
INTRODUCTION
Breast cancer, a major health concern worldwide [1], exhibits disparate clinical behaviors and patient outcomes [2]. Ductal carcinoma in situ (DCIS) is a non-invasive breast lesion that accounts for 10-25% of all breast neoplasms; DCIS is not life-threatening but a risk factor for, and potential precursor of, invasive cancers. The most common histological type of breast cancers, accounting for ~80% of the cases, is invasive ductal carcinoma (IDC). IDC has been subgrouped based on immunohistochemical detection of several markers such as estrogen receptor (ER), progesterone receptor (PR), and epidermal growth factor receptor 2 (HER2). A large fraction (70–75%) of IDCs are positive for ER and classified as luminal cancers. A small fraction of luminal cancers are positive for HER2 and called luminal HER2. PR expression usually follows the ER expression but can be low in some cases where prognoses tend to be poorer. Two major subtypes, besides luminal cancers, are HER2-enriched and triple-negative (ER-/PR-/HER2-) [2]. HER2-enriched cancers show amplification and high expression of the ERBB2 gene and several other genes of the ERBB2 amplicon. Breast cancer classification based on gene expression profiles has also been proposed [3]. Such molecular classifications of cancers are of great clinical importance, since they may provide molecular bases for predicting the tumors’ prognoses and/or responses to therapy [2].
Altered patterns of DNA methylation are found in cancer cells, and their values as potential biomarkers for diagnoses and prognoses have been explored [4]. For example, attempts to correlate genome-wide DNA methylation patterns in breast cancers and their prognoses have been made [5–7]. Since epigenetic silencing of tumor suppressor genes is involved in carcinogenesis, inhibitors of certain epigenetic regulators, such as DNA methyl transferases (DNMTs) and histone deacetylases (HDACs), have been tested in cancer therapy with promising results [8–10]. In fact, two DNMT inhibitors, 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine), have already been approved by FDA for treatment of myelodysplastic syndrome [11, 12].
RECK, initially isolated as a transformation suppressor gene by cDNA-expression cloning, encodes a GPI-anchored glycoprotein with activities to downregulate or inhibit multiple metalloproteinases [13, 14]. Reduced RECK expression is found in various types of tumors, and the levels of residual RECK expression in tumors tend to correlate with better prognoses [15, 16]. In gene-focused studies, extensive methylation of RECK CpG island has been found in cancers of the colon, stomach, liver, and lung, and its correlation with poorer prognoses detected [17–22]. Importantly, in a non-biased, genome-wide methylation study, Hill and colleagues detected RECK as one of several loci whose methylation inversely correlated with relapse-free survival among breast cancer patients [5]. They employed an assay called Combined Bisulfite Restriction Analysis (COBRA) [23] for rapid detection of CpG methylation; in the case of RECK, they used a set of PCR primers amplifying an intron-1 region of its CpG island (see Figure 1A, bottom line). We refer to the methylation of this region “RIM” (RECK Intron-1 Methylation) in this paper to distinguish it from a newly examined region (see below).
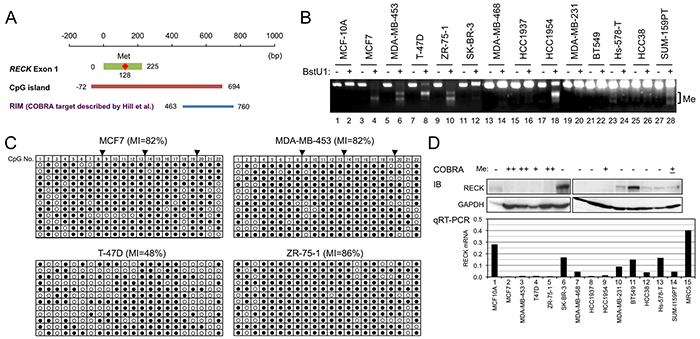
Figure 1: RECK methylation and expression in breast cancer cell lines. A. Positions of exon-1 (green line), first methionine (red diamond), CpG island (orange line), and the COBRA target region described by Hill et al. (RIM, blue line) [5] in which three BstU1 sites (vertical lines) exist. Nucleotide sequence of this region is shown in Supplementary Figure S1. B. RECK CpG methylation (RIM) in 14 cell lines detected by COBRA. Appearance of smaller bands after BstU1-digestion (even number lanes) indicates methylation of the BstU1 site (CGCG). C. RIM determined by clone-sequencing. The PCR products used for COBRA were cloned into bacterial plasmids, and 12 independent plasmid clones (represented by each line) sequenced. Methylation status of 22 CpG di-nucleotides (column 1-22) within the amplified area is shown using open (non-methylated) or filled circles (methylated). Solid triangles on the top indicate BstU1 sites (Supplementary Figure S2, red triangles). D. The levels of RECK protein detected by immunoblot assay (IB; upper autoradiographs) and RECK mRNA detected by qRT-PCR (lower bar graph). GAPDH was used as an internal control in IB; RNA from normal human fibroblasts (MRC5) was used as a positive control in qRT-PCR. Top line indicates the RIM status estimated from B and C.
Involvement of DNMT3B in oncogene-induced RECK gene silencing [24] and induction of RECK expression by DNMT inhibitors [17, 20, 21, 24–26] have been reported. RECK activation by an HDAC inhibitor, trichostatin A (TSA), has also been observed in several systems [27–32], suggesting the involvement of histone modifications. Besides these epigenetic regulations, multiple other mechanisms of RECK downregulation, such as signaling triggered by growth factors, cell density, oncoproteins, hypoxia, and post-transcriptional inhibition by microRNAs, have also been reported [29, 30, 32–34].
In the present study, we designed a new set of primers amplifying a region of RECK CpG island containing the proximal promoter and exon-1 (see Figure 3A, bottom line); we refer to the methylation of this region “RPM” (RECK Promoter/exon-1 Methylation). Our data indicate the utility of RIM and RPM as distinctive molecular markers useful for sub-grouping luminal breast cancers and their potential values in predicting the prognoses and drug-sensitivity of breast cancers.
