
INTRODUCTION
Gastrointestinal stromal tumor (GIST) is a malignancy of mesenchymal origin that arises in the gastrointestinal tract and is resistant to conventional cytotoxic chemotherapy agents[1]. KIT and platelet-derived growth factor receptor-α (PDGFRA) mutations are present in 80% and 8% of GISTs, respectively[2-4]. Approximately 13% of KIT and PDGFRA wild-type GISTs contain BRAF mutations[5]. Although receptor tyrosine kinase inhibitors, such as imatinib or sunitinib, are therapeutically active antagonists of KIT and PDGFRA in KIT- or PDGFRA-mutated GIST[6-8], effective treatments for patients with advanced BRAF-mutant GIST have not been reported.
Clinical trials of tyrosine kinase inhibitors that are highly selective for V600 BRAF mutations have demonstrated high response rates (50-80%) in BRAF-mutant melanoma, as well as improvement in overall survival and progression-free survival[9-11]. Recently, we have shown that the BRAF inhibitor dabrafenib (GSK2118436) is also active in several non-melanoma BRAF-mutated cancers[10].
Herein, we report antitumor activity in the first patient with BRAF-mutated GIST who was treated with a BRAF inhibitor. Whole exome sequencing of tumor obtained at time of progressive disease did not reveal secondary BRAF or RAS mutations, but did demonstrate a somatic gain-of-function PIK3CA mutation (H1047R) as well as a CDKN2A aberration, which may have been responsible for dabrafenib resistance.
RESULTS
A 60 year old man initially presented in September 2007 with abdominal pain and a palpable mass. Computed tomography (CT) revealed a 10 cm heterogeneous mass, and a subsequent biopsy demonstrated GIST, spindled cell histology, positive for CD34 and CD117 by immunohistochemistry with 6 mitoses per 10 high-powered fields. The patient underwent surgical resection revealing a 15 cm mass. DNA was extracted from formalin-fixed paraffin-embedded tumor tissue and subjected to polymerase chain reaction (PCR) amplifications of KIT exons 9, 11, 13, and 17 as well as PDGFRA exons 12 and 18. Sanger sequencing did not identify mutations in either the KIT or PDGFRA genes. The patient presented with a new 14 cm mass at the dome of the bladder after 10 months of adjuvant imatinib therapy (400 mg once daily). The imatinib dose was increased to 800 mg daily, followed by surgical resection of the mass. The patient received adjuvant sunitinib, a multiple tyrosine kinase inhibitor, at a dose of 50 mg on a schedule of once daily for four weeks, then off for two weeks. Nineteen months later, a PET/CT showed recurrent FDG-avid masses in the right internal iliac region and in the right abdomen extending into the rectus abdominis.
The patient enrolled on a clinical trial with an investigational KIT/PDGFRA/VEGFR tyrosine kinase inhibitor, but disease progression was noted at his first restaging (two months of treatment). Further testing of the patient’s original tumor revealed a V600E BRAF mutation. The patient was then treated with an investigational MEK inhibitor for three months, during which the tumor initially remained stable but was subsequently found to have enlarged and remained enhancing by CT imaging.
The patient was treated on a phase I trial of dabrafenib at a dose of 150 mg twice daily[10]. The patient’s baseline CT scan demonstrated multiple metastases in the lower abdomen and pelvis, with the largest tumors including a 6.3 cm mass posterior to the bladder and a 6.3 cm mass in the anterior pelvis (Figure 1, Panel A). Using the Response Evaluation Criteria in Solid Tumors (RECIST) 1.0, restaging scans revealed a 14%, 18% and 20% decrease after 6, 15 and 24 weeks of treatment, respectively. Figure 1 Panel B demonstrates response on CT scan at 24 weeks. In addition, the tumor demonstrated a marked decrease in contrast enhancement, a response criteria that has been validated in GIST[12].
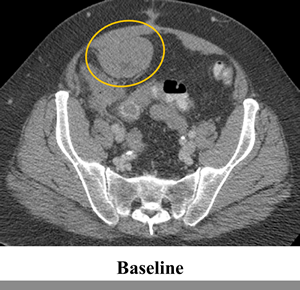
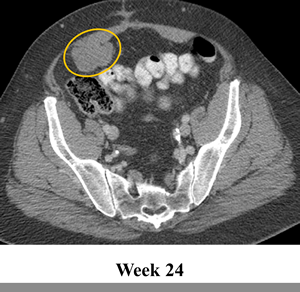
Figure 1: Tumor regression of 20% observed in abdominal and pelvic tumors on computerized tomography (CT). CT scan at (a) baseline and after (b) 24 weeks of treatment with BRAF inhibitor dabrafenib (GSK2118436).
