
INTRODUCTION
Circumventing programmed cell death (PCD) is a common action of cancer cells; hence, tumor suppressor genes (TSGs) associated with PCD are frequently inactivated by genetic or epigenetic mechanisms in human cancer cells [1]. Characterizing the molecular mechanism of TSG-mediated PCD is crucial not only for understanding the pathological process of tumorigenesis, but also for the development of novel therapeutic approaches.
The LAPTM5 (Lysosomal-associated protein multispanning transmembrane 5) gene encodes a membrane protein that localizes to intracellular vesicles, regulating vesicle trafficking such as endocytosis, and is highly expressed in lymphoid lineage cells [2–4]. Furthermore, LAPTM5 can inhibit the expression of T cell receptor (TCR), B cell receptor (BCR), and pre-B cell receptor (pre-BCR) on the cell surface via promotion of lysosomal degradation of these proteins; LAPTM5 deficiency has been shown to activate T cells or B cells due to an increase in level of their respective receptors [4, 23, 24]. Thus, previous reports have suggested that LAPTM5 may play an important role as a negative regulator of T cell or B cell receptor-mediated signaling [4, 23, 24].
We previously reported that this gene was transcriptionally down-regulated in neuroblastoma (NB) cell lines and primary tumors by DNA methylation around its transcriptional start site [5]. Interestingly, while LAPTM5 is continuously transported to lysosomes for degradation, it is accumulated in NB cells undergoing PCD within tumors with a favorable prognosis, which frequently undergo spontaneous regression. Furthermore, we have demonstrated that overexpression of LAPTM5 in NB cells induces lysosomal cell death due to lysosomal destabilization, indicated by leakage of lysosomal cathepsin D into the cytosol via lysosomal membrane permeabilization (LMP), as well as by impairment of autophagy degradation [5]. The E3 ubiquitin ligase ITCH, which is often amplified in chromosome 20q of undifferentiated thyroid tumors, is an oncogene [6] and can suppress the expression level of LAPTM5 via ubiquitination-dependent proteasomal degradation [7]. Expression of ITCH also abrogates LAPTM5-induced cell death in NB cells [7]. Thus, it has been suggested that LAPTM5 expression and accumulation may contribute to PCD during NB tumor regression, and that LAPTM5 may function as a tumor suppressor in NB cells. However, the pathological expression and role of LAPTM5 in other types of human cancers remain largely unknown.
Here, we report that expression of LAPTM5 is frequently decreased at the transcriptional level in various types of human cancer cell lines and in non-small cell lung cancer (NSCLC) and esophageal squamous cell carcinoma (ESCC) tumors, and low expression is associated with poor prognosis of patients with such tumors. Importantly, we showed that overexpression of LAPTM5 induces lysosomal cell death in KYSE170 cells, an ESCC cell line, as well as in NB cells [5]. These findings suggest that inactivation of LAPTM5 may contribute to tumorigenesis in a subset of human cancers.
RESULTS
Down-regulation of LAPTM5 expression in human cancer cells
We first examined the expression status of LAPTM5 in 333 cell lines from 18 various types of human cancers in comparison with the expression levels of each corresponding normal tissue by qRT-PCR analysis. As shown in Table 1, Figure 1A, and Figure S1, we found that the expression level of LAPTM5 mRNA was decreased in 316 of the 333 cell lines (94.9%), including in 37 of the 39 ESCC cell lines (94.9%). A qRT-PCR analysis of paired ESCC samples (primary tumors and their corresponding non-cancerous tissues) revealed a more than 30% reduction of LAPTM5 expression in primary tumor tissues compared with the corresponding non-cancerous tissues in 12 of the 32 cases (37.5%) (Figure 1B). Importantly, the Kaplan-Meier survival curve indicated that low expression of LAPTM5 was significantly associated with poor prognosis of patients with ESCC (P = 0.013) (Figure 1C). However, apart from poor prognosis, we could not show any correlation between low expression and other clinical implications such as stages, pathological grades, or lymph metastasis (Tabale S1). In addition, we could not detect aberrant DNA methylation around the transcriptional start site of LAPTM5 in the ESCC tumor samples showing low expression, as is detected in NBs (data not shown) [5]. Furthermore, we surveyed 3 gene-expression datasets of NSCLC adenocarcinomas using Oncomine, a cancer microarray database (www.oncomine.org/). LAPTM5 expression was decreased in NSCLC compared with normal lung tissues, and this difference was statistically significant in all 3 datasets (P = 1.49x10-5, P = 3.24x10-14, and P = 3.21x10-6) (Figure 1D) [13–15]. Additionally, we also showed that low expression of LAPTM5 was significantly correlated with poor prognosis in NSCLC patients by analysis of lung cancer gene expression datasets that included survival information (P = 0.043) (Figure 1E) [16]. These findings indicate that LAPTM5 is widely down-regulated in most human cancers, and notably, its down-regulation may serve as a poor prognostic factor for patients with ESCC and NSCLC.