RESULTS
RECK CpG methylation in breast cancer cell lines
Focusing on the intro-1 region of the RECK CpG island, Hill and colleagues found a significant inverse correlation between its methylation (RIM) in tumor tissues and relapse-free survival of the patients [5]. Their data also suggested the prevalence of RIM in the tumors positive for ER (P=0.013) and those at relatively early stages (P=0.022) (Supplementary Table S2).
To confirm and extend these important findings, we applied the same COBRA protocols to analyze RIM in 16 cell lines, one derived from normal mammary epithelium (MCF-10A) and 15 from breast cancers of various subtypes (Figure 1B; Supplementary Figure S1). RIM was detected in 8 of 15 cancer-derived cell lines (53%) but not in MCF-10A (Table 1). The COBRA results were confirmed by sequencing individual PCR products cloned into a bacterial plasmid (Figure 1C and data not shown). The pattern of RIM distribution among breast cancer subtypes is largely recapitulated in this small set of cell lines: significant enrichment of RIM in luminal cancers (5 of 5) and its paucity in triple negative cancers were detected (1 of 7; Table 1A, 1B, “RIM” column). RECK expression (both mRNA and protein) is low in cell lines positive for RIM (Figure 1D), suggesting the involvement of RIM in RECK silencing. These data also suggest the value of these cell lines in studying how RIM affects RECK expression and the properties of breast cancer cells.
Table 1: Summary of data on breast cancer-derived cell lines
A |
|||||||
|---|---|---|---|---|---|---|---|
Cell line |
Subtype |
Medium1) |
ER |
PR |
HER2 |
RPM (%) |
RIM (%) |
MCF10A |
Normal |
M |
- |
- |
- |
- |
- |
MCF7 |
Luminal |
D |
+ |
+ |
- |
1.1 |
82 |
T-47D |
Luminal |
R |
+ |
+ |
- |
55 |
48 |
ZR-75-1 |
Luminal |
R |
+ |
+/- |
- |
90 |
86 |
KPL-1 |
Luminal |
R |
+ |
- |
- |
- |
+2) |
KPL-3C |
Luminal |
R |
+ |
- |
- |
- |
+2) |
MDA-MB-453 |
HER2 |
D |
- |
- |
+ |
90 |
82 |
SK-BR-3 |
HER2 |
R |
- |
- |
+ |
- |
- |
HCC1954 |
HER2 |
R |
- |
- |
+ |
0.9 |
74 |
MDA-MB-468 |
TN |
R |
- |
- |
- |
- |
- |
HCC1937 |
TN |
D (HG) |
- |
- |
- |
- |
- |
MDA-MB-231 |
TN |
D |
- |
- |
- |
0.3 |
- |
BT549 |
TN |
R |
- |
- |
- |
- |
- |
HCC38 |
TN |
R |
- |
- |
- |
- |
- |
Hs578-T |
TN |
DI |
- |
- |
- |
- |
- |
SUM-159PT |
TN |
F |
- |
- |
- |
- |
17 |
1) M, MEGM; D, DME + 10% FBS, R, RPMI + 10% FBS; HG, high glucose; DI, DME + 10% FBS, 10 μg/ml bovine insulin; F, Ham’s F12 + 5%FBS, 5 μg/ml insulin,1 μg/ml hydrocortisone,10 mM HEPES
2) Analyzed only by COBRA (See Supplementary Figure S1).
B |
||||||
|---|---|---|---|---|---|---|
Sub-total |
RPM |
RIM |
||||
n |
Fraction (%) |
n |
P |
n |
P |
|
Normal |
1 |
6 |
0 |
NS (0.63) |
0 |
NS (0.32) |
Luminal |
5 |
31 |
2 |
NS (0.22) |
5 |
0.025 |
HER2 |
3 |
19 |
1 |
NS (0.52) |
2 |
NS (0.56) |
TN |
7 |
44 |
0 |
NS (0.20) |
1 |
NS (0.059) |
Total |
16 |
100 |
3 |
8 |
||
P was assessed by Pearson’s chi-squared test. NS, not significant
Effects of DNMT inhibitor and HDAC inhibitor on RECK expression
To test the effects of CpG methylation on RECK expression, we treated some of these cell lines with a DNMT inhibitor, 5-aza-2′-deoxycytidine (5-azadC), and determined the levels of RECK mRNA and protein. We also examined the effects of an HDAC inhibitor, MS275, which we found to induce RECK expression in some other cell lines, such as those derived from human fibrosarcoma (Yoshida et al., unpublished). First, we confirmed the effects of 5-azadC to reduce RIM in three luminal cell lines (MCF7, T-47D, ZR-75-1) by COBRA; the increased intensity of full-length band and decreased intensity of smaller bands in BstU1-digested samples after 5-azadC-treatment (Figure 2A–2C, compare lane 4 to lane 2) indicate successful DNA demethylation. MS275, on the other hand, exhibited little effects on RIM (Figure 2A–2C, compare lane 6 to lane 2). In these cell lines, 5-azadC alone (namely, demethylation alone) showed little effect on RECK expression (Figure 2D–2F, bar/lane 2). In two RIM-positive cell lines, T-47D and ZR-75-1, combination of two inhibitors (5-azadC and MS275) was effective in activating RECK expression (Figure 2E, 2F, bar/lane 4) while MS275 alone was not so effective (Figure 2E, 2F, bar/lane 3). In a RIM-negative cell line, MDA-MB-231, MS275 alone was sufficient to induce robust RECK expression (Figure 2G), supporting the idea that RIM may be useful in predicting whether or not demethylation is prerequisite for MS275-induced RECK expression. The response of RECK in the RIM-positive MCF7 cells, however, did not support this idea; MS275 could upregulate RECK mRNA to some extent but 5-azadC failed to enhance this effect in MCF7 (Figure 2D, bar/lane 3, 4). Survey of additional cell lines yielded another example of RIM-positive cell line, HCC1954, in which RECK could be upregulated by MS275 alone (Figure 2H) although the methylation seems to be incomplete in this case (Figure 1B, lane 18).