The patient remained on study for 8 months, after which tumor progression was noted by contrast-enhanced CT imaging. The only treatment-related adverse events were grade 2 rash and acrochrodons (skin tags), as well as grade 1 fatigue and hyperkeratosis of the plantar surface of the feet. After tumor progression was identified, the patient underwent surgical resection of all visible tumors in the abdomen and pelvis. Tissue from this resection was evaluated with whole exome sequencing.
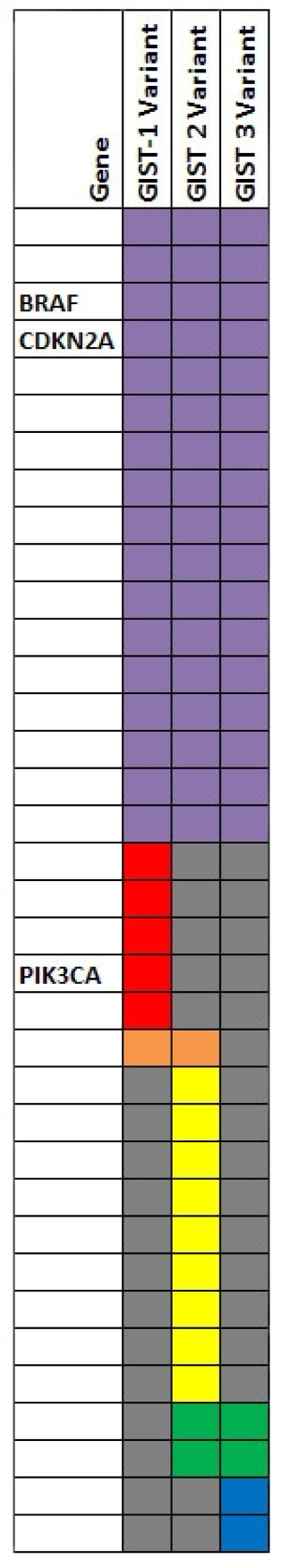
Figure 2: Three tumors were analyzed by whole exome sequencing.
To fully account for intratumor heterogeneity, which can be a factor in tumor adaptation and treatment failure[13], three lesions were analyzed by whole exome sequencing (Figure 2). All three lesions were clonally related as evidenced by identical BRAF V600E mutations, identical CDKN2A IVS1+1 G>A mutations, and fifteen other shared somatic single nucleotide variations. One of the three lesions (lesion 1), had a somatic gain-of-function PIK3CA mutation (H1047R), that has previously been reported in other human cancers[14]. Figure 3 demonstrates the PIK3CA H1047R mutation in lesion 1 (Panel A), in contrast to wild type PIK3CA in lesion 2 (Panel B), lesion 3 (Panel C), and normal tissue (Panel D). Lesions 2 and 3 appeared to be clonally related as they shared two mutations that were not present in lesion 1. Although all three lesions had a common CDKN2A mutation, lesions 1 and 3 were heterozygous for this mutation whereas lesion 2 was homozygous. This splice site mutation has been described previously as a somatic variant in melanoma[15] and glioma[16].
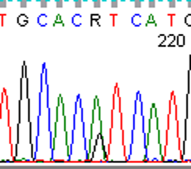
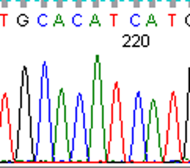
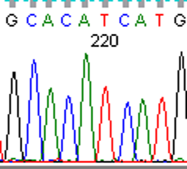
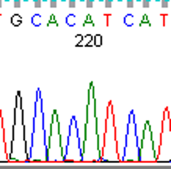
Figure 3: Lesion 1 (a) had a gain of function PIK3CA H1047R mutation while lesion 2 (b), lesion 3 (c), and normal tissue (d) were wild type for PIK3CA.
DISCUSSION
BRAF inhibitors have demonstrated antitumor activity in clinical trials of patients with BRAF mutant malignancies[9-11]. We report prolonged antitumor activity in the first patient with a BRAF-mutated GIST who was treated with a BRAF inhibitor.