Table 1: Frequency of down-regulation of LAPTM5 expression in human cancer cell lines
Tissue |
Number of down-regulated cell lines * |
Number of total cell lines |
Frequency (%) |
|---|---|---|---|
Esophageal Squamous Cell Carcinoma (ESCC) |
37 |
39 |
94.9 |
Prostate |
3 |
3 |
100 |
Anaplastic Thyroid Carcinoma (ATC) |
15 |
16 |
93.8 |
Cervical |
11 |
11 |
100 |
Hepatic Cellular Carcinoma (HCC) |
21 |
25 |
84 |
Breast |
13 |
13 |
100 |
Glioma |
21 |
23 |
91.3 |
Renal Cell Carcinoma (RCC) |
13 |
15 |
86.7 |
Endometrial |
14 |
14 |
100 |
Ovarian |
22 |
27 |
81.5 |
Bladder |
8 |
8 |
100 |
Gallbladder |
8 |
8 |
100 |
Small Cell Lung Carcinoma (SCLC) |
21 |
21 |
100 |
Gastric |
29 |
30 |
96.7 |
Mesothelial |
4 |
4 |
100 |
Myeloma |
10 |
10 |
100 |
Non-Small Cell Lung Carcinoma (NSCLC) |
35 |
35 |
100 |
Pancreatic |
31 |
31 |
100 |
Total |
316 |
333 |
94.9 |
“Down-regulation” was defined as a 50% or greater reduction in expression compared with the corresponding normal tissue.
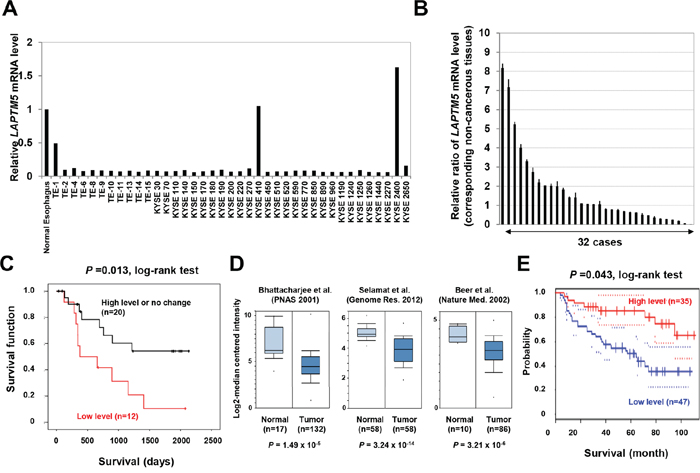
Figure 1: Down-regulation of LAPTM5 in human cancers. A and B. Expression analysis of LAPTM5 in ESCC cell lines and primary samples. The mRNA level of LAPTM5 in normal esophageal tissue, in 39 ESCC cell lines, and in 32 ESCC paired samples of primary tumors and corresponding non-cancerous tissues were measured by qRT-PCR. Expression of GAPDH was used as an internal control. The graph shows the relative expression of LAPTM5 in the ESCC cell lines (relative to that in the normal esophageal tissue) (A) or in the primary tumor (relative to that in the corresponding non-cancerous tissue) (B). Bar; standard deviation (SD). C. Kaplan–Meier curves for overall survival rates of the 32 patients with ESCC, according to LAPTM5 expression status (high or low expression). Low expression (more than 30% reduction of LAPTM5 expression) was significantly associated with worse survival by log-rank test. D. Expression levels of LAPTM5 in lung adenocarcinoma. Three lung adenocarcinoma gene expression datasets were analyzed in Oncomine and displayed as a box plot (log2 median-centered). P values were calculated using Student’s t-test. E. Correlation between LAPTM5 expression and prognosis among lung cancer patients. Correlation between LAPTM5 expression and survival was analyzed and plotted using the Kaplan–Meier method. Survival rates for patients with high (n=35) and low (n=47) LAPTM5 expression were analyzed using PrognoScan. P values were calculated by log-rank test. The 95% confidence intervals for each group are indicated by dotted lines.
