rabbit antibody (Alexa Fluor 488, 1:500 Molecular Probes) were incubated for 40 minutes at room temperature. For cell cycle assays, cells were fixed in ethanol and stained with PI. For apoptosis assays, cells were harvested and processed for Annexin V staining according to the manufacturer (Invitrogen). The cells were washed with binding buffer [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 140 mmol/l NaCl, and 5 mmol/l CaCl2 at pH 7.4] and stained with anti-Annexin V antibody (FITC) and then counterstained with PI for 15 minutes at room temperature. The results were measured using a FACS Verse laser flow cytometric analysis system (Becton Dickinson). Ten thousand cells were analyzed for each sample.
Western blotting
After the various treatments, the glioma cells were washed once with PBS and then homogenized in lysis buffer (10 mM EGTA, 2 mM MgCl2, 60 mM PIPES, 25 mM HEPES, 0.15% Triton X-100, 1 μg/ml of pepstatin A, 1 μg/ml of leupeptin, 1 mM NaF, and 1 mM phenylmethylsulfonyl fluoride). Protein samples (60 μg per lane) were electrophoresed on a 10% SDS polyacrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad). Strips from the membrane were blocked with 5% non-fat milk in Tris-buffered saline, pH 8.2, containing 0.1% Tween (TBS-Tween) and incubated overnight at 4°C with a 1:500 dilution of rabbit antibodies against phosphorylated phosphoinositide 3-kinase (p-PI3K), cyclin-dependent kinase 2 (CDK2), CDK4, CDK6, phosphorylated JNK (p-JNK), JNK, phosphorylated ERK (p-ERK), ERK, phosphorylated p38 (p-p38), p38, Poly ADP ribose polymerase (PARP), cleaved caspase-3, cleaved caspase-8, cleaved caspase-9 (Cell Signaling), phosphorylated Akt (p-Akt) (Epitomics), Akt, cyclin A, cyclin D1 (Abcam), Bcl-2, GAPDH (BioVision), and Ki-67 (Genetex). After washes, the strips were incubated with a 1:5000 dilution of HRP-conjugated anti-rabbit IgG antibodies (Promega). Then, the blots were reacted in ECL substrate developing solution (Bio-Rad). The density of the bands on the nitrocellulose membrane was quantified by densitometry using Gel Pro 3.1 (Media Cybernetics), taking the density of the control sample as 100% and expressing the density of the test sample relative to the expression of the internal control as a relative value. Phosphorylated proteins were normalized to the total protein first.
Immunocytochemistry
U87MG and LN229 cells were grown on coverslips and fixed in cold 4% paraformaldehyde for 30 minutes at room temperature. The fixed cells were permeabilized with 0.1% Triton X-100 at room temperature and blocked with 3% bovine serum albumin (BSA). Ki-67 (Genetex) primary antibody was incubated overnight at 4°C. After PBS washing three times, the conjugated goat anti-rabbit IgG secondary antibodies (Alexa Fluor 488, 1:500 Molecular Probes) were incubated for 1 hour at room temperature. The samples were mounted onto slides and visualized using a confocal microscope (Zeiss).
Animal xenograft model
BALB/cAnN.Cg-Foxn1nu/CrlNarl nude mice (20–25 g) were anesthetized with O2/isoflurane mixture. Then, 105 LN229-Luc2 cells were implanted in the right cerebral hemisphere of the mice. Gliomas were permitted to grow in the murine brain. Five days after implantation, mice were randomly assigned to five groups (n = 5) that received vehicle control, Hono, Mag, Hono-Mag or TMZ. Hono, Mag and Hono-Mag were administered via intraperitoneal injection of 20 mg/kg/day for 14 days. TMZ was administered via oral gavage of 5 mg/kg/day for 7 days. Animals were euthanized after 14 days. The bioluminescence intensity of implanted tumors was monitored using the non-invasiveIn Vivo Imaging System (IVIS) three times per week. The body weight of the mice was measured three times per week.
Histological and immunohistochemistry examination
Orthotopic tumors were excised, rinsed twice in PBS and fixed in 4% paraformaldehyde for 48 hours. Tissues were frozen and sliced into 5-μm-thick sections. Routine hematoxylin and eosin (HE) staining was performed to facilitate histological evaluation. The expression of Ki-67, cleaved caspase-3 and BrdU in brain tumors of the nude mice was detected by immunohistochemistry. Ki-67 (Genetex) and cleaved caspase-3 (Cell signaling) primary antibodies and secondary goat anti-rabbit antibodies (Jackson Laboratories) were used. The apoptotic cells were detected via Apo-BrdU-IHC In Situ DNA Fragmentation assay kit (BioVision). The expression of Ki-67, cleaved caspase-3 and BrdU was observed in 10 random fields for each group.