Activating oncogenic mutations of BRAF have been described in many malignancies, including cutaneous melanoma (67%), colorectal carcinoma (12%), non-small cell lung carcinoma (NSCLC; 3%), and KIT wild-type GIST (13%)[5,17]. The most common BRAF mutation is a substitution of valine with glutamic acid at amino acid position 600 (V600E), which locks BRAF into its active conformation, resulting in a ten-fold increase in activity over wild-type BRAF[17].
Dabrafenib is a potent ATP-competitive inhibitor of BRAF kinase and is highly selective for mutant BRAF in kinase panel screening, cell lines, and xenografts[18]. Dabrafenib has demonstrated antitumor activity in several BRAF-mutated malignancies including melanoma, colorectal carcinoma, papillary thyroid carcinoma, NSCLC, and ovarian carcinoma[10].
Kinase inhibitors targeting BRAF have the potential to be an effective therapeutic option for BRAF-mutant GIST patients[10]. The present case demonstrates proof of principle for BRAF inhibition as a therapeutic strategy for GIST patients. Tumor regression was not seen when this patient was given a multi-kinase inhibitor that did not target BRAF, or a MEK inhibitor. However, it should be noted that both of these agents were experimental, and therefore their therapeutic value has not yet been fully validated. Treatment with dabrafenib, which targets BRAF directly, resulted in tumor regression after 6 weeks, and continued decreasing in size until week 24, followed by a plateau and then progression at 8 months.
Whole exome sequencing did not reveal secondary BRAF or RAS mutations but did demonstrate a somatic gain-of-function PIK3CA mutation (H1047R), that has previously been reported in other human cancers[14]. We speculate that the PIK3CA mutation could be the cause of the acquired BRAF inhibitor resistance in lesion 1. This finding is notable, because to the best of our knowledge this is only the second PIK3CA mutation ever reported in GIST[19]. Furthermore, although PIK3CA mutations have not previously been reported as a cause of acquired resistance to BRAF inhibitors in melanoma or other malignancies, low PTEN expression and other PTEN alterations are associated with lower response rate and shorter progression-free survival in BRAF mutant melanoma patients treated with BRAF inhibitors[20,21]. We further speculate that dysregulation of cell cycle control by the homozygous CDKN2A mutation in lesion 2 may also be a molecular basis for resistance of this lesion. No obvious explanation for resistance to BRAF inhibitor treatment was seen in lesion 3. We further tested RNA from all three lesions and were unable to detect aberrant BRAF splicing as a basis for drug resistance[22]. The differences in sequencing among the three lesions highlight the prevalence of intratumor heterogeneity and the potential relevance to treatment outcomes[13].
In conclusion, we present the first patient with GIST and a V600E BRAF mutation whose tumor showed regression while receiving treatment with a BRAF inhibitor. To our knowledge, the efficacy of BRAF inhibitors in BRAF-mutant GIST has not been reported, but our case suggests that additional studies and perhaps a global clinical trial are warranted.
MATERIALS AND METHODS
Whole exome capture was performed with a SeqCap EZ Human Exome v2.0 kit (Roche NimbleGen, Madison, WI), and sequencing was carried out on a HiSeq 2000 instrument (Illumina Inc, San Diego, CA). Sequence alignment and variant calling were performed with DNAnexus software (DNAnexus Inc, Mountain View, CA). Tumor-specific variants were identified based on a minimum variant allele ratio of 20%, a minimum read depth of 20, and absence of the variant in a matched normal specimen. Nucleotide variants were translated, and non-synonymous variants were identified using SIFT[23], PolyPhen2[24], and Mutation Assessor[25]. Variants of interest were confirmed by Sanger sequence analysis.
Acknowledgments
This work was performed in the U.T. MD Anderson Cancer Center Clinical and Translational Research Center (CTRC) and was supported by the Center for Clinical and Translational Sciences, which is funded by National Institutes of Health Clinical and Translational Science Award UL1 RR024148 and by the National Institutes of Health Cancer Center Support Grant (CCSG) award CA016672 to MD Anderson Cancer Center. Short read sequencing assays were performed by the Oregon Health & Science University Massively Parallel Sequencing Shared Resource. Funded in part by philanthropic donations from the GIST Cancer Research Fund and the Life Raft Group (MCH) and a VA Merit Review Grant (MCH). We would like to thank Dr. Vicki Goodman of GlaxoSmithKline for her comments on this manuscript.