Induction of cell death by overexpression of LAPTM5 in cancer cells
To test whether overexpression of LAPTM5 can induce cell death in other types of cancer cell lines, as in NB cells [5], LAPTM5 was introduced by adenovirus into KYSE170 and KYSE770 cells (both ESCC cell lines) and NCI-H460 cells (a NSCLC cell line), all of which demonstrate down-regulation of LAPTM5 (Figure 1A and Figure S1). Western blot analysis revealed that LAPTM5 expression gradually increased with increasing adenovirus-LAPTM5 (Ad-LAPTM5) titer (Figure 2A). Overexpression of LAPTM5 for 4 days resulted in remarkable decreases in cell survival in an Ad-LAPTM5 dose-dependent manner (Figure 2B). In addition, more severe growth inhibition and a remarkable increase of dead cells were observed in cells infected with higher Ad-LAPTM5 titer, compared with adenovirus-LacZ (Ad-LacZ) (Figure 2C and 2D). Consistent with NB cells [5], there were no typical features of apoptosis, such as caspase activation or apoptotic bodies, in ESCC and NSCLC cells undergoing LAPTM5-induced cell death (data not shown). These results suggest that overexpression of LAPTM5 can effectively induce non-apoptotic cell death in human cancer cells.
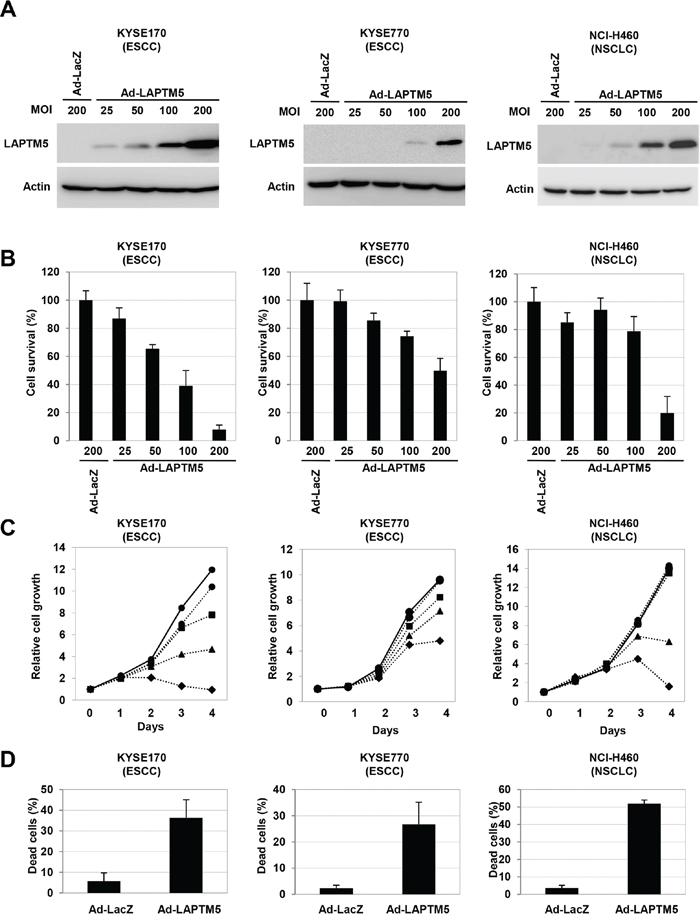
Figure 2: Overexpression of LAPTM5 induced cell death in cancer cell lines. A. Western blot analysis of LAPTM5-expressing cells. Cells were infected with Ad-LacZ or Ad-LAPTM5 for 4 days at the indicated MOI. Whole cell lysates were subjected to SDS-PAGE and immunoblotted with rabbit anti-LAPTM5 or mouse anti-β-actin antibody. B. Effect of LAPTM5 overexpression on cell survival in cancer cells. Cells were infected with Ad-LacZ or Ad-LAPTM5 for 4 days at the indicated MOI. The cell survival rate relative to that of Ad-LacZ-infected cells was assessed by the CV staining assay. Bar; standard deviation (SD) for triplicate experiments. C. Cell growth assay. Growth curves are indicated by solid lines for Ad-LacZ-infected cells or dotted lines for Ad-LAPTM5-infected cells. Ad-LacZ was infected at 200 MOI. Ad-LAPTM5 was infected at 25 (circle), 50 (square), 100 (triangular), or 200 (quarry) MOI. D. Frequency of dead cells. KYSE170 cells were infected with Ad-LacZ or Ad-LAPTM5 at 200 MOI. Four days after infection, dead cells were counted using the trypan blue exclusion method, and indicated as percentages. Bar; standard deviation (SD) for triplicate experiments.