Statistical analysis
All experiments were performed at least three times, and the results were expressed as the mean ± SEM for the total number of experiments. Differences between means were assessed using the Kruskal-Wallis test. The Mann-Whitney test was used for post hoc analysis. Statistical significance was set at p < 0.05.
CONFLICTS OF INTEREST
None.
GRANT SUPPORT
MOST 103-2320-B016-003-MY3 and Medical Research Grants of National Defense Medical Center 103-M063.
REFERENCES
1. Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol. 2012; 108:11–27.
2. Sehgal A. Molecular changes during the genesis of human gliomas. Semin Surg Oncol. 1998; 14:3–12.
3. Bredel M, Piribauer M, Marosi C, Birner P, Gatterbauer B, Fischer I, Strobel T, Rossler K, Budka H, Hainfellner JA. High expression of DNA topoisomerase IIalpha and Ki-67 antigen is associated with prolonged survival in glioblastoma patients. Eur J Cancer. 2002; 38:1343–1347.
4. Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012; 107:359–364.
5. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010; 60:166–193.
6. Ho NK. Traditional Chinese medicine and treatment of neonatal jaundice. Singapore Med J. 1996; 37:645–651.
7. Hibasami H, Achiwa Y, Katsuzaki H, Imai K, Yoshioka K, Nakanishi K, Ishii Y, Hasegawa M, Komiya T. Honokiol induces apoptosis in human lymphoid leukemia Molt 4B cells. Int J Mol Med. 1998; 2:671–673.
8. Hirano T, Gotoh M, Oka K. Natural flavonoids and lignans are potent cytostatic agents against human leukemic HL-60 cells. Life Sci. 1994; 55:1061–1069.
9. Chen LC, Liu YC, Liang YC, Ho YS, Lee WS. Magnolol inhibits human glioblastoma cell proliferation through upregulation of p21/Cip1. J Agric Food Chem. 2009; 57:7331–7337.
10. Chen LC, Lee WS. P27/Kip1 is responsible for magnolol-induced U373 apoptosis in vitro and in vivo. J Agric Food Chem. 2013; 61:2811–2819.
11. Jeong JJ, Lee JH, Chang KC, Kim HJ. Honokiol exerts an anticancer effect in T98G human glioblastoma cells through the induction of apoptosis and the regulation of adhesion molecules. Int J Oncol. 2012; 41:1358–1364.
12. Liang WZ, Chou CT, Chang HT, Cheng JS, Kuo DH, Ko KC, Chiang NN, Wu RF, Shieh P, Jan CR. The mechanism of honokiol-induced intracellular Ca(2+) rises and apoptosis in human glioblastoma cells. Chem Biol Interact. 2014; 221:13–23.
13. Kumar S, Guru SK, Pathania AS, Kumar A, Bhushan S, Malik F. Autophagy triggered by magnolol derivative negatively regulates angiogenesis. Cell Death Dis. 2013; 4:e889.
14. Crane C, Panner A, Pieper RO, Arbiser J, Parsa AT. Honokiol-mediated inhibition of PI3K/mTOR pathway: a potential strategy to overcome immunoresistance in glioma, breast, and prostate carcinoma without impacting T cell function. J Immunother. 2009; 32:585–592.
15. Wang X, Duan X, Yang G, Zhang X, Deng L, Zheng H, Deng C, Wen J, Wang N, Peng C, Zhao X, Wei Y, Chen L. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS One. 2011; 6:e18490.
16. Wolf I, O’Kelly J, Wakimoto N, Nguyen A, Amblard F, Karlan BY, Arbiser JL, Koeffler HP. Honokiol, a natural biphenyl, inhibits in vitro and in vivo growth of breast cancer through induction of apoptosis and cell cycle arrest. Int J Oncol. 2007; 30:1529–1537.
17. Hahm ER, Sakao K, Singh SV. Honokiol activates reactive oxygen species-mediated cytoprotective autophagy in human prostate cancer cells. Prostate. 2014; 74:1209–1221.
18. Amblard F, Govindarajan B, Lefkove B, Rapp KL, Detorio M, Arbiser JL, Schinazi RF. Synthesis, cytotoxicity, and antiviral activities of new neolignans related to honokiol and magnolol. Bioorg Med Chem Lett. 2007; 17:4428–4431.
19. Hwang ES, Park KK. Magnolol suppresses metastasis via inhibition of invasion, migration, and matrix metalloproteinase-2/-9 activities in PC-3 human prostate carcinoma cells. Biosci Biotechnol Biochem. 2010; 74:961–967.
20. Luo H, Zhong Q, Chen LJ, Qi XR, Fu AF, Yang HS, Yang F, Lin HG, Wei YQ, Zhao X. Liposomal honokiol, a promising agent for treatment of cisplatin-resistant human ovarian cancer. J Cancer Res Clin Oncol. 2008; 134:937–945.
21. Hahm ER, Singh SV. Honokiol causes G0-G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol Cancer Ther. 2007; 6:2686–2695.