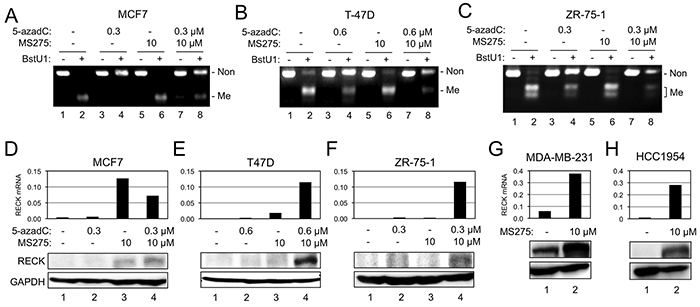
Figure 2: Effects of 5-azadC and MS275 on RIM and RECK expression. A–C. RIM detected by COBRA in three breast cancer cell lines [MCF7 (A), T-47D (B), or ZR-75-1 (C)] after incubation under indicated conditions. D–F. Effects of 5-azadC and/or MS275 on the level of RECK mRNA detected by qRT-PCR (upper bar graph) and RECK protein detected by immunoblot assay (lower autoradiographs) in MCF7 (D), T-47D (E), or ZR-75-1 (F). G, H. Effects of MS275 on the level of RECK mRNA and protein in two breast cancer cell lines: MDA-MB-231 (G) and HCC1954 (H).
RECK promoter/exon-1 methylation (RPM) in breast cancer cell lines
Why MS275 could upregulate RECK expression in some RIM-positive cells but not others? To test the possibility that we were focusing on an area inappropriate for predicting cell’s response to MS275, we designed a new set of COBRA primers to monitor methylation of more upstream region of RECK CpG island (RPM; Figure 3A, bottom line). Among the 15 breast cancer cell lines, three (T-47D, ZR-75-1, MDA-MB-453) were positive for RPM (Figure 3B; Table 1); the results were confirmed by clone-sequencing (Figure 3C). As expected, RPM could be reduced by 5-azadC in these cell lines (Figure 3D–3F, compare lane 4 to lane 2) but not by MS275 (Figure 3D–3F, compare lane 6 to lane 2).
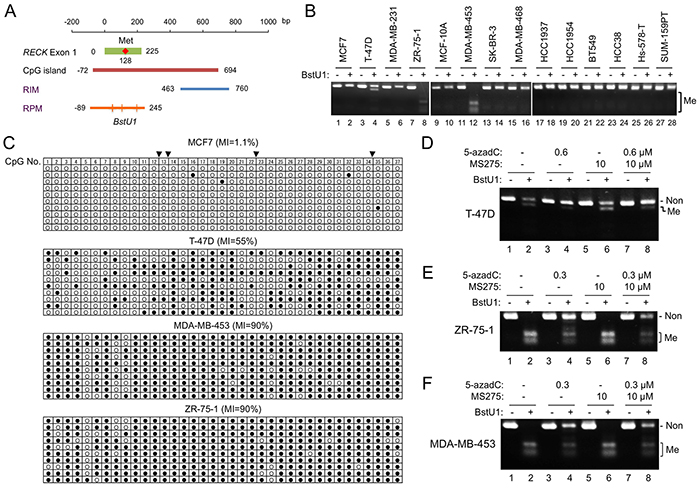
Figure 3: RECK gene methylation detected by new COBRA primers. A. Position of RPM (orange line) with respect to the RECK CpG island (brown line) and the original COBRA target (RIM; blue line). Four BstU1 sites (vertical lines), two of which are tandem, exist within the RPM region. Nucleotide sequence of this region is shown in Supplementary Figure S3. B. RPM status in 14 cell lines detected by COBRA. C. RPM status in four cell lines determined by clone-sequencing. Methylation status of 37 CpG di-nucleotides (column 1-37) within the amplified area is shown. Solid triangles on the top indicate BstU1 sites (Supplementary Figure S3, red triangles). D–F. Effects of 5-azadC and/or MS275 on RPM status detected by COBRA in T-47D (D), ZR-75-1 (E), and MDA-MB-453 (F). Note the increased full-length band (representing non-methylated allele) and decreased smaller bands (methylated allele) in BstU1-digested samples (even-numbered lanes) after treatment with 5-azadC (compare lanes 4, 8 to lane 2) but not with MS275 (compare lane 6 to lane 2). Non, band representing to non-methylated DNA; Me, bands representing methylated DNA; MI, ratio of methylated CpG to all CpG.
Importantly, the extent of RPM at the sequence level (Figure 3C) differs dramatically between MCF7 (1.1%) and two other luminal cell lines, T-47D (55 %) and ZR-75-1 (90%). The other MS275-responsive cell line HCC1954 (Figure 2H) was also RPM-negative (Figure 3B, lane 20). Thus, RPM better predicts the cells’ response to MS275 than RIM. In this small set of cell lines, however, no significant association of RPM to certain tumor subtype could be detected (Table 1B, “RPM” column).