Induction of lysosomal destabilization during LAPTM5-mediated cell death
We next examined whether lysosomal destabilization is induced by overexpression of LAPTM5 in other types of cancer cell lines, as is in NB cells [5]. Because p62/SQSTM1 (referred to as p62) and ubiquitinated proteins are continuously degraded in the lysosome via an autophagic process, lysosomal dysfunction or autophagy inhibition leads to an accumulation of these proteins in the Triton-X-insoluble fraction [17–22]. As shown in Figure 3A, levels of p62 and ubiquitinated proteins were clearly increased in the Triton-X-insoluble fraction from LAPTM5-expressing cells, compared with that of the Triton-X-soluble fraction. Additionally, we showed that overexpression of LAPTM5 induced leakage of lysosomal cathepsin D into the cytoplasm in KYSE170 cells, indicated by a change in cathepsin D localization from a punctate to a diffusive pattern, suggesting lysosomal membrane permeabilization (LMP), an indicator of lysosomal destabilization (Figure 3B). Taken together, these findings suggest that overexpression of LAPTM5 protein induces lysosomal destabilization, accompanied by autophagy impairment and cathepsin D leakage, resulting in the lysosomal cell death of ESCC and NSCLC cells.
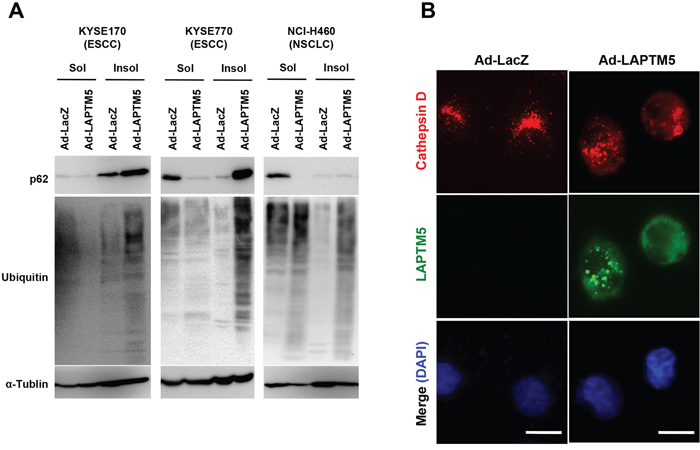
Figure 3: Induction of lysosomal destabilization during LAPTM5-mediated cell death. A. Western blotting for ubiquitinated proteins. Four days after infection in KYSE170, KYSE770, or NCI-H460 cells, cells were lysed in 0.1% Triton-X solution. Triton-insoluble pellets were solubilized in 2% SDS buffer. Triton-X soluble or -insoluble lysates were loaded onto a 6% SDS-PAGE and analyzed by immunoblotting with antibodies against p62, ubiquitin, and α-tubulin. In both soluble and insoluble samples, greater accumulation of p62 and ubiquitinated proteins were observed in Ad-LAPTM5-infected cells compared with Ad-LacZ-infected cells. B. Representative images of cathepsin D staining. Four days after infection, KYSE170 cells were fixed in 10% TCA, reacted with anti-cathepsin D and anti-LAPTM5 antibodies, and visualized with Alexa Fluor 594-conjugated mouse IgG and Alexa Fluor 488-conjugated rabbit IgG antibodies, respectively. Bar, 10 μm.