22. Kaushik G, Ramalingam S, Subramaniam D, Rangarajan P, Protti P, Rammamoorthy P, Anant S, Mammen JM. Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am J Surg. 2012; 204:868–873.
23. Funa NS, Reddy K, Bhandarkar S, Kurenova EV, Yang L, Cance WG, Welsh M, Arbiser JL. Shb gene knockdown increases the susceptibility of SVR endothelial tumor cells to apoptotic stimuli in vitro and in vivo. J Invest Dermatol. 2008; 128:710–716.
24. Lee DH, Szczepanski MJ, Lee YJ. Magnolol induces apoptosis via inhibiting the EGFR/PI3K/Akt signaling pathway in human prostate cancer cells. J Cell Biochem. 2009; 106:1113–1122.
25. Cai H, Xue Y, Li Z, Hu Y, Wang Z, Liu W, Li Z, Liu Y. Round about 4 suppresses glioma-induced endothelial cell proliferation, migration and tube formation in vitro by inhibiting VEGR2-mediated PI3K/AKT and FAK signaling pathways. Cell Physiol Biochem. 2015; 35:1689–1705.
26. Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW. Mechanisms regulating glioma invasion. Cancer Lett. 2015; 362:1–7.
27. Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003; 4:257–262.
28. Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003; 2:339–345.
29. Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, Wan L, Singh A, Zhai B, et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014; 508:541–545.
30. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999; 13:2905–2927.
31. Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000; 275:10761–10766.
32. Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005; 15:36–42.
33. Aderca I, Moser CD, Veerasamy M, Bani-Hani AH, Bonilla-Guerrero R, Ahmed K, Shire A, Cazanave SC, Montoya DP, Mettler TA, Burgart LJ, Nagorney DM, Thibodeau SN, et al. The JNK inhibitor SP600129 enhances apoptosis of HCC cells induced by the tumor suppressor WWOX. J Hepatol. 2008; 49:373–383.
34. Greenberg AK, Basu S, Hu J, Yie TA, Tchou-Wong KM, Rom WN, Lee TC. Selective p38 activation in human non-small cell lung cancer. Am J Respir Cell Mol Biol. 2002; 26:558–564.
35. Varghese J, Chattopadhaya S, Sarin A. Inhibition of p38 kinase reveals a TNF-alpha-mediated, caspase-dependent, apoptotic death pathway in a human myelomonocyte cell line. J Immunol. 2001; 166:6570–6577.
36. Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003; 23:8306–8315.
37. Vo VA, Lee JW, Lee HJ, Chun W, Lim SY, Kim SS. Inhibition of JNK potentiates temozolomide-induced cytotoxicity in U87MG glioblastoma cells via suppression of Akt phosphorylation. Anticancer Res. 2014; 34:5509–5515.
38. Kumar D, Das B, Sen R, Kundu P, Manna A, Sarkar A, Chowdhury C, Chatterjee M, Das P. Andrographolide Analogue Induces Apoptosis and Autophagy Mediated Cell Death in U937 Cells by Inhibition of PI3K/Akt/mTOR Pathway. PLoS One. 2015; 10:e0139657.
39. Zhang ZS, Wang J, Shen YB, Guo CC, Sai KE, Chen FR, Mei X, Han FU, Chen ZP. Dihydroartemisinin increases temozolomide efficacy in glioma cells by inducing autophagy. Oncol Lett. 2015; 10:379–383.
40. Rasul A, Yu B, Khan M, Zhang K, Iqbal F, Ma T, Yang H. Magnolol, a natural compound, induces apoptosis of SGC-7901 human gastric adenocarcinoma cells via the mitochondrial and PI3K/Akt signaling pathways. Int J Oncol. 2012; 40:1153–1161.
41. Lv X, Liu F, Shang Y, Chen SZ. Honokiol exhibits enhanced antitumor effects with chloroquine by inducing cell death and inhibiting autophagy in human non-small cell lung cancer cells. Oncol Rep. 2015; 34:1289–1300.
42. Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004; 4:592–603.
43. Yeh PS, Wang W, Chang YA, Lin CJ, Wang JJ, Chen RM. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2015.
44. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
45. Newlands ES, Blackledge GR, Slack JA, Rustin GJ, Smith DB, Stuart NS, Quarterman CP, Hoffman R, Stevens MF, Brampton MH, et al. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br J Cancer. 1992; 65:287–291.
46. Cuperlovic-Culf M, Touaibia M, St-Coeur PD, Poitras J, Morin P, Culf AS. Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites. 2014; 4:807–830.
47. Cheng Y, Sk UH, Zhang Y, Ren X, Zhang L, Huber-Keener KJ, Sun YW, Liao J, Amin S, Sharma AK, Yang JM. Rational incorporation of selenium into temozolomide elicits superior antitumor activity associated with both apoptotic and autophagic cell death. PLoS One. 2012; 7:e35104.