RPM and RIM in breast cancer tissues
To better understand the biological and clinical significance of above findings in cell lines, we compared RPM, RIM, and the level of RECK mRNA in tissue samples from 62 breast cancer patients (results summarized in Figure 4 and Table 2; representative data shown in Supplementary Figures S4 and S5). Several findings of potential importance were noted. First, comparison between matched pairs of normal and tumor tissues (n=13) indicated significant downregulation of RECK mRNA (P=0.001; Figure 4A) and prevalence of RECK CpG methylation, both RPM and RIM, in tumor tissues (Figure 4B, bar 2, 4). Second, when 62 tumor samples were divided into two groups based on RECK methylation status (RPM or RIM), the average level of RECK mRNA was lower in the RIM+ group than the RIM- group, although the difference was not statistically significant (P=0.055; Figure 4C, bar 3, 4). RECK mRNA was relatively abundant in luminal tumors (Supplementary Figure S6A), and within this subtype, significant correlation between RIM and lower RECK expression was detected (P=0.041; Figure 4C, bar 5). Third, as we found in cell lines (Table 1B, 1A), frequency of RIM among tumor samples (33 of 62 cases; 53%) was higher than that of RPM (16 of 62; 26%), and majority of RPM+ tumors (15 of 16; 94%) were also RIM+ (Figure 4D). Fourth, both RPM and RIM were prevalent in luminal/ER+ tumors (Figure 4E; Table 2). Fifth, RECK CpG methylation (both RPM and RIM) was negative, althogh RECK mRNA was minimal, in DCIS (n=6; Table 2 ; Supplementary Figure S6A). Sixth, all the ER+PR- tumors were positive for RIM (10 of 10; Table 2). Seventh, RECK CpG methylation, especially RIM, correlates with higher tumor stages and histological grades (Table 2), supporting the idea that the RECK silencing (associated with RIM) contributes to the malignant behaviors of breast cancer cells.
Table 2: Clinicopathological features and methylation at two regions of RECK CpG island (RPM and RIM) in breast cancer tissues
n |
RPM |
RIM |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Number |
Ratio |
P |
Number |
Ratio |
P |
||||||
+ |
- |
RPM+/n (%) |
+ |
- |
RIM+/n (%) |
||||||
Total |
62 |
16 |
46 |
26 |
33 |
29 |
53 |
||||
Age |
<55 |
20 |
4 |
16 |
20 |
NS (0.47) |
10 |
10 |
50 |
NS (0.73) |
|
>55 |
42 |
12 |
30 |
29 |
23 |
19 |
55 |
||||
Subtype |
DCIS |
6 |
0 |
6 |
0 |
NS (0.13) |
0 |
6 |
0 |
0.00014 |
|
Lum (ER+) |
PR+ |
28 |
9 |
19 |
32 |
16 |
12 |
57 |
|||
PR- |
10 |
4 |
6 |
40 |
10 |
0 |
100 |
||||
HER2 |
5 |
2 |
3 |
40 |
4 |
1 |
80 |
||||
TN |
15 |
1 |
14 |
7 |
3 |
12 |
20 |
||||
ER |
+ |
40 |
15 |
25 |
38 |
0.014 |
29 |
11 |
73 |
0.00087 |
|
- |
16 |
1 |
15 |
6 |
4 |
12 |
25 |
||||
PR |
+ |
30 |
10 |
20 |
33 |
NS (0.32) |
19 |
11 |
63 |
NS (0.27) |
|
- |
26 |
6 |
20 |
23 |
14 |
12 |
54 |
||||
HER2-enrichied |
+ |
5 |
2 |
3 |
40 |
NS (0.44) |
4 |
1 |
80 |
NS (0.19) |
|
- |
51 |
14 |
37 |
27 |
29 |
22 |
57 |
||||
Menopause |
Pre |
17 |
4 |
13 |
24 |
NS (0.80) |
9 |
8 |
53 |
NS (0.98) |
|
Post |
45 |
12 |
33 |
27 |
24 |
21 |
53 |
||||
T stage |
Tis |
7 |
1 |
6 |
14 |
0.037 |
1 |
6 |
14 |
0.018 |
|
T1/2 |
47 |
10 |
37 |
21 |
25 |
22 |
53 |
||||
T3/4 |
8 |
5 |
3 |
63 |
7 |
1 |
88 |
||||
Nodal status |
+ |
28 |
5 |
23 |
18 |
NS (0.10) |
15 |
13 |
54 |
NS (0.48) |
|
- |
27 |
10 |
17 |
37 |
17 |
10 |
63 |
||||
Distant Metastasis |
+ |
3 |
1 |
2 |
33 |
NS (0.61) |
3 |
0 |
100 |
NS (0.14) |
|
- |
53 |
15 |
38 |
28 |
30 |
23 |
57 |
||||
Grade |
DCIS |
6 |
0 |
6 |
0 |
NS (0.15) |
0 |
6 |
0 |
0.0055 |
|
1 |
13 |
2 |
11 |
15 |
5 |
8 |
38 |
||||
2/3 |
43 |
14 |
29 |
33 |
28 |
15 |
65 |
||||
P was assessed by Pearson’s chi-squared test. NS, not significant; Tis, tumor in situ
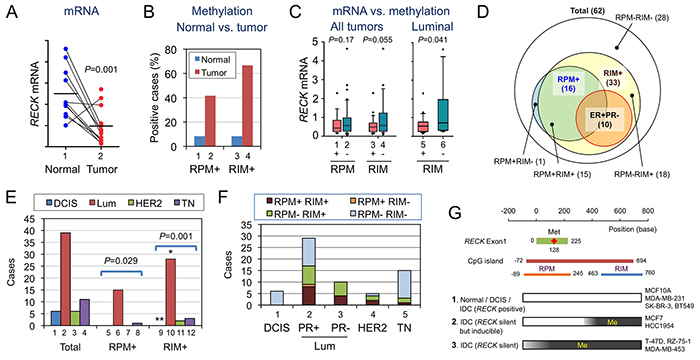
Figure 4: Statuses of RECK mRNA, RPM, and RIM in human breast tissues. A. RECK mRNA in normal and tumor tissues from the same patients (n=14). B. Proportions of RPM+ and RIM+ cases among the normal (blue) and tumor (red) tissues. C. Relationship between the status of RPM or RIM and the level of RECK mRNA. D. Relationship among breast cancer subgroups exhibiting different RPM/RIM statuses. Their relationship with ER+PR- tumors is also shown. E. Distribution of molecular subtypes [DCIS, luminal (Lum), HER2-enriched (HER2), and triple negative (TN)] among total (bars 1-4), RPM-positive (bars 5-8), or RIM-positive (bars 9-12) breast cancer cases. F. Distribution of tumors with different RPM/RIM statuses in each molecular subtype. G. Three classes of cells with different RPM/RIM statuses. [1] When RPM and RIM are negative, RECK is likely to be expressed, albeit at lower levels in tumors. [2] When methylation is confined to RIM, RECK is silent but inducible by MS275. [3] When both RPM and RIM are positive, RECK is silent and insensitive to MS275 unless the CpG island has been demethylated. Some cell lines of each class are listed on the right-hand side.