DISCUSSION
We showed that overexpression of LAPTM5 induces lysosomal cell death, demonstrated by cathepsin D release from the lysosome into the cytoplasm, as well as impairment of autophagy via lysosomal destabilization in ESCC and NSCLC cells, as was previously observed in NB cells [5]. This suggests that LAPTM5-induced cell death may widely occur in various human cancers. However, the detailed molecular mechanism underlying LAPTM5-induced cell death remains unknown. We previously demonstrated that LAPTM5 protein levels were negatively regulated not only by lysosomal degradation, but also by proteasomal degradation via ITCH, an E3 ubiquitin ligase, in NB cells [5, 7]. We demonstrated that accumulation of LAPTM5 due to dysfunction of these degradation pathways was critical for the induction of cell death in NB cells [5, 7]. We also showed that LAPTM5 was accumulated in dying NB cells within tumors undergoing spontaneous regression [5]. In the present study, we showed that overexpression of LAPTM5 protein by a high-dose of adenovirus could induce cell death in ESCC and NSCLC cells. Thus, these findings suggest that excessive accumulation of LAPTM5 may be a triggering event for induction of cell death in human cancers.
Moreover, we observed that LAPTM5 expression was frequently decreased at the transcriptional level in various types of human cancer cells, similar to NB cells [5]. In particular, LAPTM5 down-regulation was significantly correlated with poor prognosis of patients with ESCC and NSCLC. We have previously reported that aberrant DNA methylation around the transcriptional start site (TSS) of LAPTM5 may be a possible mechanism for down-regulation of LAPTM5 in NB cells [5]. However, we were unable to detect aberrant methylation in this region in ESCC tumors that showed low expression of LAPTM5 (data not shown), suggesting that other mechanisms, such as chromatin modification around the TSS, chromosomal deletion, or loss of transcriptional factors, may be closely associated with the down-regulation of LAPTM5 expression in ESCC tumors. Thus, further validations including somatic mutation will be required for understanding molecular mechanism for inactivation of LAPTM5 gene in human cancers.
Finally, our findings suggest that the transcriptional down-regulation of LAPTM5 may lead to the repression of LAPTM5 accumulation-mediated cell death in human cancers, including ESCC and NSCLC. Although we have observed accumulation of LAPTM5 in dying cells within regressing NB tumors, it is unknown whether this accumulation actually occurs in other tumors, including ESCC. Thus, identifying the physiological and cellular conditions of LAPTM5 accumulation-mediated cell death will be essential to further understand the biological significance of this type of cell death in human cancers.
MATERIALS AND METHODS
Cell culture and primary tumor samples
Cell lines were obtained from the American Type Culture Collection (ATCC) or from the Japanese Collection of Research Bioresources (JCRB). ESCC cell lines, which were given by Dr. Yataka Shimada of Kyoto University [8–10], and NCI-H460 (an NSCLC cell line) were cultured in RPMI1640 medium containing 10% FBS. All cell lines were maintained at 37°C with 5% CO2.
A total of 32 primary ESCC tumor samples and their corresponding non-cancerous esophageal mucosa were investigated in this study. All samples were obtained from patients treated at the Tokyo Medical and Dental University Hospital between November 2007 and October 2012, were frozen immediately in liquid nitrogen, and stored at -80°C until total RNA was extracted. The collection and analysis of patient samples were approved by the Tokyo Medical and Dental University Institutional Review Board (approval #2010-5-4), and written consent was obtained from all patients.
Antibodies
Antibodies against β-actin (A5441, Sigma), α-tubulin (Sigma), p62 (SQSTM1; sc-28359, Santa Cruz Biotechnology), Ubiquitin (Wako), Cathepsin D (IM03, Calbiochem), and LAPTM5 (H-178, Santa Cruz Biotechnology) were used for western blotting and immunofluorescence analysis.
Recombinant adenovirus
Replication-defective recombinant adenoviruses were constructed with the Adenovirus Expression Vector Kit (Takara) following the manufacturer’s recommendations [5]. Viral titers were measured in pfu/ml by a serial dilution method using HEK293 cells. Cells were infected with each indicated MOI (multiplicity of infection; PFU/cell).
RNA isolation and quantitative reverse transcription (qRT)-PCR
RNA isolation and qRT-PCR were performed as described in previous reports [11] for the expression analysis of 338 cell lines and the corresponding normal tissues, as shown in Supplementary Figure 1. Total RNA was isolated using TRIzol® reagent (Invitrogen) according to standard procedures. Single-stranded cDNA generated from total RNA was amplified with primer sets specific for each gene. For quantitative real-time RT–PCR, probes for LAPTM5 were purchased from the KAPA PROBE FAST system (KAPA BIOSYSTEMS). RT–PCR was performed using an ABI PRISM 7500 sequence detection system (Applied Biosystems) according to the manufacturer’s instructions. Gene expression values are given as ratios (differences between the Ct values) between the gene of interest and an internal reference (GAPDH) that provides a normalization factor for the amount of RNA isolated from a specimen, which was then normalized by the control cell value (relative expression level).
