
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common deadly tumors worldwide [1]. HCC can result from chronic viral hepatitis, mainly after hepatitis B virus (HBV) or hepatitis C virus (HCV) infections. HCC may also result from alcoholic liver disease, aflatoxin exposure, and metabolic diseases, such as hereditary hemochromatosis, alpha-1 antitrypsin deficiency or non-alcoholic fatty liver disease [2]. Nevertheless, chronic HBV infections remain the main etiology of HCC in Asia. The risk of HCC development for HBV carriers is 223-fold higher than for non-carriers [3].
HBV has a partially double-stranded, 3.2-kb genome. It is a non-cytopathic hepatotropic DNA virus classified as a member of the hepadnaviridae family [4][5][6]. The 3.2-kb genome can be unidirectionally transcribed from four promoters to produce various HBV proteins. The first promoter, called the basal core, produces precore (HBeAg) and pregenomic (HBV-core and HBV-polymerase [Pol] proteins). The second promoter, SPI, produces a large HBV surface antigen (HBsAg). The third promoter, SPII, produces intermediate and small HBsAgs. The fourth promoter, X, produces the X protein, whose function is not fully understood [7][8][9]. The HBV genome replication cycle in the host can be divided broadly into three phases. First, a partially double-stranded, relaxed circular DNA (RC-DNA) is produced; second, the RC-DNA is converted to a covalently closed circular DNA (cccDNA); and third, the cccDNA is transcribed by the cellular RNA polymerase II to produce pregenomic RNA (pgRNA) and subgenomic RNA. In the active HBV replication state, the HBV-core protein is transcribed from the pgRNA; then, HBV-core selectively packages pgRNA into progeny capsids. Next, the pgRNA is reverse-transcribed by the co-packaged Pol to produce new RC-DNA. Individual copies of mature RC-DNA are enveloped by HBsAg to form progeny virions that are released into the circulation. Clinically, detection of HBsAg is the hallmark of an HBV infection; detection of HBeAg, which is processed from the precore protein, is a marker of infectivity; and detection of circulating HBV DNA is an indicator of active replication [7]. HBsAg becomes undetectable in most patients four to six months after acute HBV exposure. In less than 1% of patients infected with HBV, HBsAg persists for more than six months, indicating a chronic infection and integration of the HBV genome into the host chromosome [8][9]. In an inactive carrier state, the clearance rate of HBsAg is slow (~0.5% per year), regardless of the expression levels of HBV-derived RNA and DNA and other viral products [10].
Recent studies have shown that, upon viral entry, sensors and/or receptors in the infected host cells can detect virus-derived RNAs, DNAs, and proteins. These sensors activate intracellular signaling pathways that induce inflammatory cytokines, chemokines, interferons, gene transcription, and protein modifications, which exert innate and adaptive immune responses against viral replication [11]. When the virus cannot be eradicated, the accumulated viral proteins will induce endoplasmic reticulum (ER) stress and unfolded-protein responses that inhibit protein synthesis, induce protein degradation, trigger apoptosis, and facilitate cell transformation [12][13]. Although there is significant evidence for the effects of HBsAg-induced ER stress on hepatocarcinogenesis in HBV carriers [13][14], no specific genes are universally altered by the HBsAg-induced oxidative damage and HBV DNA integration. We hypothesized that hepatocarcinogenesis involves interplay between HBV and host hepatocytes. Therefore, the present study aimed to identify host tumor suppressors and/or oncogenic proteins that interact with HBsAg during HBV-associated pathogenesis. Identifying such proteins might facilitate the development of treatments for HCC.