Effects of MS275 on RECK promoter
How does MS275 upregulate RECK in RPM-negative breast cancer cells? We confirmed that MS275 increased the level of acetylated histones, as monitored using acetylated H4 (Ac-H4), in three cell lines: MCF7 (RPM-RIM+), T-47D (RPM+RIM+), and MDA-MB-231 (RPM-RIM-) (Supplementary Figure S7B–S7D, lanes 1, 2). As detected by chromatin immunoprecipitation (ChIP) assay, MS275 increased the amount of Ac-H4 bound to three regions (termed P1, P2, and E1; Supplementary Figure S7A) upstream of, or containing, RECK exon-1 (Supplementary Figure S7B–S7D, lanes 5, 6). The increase (especially in E1) seems to be modest in T-47D cells (RPM+) (Supplementary Figure S7C, lanes 5, 6). Second, association of HDAC1, a known target of MS275, to the RECK promoter was confirmed by ChIP assay (Supplementary Figure S7E, S7F). Relative intensity of 3 ChIP bands differed between cell lines: P1 was most intense in T-47D while E1 was most intense in MDA-MB-231, although biological significance of this difference is unclear. Third, knockdown of HDAC1 using two siRNAs (#1, #2), resulted in RECK-upregulation in MDA-MB-231 cells (Supplementary Figure S7G, lanes 4, 5). These data are consistent with the idea that MS275 upregulates RECK (in RPM-negative breast cancer cell lines) by inhibiting HDAC1, thereby increasing the acetylation of its targets.
Biological significance of RECK-inducibility
RPM was inversely correlated with the cells’ ability to express RECK in response to MS275 (Figure 2; Table 1). Is there any biological significance or clinical relevance in this finding? To address this issue, we first assessed the effects of MS275, 5-azadC, or both on the growth/surival of T-47D cells (RPM+RIM+; Figure 5A, 5B). Although the growth was suppressed to some extent by treatment with MS275 alone or 5-azadC alone (Figure 5A, blue and red lines; Figure 5B-2, 3), almost complete cell killing was observed only after combined treatment (Figure 5A, green line; Figure 5B-4).
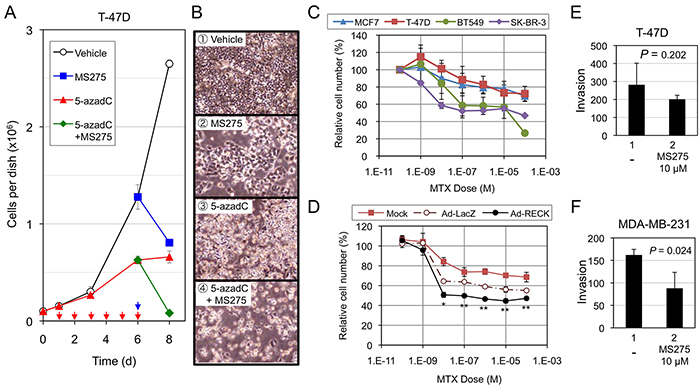
Figure 5: Effects of RECK-activation on the growth/survival of breast cancer cell lines. A, B. Effects of MS275, 5-azadC, or both on the growth/survival of T-47D cells. The cells (1×105/35-mm dish) plated on day 0 were incubated from day 1 to day 6 in growth medium or the medium containing 5-azadC (0.6 μM) with daily medium changes (red arrows), and then exposed to the same medium without or with MS275 (10 μM; blue arrow) from day 6 to day 8. Cell number was estimated by SF assay on day 1, 3, 6, and 8. Bar graph (A) represents mean ± s.e.m. (n=3). Representative micrographs at day 8 are also shown (B). C. Effects of MTX on four breast cancer cell lines. The cells were exposed to indicated concentrations of MTX for 48 h, and their number estimated by SF assay. The data represent mean ± s.e.m. (n=3). D. Effects of acute RECK expression on MTX-sensitivity of T-47D cells. T-47D cells were kept uninfected (Mock) or infected (MOI=600) with either control adenovirus (Ad-LacZ) or RECK-expressing virus (Ad-RECK). After 24 h, the cells were replated onto 96-well plates (1 × 104 cell/well), incubated for 16 h, exposed to MTX at various concentrations (0-10−4 M; triplicate) for 48 h, and then subjected to SF assay. Data (mean±SD) are presented as their ratios to the vehicle-treated controls. E, F. Effects of MS275 on Matrigel invasion of T-47D (E; 20-h incubation) or MDA-MB-231 cells (F; 16-h incubation). The graph represents mean±s.e.m. (n=3).
Next, we treated four cell lines that exhibit different RECK methylation patterns [T-47D (RPM+RIM+), MCF7 (RPM-RIM+), BT549 and SKBR3 (RPM-RIM-)] with a RECK-activating anticancer drug, methotrexate (MTX) [35]. T-47D (RPM+) showed lowest sensitivity to this drug (Figure 5C; red line). Acute expression of RECK using adenoviral vector made T-47D cells susceptible to MTX (Figure 5D, black line), suggesting possible contribution of RECK in MTX-induced growth inhibition.
Finally, we subjected two cell lines, T-47D and MDA-MB-231, to Matrigel invasion assay in the absence or presence of MS275 (Figure 5E, 5F) and found significant suppression of invasion by MS275 in MDA-MB-231 cells (RPM-; Figure 5F) but not in T-47D cells (RPM+; Figure 5E). Taken together, these results support the idea that RECK-inducibility, as predicted by RPM, may be useful in predicting effectiveness of certain drugs, especially those activating RECK expression, in suppressing malignant growth and behaviors of breast cancer cells.