Western blotting analysis
Western blotting was performed as described in previous reports [5, 12]. Whole cell lysates were subjected to SDS-PAGE, and proteins were transferred to PVDF membranes (GE Healthcare). After blocking with TBS containing 0.05% Tween-20 and 5% non-fat dry milk for 1 hour, the membrane was reacted with an antibody overnight. The dilutions for primary antibodies were: 1/5,000 for β-actin, 1/2,000 for p62, and 1/1,000 for LAPTM5 and Ubiquitin. The membrane was washed and exposed to horseradish peroxidase (HRP)-conjugated anti-mouse or rabbit IgG antibodies (both at 1/4,000) for 2 hours. The bound antibodies were visualized in LAS3000 (GE Healthcare) using a Pierce ECL Western detection kit according to the manufacturer’s instructions (Thermo Scientific). Alternatively, cells were lysed in 0.1% Triton-X solution and the Triton-X-insoluble pellet was solubilized in 2% SDS buffer. Triton-X soluble or -insoluble lysates were loaded onto a 6% SDS-PAGE and analyzed by immunoblotting with p62, ubiquitin, or α-tubulin antibodies.
Cell survival and cell death assays
Cell survival and cell death assays were performed as previously described [5, 12]. Cell survival was assessed by the crystal violet (CV) staining assay. Cells were washed in PBS and fixed with 0.1% CV in 10% formaldehyde in PBS for 10 minutes. After excess CV solution was discarded, stained cells were completely air-dried, and then lysed with a 2% SDS solution with shaking for 2 hours. Optical density (OD) absorbance was measured at 560 nm using a microplate reader (ARVOmx; PerkinElmer), and the percent absorbance of every well was determined. The OD absorbance of cells in control wells was arbitrarily set at 100% to determine percentages of viable cells. Dead cells were counted by trypan blue staining using the TC20™ Automated Cell Counter (BIO-RAD), and the percent dead cells of total cells was calculated.
Immunofluorescence (IF) analysis
IF analysis was performed as described from previous reports [5, 7]. Cells were fixed in 10% trichloroacetic acid and permeabilized with 0.5% Triton X-100 in PBS. After blocking with PBS containing 1% bovine serum albumin and 0.01% Triton X-100 for 1 h at 4°C, the cells were incubated with mouse anti-Cathepsin D (dilution: 1/1000) overnight at 4°C. Bound antibody was visualized using Alexa Fluor 488 anti-mouse IgG antibody (Invitrogen, dilution: 1/2000). The cells were mounted in VECTASHIELD Mounting Medium with DAPI (Vector Laboratories) and observed under a fluorescence microscope (DM6000 B, Leica, Wetzlar, Germany).
Analysis of gene expression datasets
Three gene expression datasets of human lung cancers [13–15] were analyzed in Oncomine, a cancer microarray database (www.oncomine.org/). Kaplan–Meier analysis of 82 lung cancer patients was based on the PrognoScan database (http://www.prognoscan.org/) using the publicly available Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo), with the accession number GSE68465 [16]. P values were calculated by log-rank test.
Statistical analysis
Differences between subgroups were tested by Student’s t-test. A P value of < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Ayako Takahashi and Rumi Mori for technical assistance.
CONFLICTS OF INTEREST
There are no conflicts of interest to disclose.
FINANCIAL SUPPORT
This study was supported in part by Grants-in-Aid for Scientific Research (A; 25250019 and C; 24590372) and Grants-in-Aid for Scientific Research on Innovative Areas “Integrative Systems Understanding of Cancer for Advanced Diagnosis, Therapy and Prevention” (22134002) from the Japan Society for the Promotion of Science (JSPS), a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct) and the Tailor-Made Medical Treatment with the BioBank Japan Project (BBJ) from The Japan Agency for Medical Research and Development, the Global Center of Excellence (GCOE) Program from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan, and Foundation for Promotion of Cancer Research for the 3rd Term Comprehensive 10-year-Strategy (H24-the 3rd Term-Young-002) from the Ministry of Health, Labour and Welfare (MHLW), Japan.