RESULTS
Spatiotemporally reciprocal negative interactions between human PML and HBsAg during human HBV-related pathogenesis
Ubiquitin-mediated degradation appears to be the common mechanism accounting for loss of the tumor suppressor promyelocytic leukemia (PML) in virus infection and cancer [15][16]. The ubiquitin E3-ligases, SIAH-1 and -2 (SIAH-1/2), are implicated in tumorigenesis through physical interaction with PML triggering its degradation [17][18][19][20]. In addition, our earlier study in 155 HBV-infected patients demonstrated that suppression of PML persisted until HBsAg expression was gradually down-regulated [21]; and our analysis of each HBV component effect by transfection assays revealed that HBsAg induced proteasome-mediated PML degradation [22]. These findings led us to ask whether the tumorigenesis of HBsAg was associated with SIAH-1/2-mediated PML loss [23]. Therefore, in order to examine the interactive expression patterns of PML, SIAH-1/2 and HBsAg during the HBV-related pathogenesis, the liver tissue arrays from our previously studied 155 HBV-infected patients were further stratified by their clinical phases of HBV infection for immunohistochemistry [21][24]. Clinically, chronic HBV pathogenesis can be subdivided into several phases: the immune tolerance (acute) phase, the immune clearance (chronic hepatitis) phase, the inactive carrier phase and HCC formation. To show expression patterns of HBV surface antigen (HBsAg) and the host growth-regulated proteins, PML and SIAH-1/2, liver tissue arrays from different phases were immunostained with anti-HBsAg, anti-PML and anti-SIAH-1/2. We found that, in acute HBV infections, diffuse, strong HBsAg expression correlated with complete suppression of PML expression (Figure 1A). Conversely, in the subsequent phases of chronic hepatitis, inactive carrier and HCC development, a gradual-to-total disappearance or downregulation of HBsAg was accompanied by re-appearance of nuclear PML (Figures 1A, 1B and 1C). In contrast, SIAH-1/2 expression did not concordantly change in response to the dynamics of HBsAg or inversely correlate with fluctuating PML expression (Figures 1B and 1C). Thus, the consistent spatiotemporal findings of mutual exclusion (Figure 1A) and reciprocal negative interaction (Figure 1B) between HBsAg and PML during the whole course of HBV-induced pathogenesis and HCC development prompted us to investigate whether different biological consequences progress from HBsAgextensivePMLsuppression to HBsAglostPMLre-appearance conditions (Figure 1C).
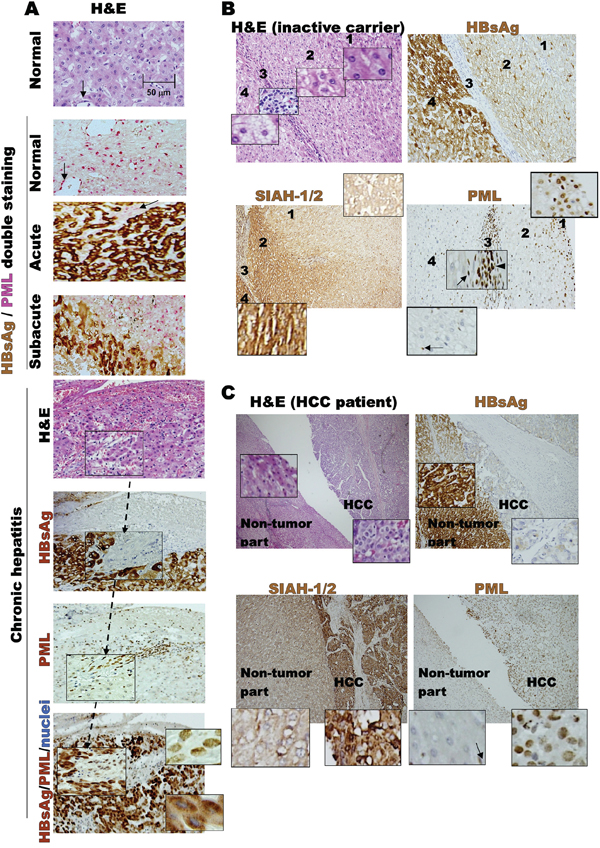
Figure 1: Dynamic interaction between HBV and host factors during chronic HBV-related pathogenesis. A. Liver biopsies from initial normal state, acute HBV infection and chronic HBV hepatitis patients. Double-stainings of cytoplasmic HBsAg (brown) and nuclear PML (pink) are shown at different disease stages, exhibiting mutual exclusion and reciprocal negative expression patterns. Immunohistochemistry of chronic hepatitis was counterstained with Mayer's Hematoxylin (blue) for nuclei visualization in tissue sections. PML-positive HBsAg-negative sinusoidal endothelial cells (arrow), and mononuclear infiltrating cells in chronic hepatitis or septum lymphocytes (arrowhead) in the HBV carrier are internal staining controls. B. Tissues from an HBV carrier with zones 1, 2, 3 (the septum) and 4 in the same liver section exhibiting differential expression patterns of SIAH-1/2, PML and HBsAg, as well as distinct changes in fat distribution. C. Expression patterns of SIAH-1/2, PML and HBsAg in tissues from patient with HBV-related HCC, compared to those of the surrounding non-tumor liver.