DISCUSSION
Previous study by Hill et al. [5] revealed a strong correlation between RIM found in tumor tissue and relapse of the disease, which underscored the critical role for RECK in tumor suppression. Practical value of RIM as a prognostic marker, however, depends on the availability of therapeutic options for subgroups of the disease to be identified, especially the one with higher risk. RPM developed in this study may be of value in this respect, since it provides information on drug sensitivity. For instance, RIM was positive in all cases (n=10) of ER+PR- tumors examined in this study, while RPM was positive only in 4 cases (Table 2). Similarly, a half of all RIM-positive luminal tumors (n=26) were also RPM-positive. According to our findings in vitro (Figure 2), treatment with both 5-azadC and MS275 may be required to upregulate RECK in RPM-positive tumors whereas MS275 alone may be sufficient to induce RECK in RPM-negative tumors. Although the exact biological responses of these particular tumors are unknown, our data in vitro (Figure 5) as well as several previous studies indicate its beneficial effects (tumor suppression). Hence, the tumors that require extensive therapies (about a half of RIM-positive cases) may be identified using RPM, in conjunction with RIM and conventional molecular markers (ER, PR, and HER2).
Two lines of new evidence indicate the involvement of CpG methylaiton, especially RIM, in RECK silencing. First, RIM was inversely correlated with the level of RECK mRNA/protein among the cell lines (Figure 1; Table 1) and tissue samples (Figure 4A–4C). Second, 5-azadC could convert RIM+ cell lines susceptible to the activity of MS275 to induced RECK expression (Figure 2). This mechanism of silencing, however, probably operate only in later stages of mammary carcinogenesis, since RIM was negative in MCF-10A (Figure 1B), most normal tissues (Figure 4B), and DCIS (Table 2) while it was more frequent in high-grade carcinomas (28 of 43; 65%; Table 2).
ER+PR- breast cancers are known to be refractory to anti-estrogen treatment and prone to relapse [36]. A recent study indicate that progesterone inhibits estrogen-induced growth of ER+ breast tumors and that copy number loss of the PR gene is not rare among ER+ breast cancers [37]. The use of DNMT inhibitors in conjunctions with other therapeutic agents, such as anti-estrogens, Cdk inhibitors [38], HDAC inhibitors, or other RECK-activating agents [35], may have potential value in treating such tumors.
Our data (Supplementary Figure S7) suggest the involvement of HDAC1 in RECK gene repression. Our data also suggest the dominant nature of CpG methylation over this repression (Figure 2). We interpret that although both RPM and RIM are involved in RECK gene silencing, RPM probably represent a higher degree of silencing than RIM alone (Figure 4G). The RPM area spans the exon-1 and proximal promoter which harbors numerous potential cis-regulatory elements and has a feature of “CpG island shore” in which the degree of methylation can be influenced by external stimuli, tissue of origin, etc. [4, 39]. This may explain why RPM status varies among RIM-positive tumors and better predicts the cells’ ability to express RECK in response to MS275. It remains unknown, however, why two groups (RPM+ and RPM-) of RIM-positive luminal cancers arise.
MCF7 and T-47D are commonly used cell lines derived from luminal breast cancer. We found, however, that they differed in the pattern of RECK CpG methylation and RECK inducibility by chemicals. Molecular mechanisms by which these differences arise are important issues to be addressed in future studies. Some RIM-negative cell lines show different basal levels of RECK expression (Figure 1D). For instance, in MDA-MB-231, basal RECK expression was moderate (Figure 1D, lane/bar 10) and further upregulated by MS275 (Figure 2H), resulting in reduced Matrigel invasion (Figure 5F). Effects of MS275 and other HDAC inhibitors on RECK-expression in, and the behaviors of, other RIM-negative cells may also yield important insights. Studies on the mechanism of action of MS275 in RECK-upregulation may also provide some clues to better understanding how RECK can be repressed in cancer cells.
MATERIALS AND METHODS
Cell culture
The mammary epithelial cell line (MCF-10A) and 13 breast cancer cell lines (MCF7, MDA-MB-453, T-47D, ZR-75-1, SK-BR-3, MDA-MB-468, HCC1937, HCC1954, MDA-MB-231, BT549, Hs-578-T, HCC38, SUM-159PT) were obtained from ATCC. Two breast cancer cell lines, KPL-1 and KPL-3C, were described previously [40, 41]. Growth media used for maintainance of these cell lines are shown in Table 1 and were supplemented with fetal bovine serum (FBS; 5% for SUM-159PT and 10% for others).
Tissue samples
Breast cancer tissues from primary tumor sites were collected through needle biopsy or surgical resection performed in Department of Breast Surgery, Kyoto University Hospital. Prior written informed consents were obtained from all patients. The study protocols were approved by the Ethics Committee for Clinical Research, Kyoto University Hospital (authorization numbers G424). Obtained sample was quickly frozen in liquid nitrogen and stored under −80°C or liquid nitrogen until analyses. Samples collected between May 2011 and June 2014 were used in this study. Pathological characterization was performed as described previously [42]. For molecular analyses, frozen tissues were homogenized using Bead Smash 12 (Waken-yaku, Kyoto, Japan), and the total DNA and RNA was extracted using QIAamp DNA Mini Kit (QIAGEN) and RNeasy Mini Kit (QIAGEN), respectively.
Methylation analysis of RECK gene
COBRA [23] was used to assess the methylation status of RECK gene. Bisulfite conversion of genomic DNA was performed using EpiTect Bisulfite kit (Qiagen). Primers for semi-nested PCRs are listed in Supplemental Table S1. Primary PCRs (reaction volume, 25 μl) were performed under the following conditions: 40 cycles of PCR [94°C, 30 s; 50°C, 30 s; 72°C, 60 s] followed by a final extension at 72°C for 5 min. Secondary PCRs were performed using 3 μl of the first PCR product as a template as follows: 45 cycles of PCR [94°C, 30 s; 53°C, 30 s; 72°C, 60 s] followed by a final extension at 72°C, 5 min. PCR products were digested with BstUI (Fermentas, UK) at 37°C for 4 h and resolved by agarose gel electrophoresis (1.5% agarose). For clone-sequencing, PCR products were cloned into pTA2 vector (TOYOBO), and 12 independent clones isolated were sequenced using T3 and T7 primers. Methylation index is defined as the ratio (%) of the number of methylated CpGs to the number all CpGs present in the fragment.