Author contributions
The authors have contributed equally to this work.
REFERENCES
1. Hanahan D, Weinberg R. Hallmarks of Cancer: The Next Generation. Cell. 2011; 144:646–674.
2. Salah Z, Melino G, Aqeilan R. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Research. 2011; 71:2010-2020.
3. Pak Y, Glowacka WK, Bruce MC, Pham N, Rotin D. Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination. Journal of Cell Biology. 2006; 175:631–645.
4. Ouchida R, Kurosaki T, Wang J. A role for lysosomal-associated protein transmembrane 5 in the negative regulation of surface B cell receptor levels and B cell activation. Journal of Immunology. 2010; 85:294–301.
5. Inoue J, Misawa A, Tanaka Y, Ichinose S, Sugino Y, Hosoi H, Sugimoto T, Imoto I, Inazawa J. Lysosomal-associated protein multispanning transmembrane 5 gene (LAPTM5) is associated with spontaneous regression of neuroblastomas. PloS One. 2009; 4:e7099.
6. Ishihara T, Tsuda H, Hotta A, Kozaki K, Yoshida A, Noh JY, Ito K, Imoto I, Inazawa J. ITCH is a putative target for a novel 20q11.22 amplification detected in anaplastic thyroid carcinoma cells by array-based comparative genomic hybridization. Cancer Science. 2008; 99:1940-1949.
7. Ishihara T, Inoue J, Kozaki K, Inazawa J. HECT-type ubiquitin ligase ITCH targets lysosomal-associated protein multispanning transmembrane 5 (LAPTM5) and prevents LAPTM5-mediated cell death. Journal of Biological Chemistry. 2011; 286:44086-44094.
8. Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992; 69:277-284.
9. Tanaka H, Shibagaki I, Shimada Y, Wagata T, Imamura M, Ishizaki K. Characterization of p53 gene mutations in esophageal squamous cell carcinoma cell lines: increased frequency and different spectrum of mutations from primary tumors. International Journal of Cancer. 1996; 65:372-376.
10. Tanaka H, Shimada Y, Imamura M, Shibagaki I, Ishizaki K. Multiple types of aberrations in the p16 (INK4a) and the p15 (INK4b) genes in 30 esophageal squamous-cell-carcinoma cell lines. International Journal of Cancer. 1997; 70:437-442.
11. Bai H, Inoue J, Kawano T, Inazawa J. A transcriptional variant of the LC3A gene is involved in autophagy and frequently inactivated in human cancers. Oncogene. 2012; 31:4397-4408.
12. Fujiwara N, Inoue J, Kawano T, Tanimoto K, Kozaki K, Inazawa J. MiR-634 activates the mitochondrial apoptosis pathway and enhances chemotherapy-induced cytotoxicity. Cancer Research. 2015; 75:3890-3901.
13. Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:13790-13795.
14. Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, Laird-Offringa IA, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Research. 2012; 22:1197-1211.
15. Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature Medicine. 2002; 8:816-824.
16. Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma, Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, Chang AC, Zhu CQ, Strumpf D, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nature Medicine. 2008; 14:822-827.
17. Mizushima N. Autophagy: process and function. Genes and Development. 2007; 21:2861-2873.
18. Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. Journal of Cell Biology. 2005; 171: 603-614.
19. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. Journal of Biological Chemistry. 2006; 282:24131-24145.
20. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006; 441:880–884.
21. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006; 441:885–889.
22. Adra CN, Zhu S, Ko JL, Guillemot JC, Cuervo AM, Kobayashi H, Horiuchi T, Lelias JM, Rowley JD, Lim B. LAPTM5: a novel lysosomal-associated multispanning membrane protein preferentially expressed in hematopoietic cells. Genomics. 1996; 35:328–337.
23. Ouchida R, Yamasaki S, Hikida M, Masuda K, Kawamura K, Wada A. A lysosomal protein negatively regulates surface T cell antigen receptor expression by promoting CD3 zeta-chain degradation. Immunity. 2008; 9:33-43.
24. Kawano Y, Ouchida R, Wang JY, Yoshikawa S, Yamamoto M, Kitamura D. A novel mechanism for the autonomous termination of pre-B cell receptor expression via induction of lysosome-associated protein transmembrane 5. Molecular Cell Biology. 2012; 32:4462-4471.