Phenotypes and pathogenesis of HBsAg-transgenic mice with or without PML deletion
In order to examine the influence of PML on HBsAg-driven hepatocarcinogenesis, we crossed PML-/- knockout mice with a liver-specific, HBsAg-transgenic mice to generate wild-type, PML-/-, PML+/+HBsAgtg/0, PML+/-HBsAgtg/0 andPML-/-HBsAgtg/0 mice with the same genetic backgrounds (Figure 2A). The PML-/- mice showed body fat accumulation, pale livers, but no liver tumor development even at two years of age. Complete loss of PML in the HBsAg-transgenic mice (PML-/-HBsAgtg/0) conferred a strong growth advantage and altered lipid metabolism, predisposing the mice to obesity and accelerating the development of multiple hyperplasial foci and large fatty adenomas, which appeared at 12-20 weeks of age, followed by formation of solid adipose-like HCCs by one year of age. In contrast, almost all of the PML+/+HBsAgtg/0 mice exhibited a protracted two-year course of hepatocarcinogenesis, which started with ground-glass hepatocytes and progressed to steatohepatitis, dysplastic adenomas, and finally, angiogenic trabecular HCC formation (Figure 2B). Interestingly, PML appeared to act in a haploinsufficient manner in PML+/-HBsAgtg/0 mice that showed mixed phenotypic features of PML-/-HBsAgtg/0 and PML+/+HBsAgtg/0 mice. Moreover, PML and HBsAg patterns in the liver cells of HBsAg-transgenic mice recapitulated the mutual exclusion and reciprocal negative interaction observed in chronic human HBV-infected liver cells (Figures 1 and 3A). Although the HBsAg transgene has been found to be intact at the integrated site and can be reactivated, progressive loss of HBsAg is a characteristic common to human and mouse HBV-related pathogenesis [10][25]. However, we found that when HBsAg decayed over time, the reappearance of the bona fide tumor suppressor PML did not retard the late-onset progression of HCC in PML+/+HBsAgtg/0 mice (Figure 3B).