Quantitative reverse transcription-PCR
Total RNA was extracted using RNeasy Mini Kit (QIAGEN). The levels of specific mRNA were determined using SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen) with the Mx 3005P RealTime OCR System and Mx Pro software (Stratagene). Primers for RECK mRNA and HPRT mRNA (internal standard) are listed in Supplemental Table S1. The reaction conditions: an initial reverse transcription [50°C, 5 min; 95°C, 5 min] followed by 45 cycles of PCR [94 °C, 15 s; 54 °C (HPRT) or 64°C (hRECK), 40 s; 72°C, 20 s].
Immunoblot assay
Immunoblot assay was performed as described by Oh et al. using the following antibodies [14]. Primary antibodies: RECK (5B11D12) [13], acetyl-Histone H4 (06-866, Millipore), HDAC1 (5356, CST), and GAPDH (AM4300, Ambion). The secondary antibodies: HRP-conjugated anti-mouse IgG-F(ab’)2 monoclonal antibody (A4416, Sigma) and HRP-conjugated anti-Rabbit antibody (7074, CST).
siRNA transfection
Validated siRNAs for HDAC1 [HSS104725 (#1), HSS104726 (#2)] and a control siRNA (GC Duplex, 462001) were obtained from Invitrogen. The cells (2.5×105/well) were seeded onto six-well plates with DMEM supplemented with 10% FBS without antibiotics. Sixteen hours after plating, siRNA was transfected using Lipofectamine RNAiMAX (Invitrogen) at a final RNA concentration of 100 nM.
Matrigel invasion assay
FluoroBlok Transwell insert of 8-μm pore size (Corning) were coated by adding 30 ul diluted Matrigel (BD Biosciences; 9.7 mg/ml) and air-dried overnight. The coated inserts were placed on a 24-well plate containing DMEM supplemented with 10% FBS as a chemo-attractant. The cells (2.5×104) were suspended in DMEM containing 0.1% FBS and plated onto the insert. After 16-h incubation, the cells invaded to the lower side of the membrane were stained with Calcein-AM (Dojindo, Japan) for 10 min, and then recorded using Microplate Fluorescence Reader (Molecular Devices, USA).
Adenovirus-mediated gene transfer
Construction of the control adenoviral vector (Ad-LacZ) and the vector expressing human RECK (Ad-RECK) have been described [43, 44]. Replication-defective viruses were produced from these recombinant cosmids following the protocols of Miyake et al. [45].
Statistical analyses
Student’s t test was used to assess the significance of difference in RECK mRNA levels among different groups of tumor samples and in matrix invasion. Relationships between the RECK methylation status and clinicopathological parameters were evaluated by Pearson Chi-square tests using JMP software (ver.10, SAS Institute). The difference was considered significant when the P-value was smaller than 0.05.
ACKNOWLEDGMENTS
We are grateful to Emi Nishimoto, Hai-Ou Gu, Mika Fujiwara, and Kumi Kawade for technical assistance and Aki Miyazaki for secretarial assistance.
CONFLICTS OF INTEREST
The authors have no conflict of interest to declare.
GRANT SUPPORTS
This work was supported by KAKENHI [Grants-in-Aid for Creative Scientific Research, Scientific Research on Priority Areas, and Scientific Research on Innovative Areas]. G. S. was supported by fellowships from Global COE Program and LIMS Program, Kyoto University Graduate School of Medicine.
REFERENCES
1. Coughlin SS, Ekwueme DU. Breast cancer as a global health concern. Cancer Epidemiol. 2009; 33: 315-318.
2. Rivenbark AG, O’Connor SM, Coleman WB. Molecular and cellular heterogeneity in breast cancer: challenges for personalized medicine. Am J Pathol. 2013; 183: 1113-1124.
3. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000; 406: 747-752.
4. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11: 726-734.
5. Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, Maher ER, Latif F. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011; 71: 2988-2999.
6. Li L, Lee KM, Han W, Choi JY, Lee JY, Kang GH, Park SK, Noh DY, Yoo KY, Kang D. Estrogen and progesterone receptor status affect genome-wide DNA methylation profile in breast cancer. Hum Mol Genet. 2010; 19: 4273-4277.
7. Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, Baylin SB, Ahuja N, Viale A, Massague J, Norton L, Vahdat LT, Moynahan ME, Chan TA. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011; 3: 75ra25.
8. Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007; 1: 19-25.
9. Connolly R, Stearns V. Epigenetics as a therapeutic target in breast cancer. J Mammary Gland Biol Neoplasia. 2012; 17: 191-204.
10. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012; 11: 384-400.
11. Gore SD, Jones C, Kirkpatrick P. Decitabine. Nat Rev Drug Discov. 2006; 5: 891-892.
12. Issa JP, Kantarjian HM, Kirkpatrick P. Azacitidine. Nat Rev Drug Discov. 2005; 4: 275-276.
13. Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, Ikawa Y, Ratzkin BJ, Arakawa T, Noda M. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad Sci U S A. 1998; 95: 13221-13226.
14. Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, Ide C, Horan TP, Arakawa T, Yoshida H, Nishikawa S, Itoh Y, Seiki M, Itohara S, Takahashi C, Noda M. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001; 107: 789-800.
15. Clark JC, Thomas DM, Choong PF, Dass CR. RECK--a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007; 26: 675-683.
16. Noda M, Takahashi C. Recklessness as a hallmark of aggressive cancer. Cancer Sci. 2007; 98: 1659-1665.
17. Cho CY, Wang JH, Chang HC, Chang CK, Hung WC. Epigenetic inactivation of the metastasis suppressor RECK enhances invasion of human colon cancer cells. J Cell Physiol. 2007; 213: 65-69.