Conflict of interest statement
Gerald Falchook received research funding and travel reimbursement for conference attendance from GlaxoSmithKline. Razelle Kurzrock received research funding from GlaxoSmithKline, Novartis, and Pfizer. Samuel Blackman was an employee of GlaxoSmithKline at the time of the study. The other coauthors report no conflicts of interest.
References
1. Trent JC, Beach J, Burgess MA, Papadopolous N, Chen LL, Benjamin RS, Patel SR. A two-arm phase II study of temozolomide in patients with advanced gastrointestinal stromal tumors and other soft tissue sarcomas. Cancer. 2003; 98(12):2693-2699.
2. Gomes AL, Bardales RH, Milanezi F, Reis RM, Schmitt F. Molecular analysis of c-Kit and PDGFRA in GISTs diagnosed by EUS. Am J Clin Pathol. 2007; 127(1):89-96.
3. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003; 299(5607):708-710.
4. Steigen SE, Eide TJ, Wasag B, Lasota J, Miettinen M. Mutations in gastrointestinal stromal tumors--a population-based study from Northern Norway. APMIS. 2007; 115(4):289-298.
5. Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, Bringuier PP, Scoazec JY, Coindre JM. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010; 133(1):141-148.
6. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Henrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347(7):472-480.
7. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006; 368(9544):1329-1338.
8. McAuliffe JC, Hunt KK, Lazar AJ, Choi H, Qiao W, Thall P, Pollock RE, Benjamin RS, Trent JC. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol. 2009; 16(4):910-919.
9. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364(26):2507-2516.
10. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, Lebowitz P, Ma B, Ouellet D, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012; 379(9829):1893-1901.
11. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363(9):809-819.
12. Choi H, Charnsangavej C, de Castro Faria S, Tamm EP, Benjamin RS, Johnson MM, Macapinlac HA, Podoloff DA. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR Am J Roentgenol. 2004; 183(6):1619-1628.
13. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366(10):883-892.
14. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304(5670):554.
15. Casula M, Muggiano A, Cossu A, Budroni M, Caraco C, Ascierto PA, Pagani E, Stanganelli I, Canzanella S, Sini M, Palomba G, Palmieri G. Role of key-regulator genes in melanoma susceptibility and pathogenesis among patients from South Italy. BMC Cancer. 2009; 9:352.
16. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol. 2009; 11(4):341-347.
17. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417(6892):949-954.
18. Laquerre S, Arnone M, Moss K, Yang J, Fisher K, Kane-Carson LS, Smitheman K, Ward J, Heidrich B, Rheault T, Adjabeng G, Hornberger K, Stellwagen J, Waterson A, Han C, Mook RA, Jr, et al. A selective Raf kinase inhibitor induces cell death and tumor regression of human cancer cell lines encoding B-RafV600E mutation. Mol Cancer Ther. 2009; 8(12, Suppl 1):abstr B88.
19. Daniels M, Lurkin I, Pauli R, Erbstosser E, Hildebrandt U, Hellwig K, Zschille U, Luders P, Kruger G, Knolle J, Stengel B, Prall F, Hertel K, Lobeck H, Popp B, Theissig F, et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011; 312(1):43-54.
20. Sosman JA, Pavlick AC, Schuchter LM, Lewis KD, McArthur GA, Cowey CL, Moschos SJ, Flaherty KT, Kim KB, Weber J, Hersey P, Long GV, Lawrence MK, Spleiss O, Koehler A, Bollag G, et al. Analysis of molecular mechanisms of response and resistance to vemurafenib (vem) in BRAFV600E melanoma. J Clin Oncol. 2012; 30(Suppl):504s, abstr 8503.
21. Nathanson KL, Martin A, Letrero R, D’Andrea KP, O’Day S, Infante JR, Falchook GS, Millward M, Curtis CM, Ma B, Gagnon RC, Lebowitz PF, Long GV, Kefford RF. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor GSK2118436 (GSK436). J Clin Oncol. 2011; 29(15S):526s abstract 8501.
22. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011; 480(7377):387-390.
23. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4(7):1073-1081.
24. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7(4):248-249.
25. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39(17):e118.