18. Du YY, Dai DQ, Yang Z. Role of RECK methylation in gastric cancer and its clinical significance. World J Gastroenterol. 2010; 16: 904-908.
19. Zhang C, Ling Y, Xu Y, Gao L, Li R, Zhu J, Fan L, Wei L. The silencing of RECK gene is associated with promoter hypermethylation and poor survival in hepatocellular carcinoma. Int J Biol Sci. 2012; 8: 451-458.
20. Pesta M, Kulda V, Topolcan O, Safranek J, Vrzalova J, Cerny R, Holubec L. Significance of methylation status and the expression of RECK mRNA in lung tissue of patients with NSCLC. Anticancer Res 2009; 29: 4535-4539.
21. Chang HC, Cho CY, Hung WC. Downregulation of RECK by promoter methylation correlates with lymph node metastasis in non-small cell lung cancer. Cancer Sci. 2007; 98: 169-173.
22. Long NK, Kato K, Yamashita T, Makita H, Toida M, Hatakeyama D, Hara A, Mori H, Shibata T. Hypermethylation of the RECK gene predicts poor prognosis in oral squamous cell carcinomas. Oral Oncol. 2008; 44: 1052-1058.
23. Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997; 25: 2532-2534.
24. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res .2006; 66: 8413-8420.
25. Huang YC, Hung WC, Chen WT, Yu HS, Chai CY. Effects of DNMT and MEK inhibitors on the expression of RECK, MMP-9, -2, uPA and VEGF in response to arsenite stimulation in human uroepithelial cells. Toxicol Lett. 2010; 201: 62-71.
26. Zhou XQ, Huang SY, Zhang DS, Zhang SZ, Li WG, Chen ZW, Wu HW. Effects of 5-aza-2′deoxycytidine on RECK gene expression and tumor invasion in salivary adenoid cystic carcinoma. Braz J Med Biol Res. 2014; 48: 254-260.
27. Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003; 63: 3069-3072.
28. Chang HC, Liu LT, Hung WC. Involvement of histone deacetylation in ras-induced down-regulation of the metastasis suppressor RECK. Cell Signal. 2004; 16: 675-679.
29. Hsu MC, Chang HC, Hung WC. HER-2/neu represses the metastasis suppressor RECK via ERK and Sp transcription factors to promote cell invasion. J Biol Chem. 2006; 281: 4718-4725.
30. Hatta M, Matsuzaki T, Morioka Y, Yoshida Y, Noda M. Density- and serum-dependent regulation of the Reck tumor suppressor in mouse embryo fibroblasts. Cell Signal. 2009; 21: 1885-1893.
31. Jeon HW, Lee YM. Inhibition of histone deacetylase attenuates hypoxia-induced migration and invasion of cancer cells via the restoration of RECK expression. Mol Cancer Ther. 2010; 9: 1361-1370.
32. Lee KJ, Lee KY, Lee YM. Downregulation of a tumor suppressor RECK by hypoxia through recruitment of HDAC1 and HIF-1alpha to reverse HRE site in the promoter. Biochim Biophys Acta. 2010; 1803: 608-616.
33. Sasahara RM, Takahashi C, Noda M. Involvement of the Sp1 site in ras-mediated downregulation of the RECK metastasis suppressor gene. Biochem Biophys Res Commun. 1999; 264: 668-675.
34. Loayza-Puch F, Yoshida Y, Matsuzaki T, Takahashi C, Kitayama H, Noda M. Hypoxia and RAS-signaling pathways converge on, and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs. Oncogene. 2010; 29: 2638-2648.
35. Murai R, Yoshida Y, Muraguchi T, Nishimoto E, Morioka Y, Kitayama H, Kondoh S, Kawazoe Y, Hiraoka M, Uesugi M, Noda M. A novel screen using the Reck tumor suppressor gene promoter detects both conventional and metastasis-suppressing anticancer drugs. Oncotarget. 2010; 1: 252-264. doi: 10.18632/oncotarget.136.
36. Rakha EA, El-Sayed ME, Green AR, Paish EC, Powe DG, Gee J, Nicholson RI, Lee AH, Robertson JF, Ellis IO. Biologic and clinical characteristics of breast cancer with single hormone receptor positive phenotype. J Clin Oncol. 2007; 25: 4772-4778.
37. Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, Serandour AA, Birrell SN, Bruna A, Saadi A, Menon S, Hadfield J, Pugh M, Raj GV, Brown GD, D’Santos C, Robinson JL, Silva G, Launchbury R, Perou CM, Stingl J, Caldas C, Tilley WD, Carroll JS. Progesterone receptor modulates ERalpha action in breast cancer. Nature. 2015; 523: 313-317.
38. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009; 11: R77.
39. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012; 150: 12-27.
40. Kurebayashi J, Kurosumi M, Sonoo H. A new human breast cancer cell line, KPL-1 secretes tumour-associated antigens and grows rapidly in female athymic nude mice. Br J Cancer. 1995; 71: 845-853.
41. Kurebayashi J, Kurosumi M, Sonoo H. A new human breast cancer cell line, KPL-3C, secretes parathyroid hormone-related protein and produces tumours associated with microcalcifications in nude mice. Br J Cancer. 1996; 74: 200-207.
42. Tsuji W, Teramukai S, Ueno M, Toi M, Inamoto T. Prognostic factors for survival after first recurrence in breast cancer: a retrospective analysis of 252 recurrent cases at a single institution. Breast Cancer 2012; 21: 86-95.
43. Yoshida Y, Hamada H. Adenovirus-mediated inducible gene expression through tetracycline-controllable transactivator with nuclear localization signal. Biochem Biophys Res Commun. 1997; 230: 426-430.
44. Yoshida Y, Ninomiya K, Hamada H, Noda M. Involvement of the SKP2-p27(KIP1) pathway in suppression of cancer cell proliferation by RECK. Oncogene. 2012; 31: 4128-4138.
45. Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci U S A. 1996; 93: 1320-1324.