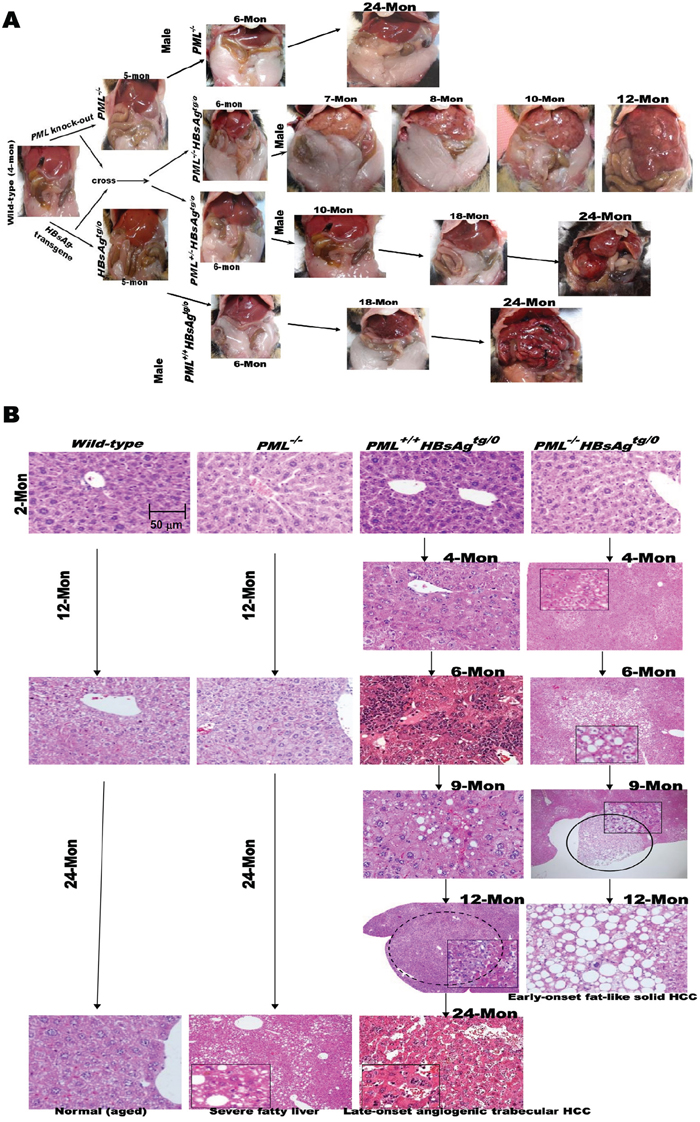
Figure 2: Phenotypes and pathogenesis of liver-specific HBsAg-transgenic mice with or without PML deletion. A. Open abdominal cavities of PML-knockout mice (129/SV-Pmltm/Ppp) crossed with liver-specific HBsAg-transgenic mice (C57BL/6J-Tg(Alb1HBV)44Bri/J), and of offspring crossed to generate different genotypes, including wild-type, PML-/-, PML+/+HBsAgtg/0, PML+/-HBsAgtg/0 and PML-/-HBsAgtg/0. Each genotype shows gross changes over time in the liver and body fat masses with HCC progression. Once liver tumors develop, the body fat mass seems to be consumed, a sign of cachexia. Mon = months. B. Representative histological images of liver sections from wild-type, PML-/-, PML+/+HBsAgtg/0 and PML-/-HBsAgtg/0 mice sacrificed at 2 -24 months of age. Adenomas are indicated by circles.
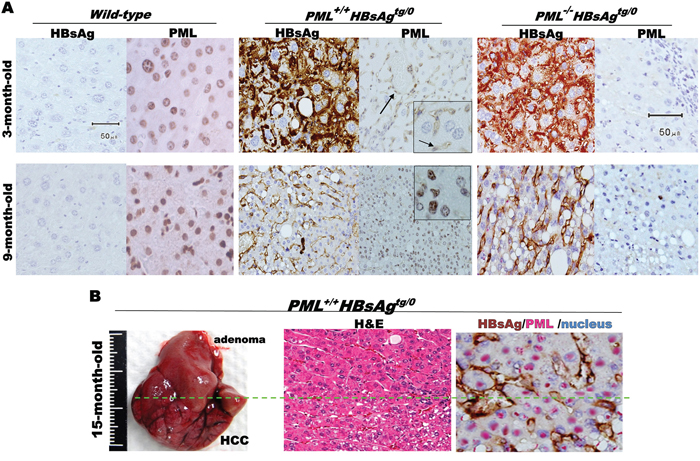
Figure 3: Mutually exclusive interactive patterns between PML and HBsAg in HBsAg-transgenic mice. A. Immunohistochesmistry with anti-PML and/or anti-HBsAg antibodies counterstained with Mayer's Hematoxylin (blue) for visualization of nuclei in liver sections from wild-type, PML+/+HBsAgtg/0 and PML-/-HBsAgtg/0 mice at 3-9 months of age. PML-positive HBsAg-negative sinusoidal endothelial cells (arrow) highlighted as internal positive staining controls. B. Liver of PML+/+HBsAgtg/0 mouse with late-onset HCC (left) at 15 months of age and double-staining of tissue sections (middle and right) showing a mutual exclusion pattern between the fading cytoplasmic or membrane HBsAg (brown) and the restored nuclear PML (pink).
Mutation landscape of HBsAg-transgenic mice with or without PML deletion
Given that PML is a genome guardian [21][22][26], we performed whole-exome sequencing of DNA derived from liver cells of wild-type, PML-/-, PML+/+HBsAgtg/o, and PML-/-HBsAgtg/o mice, with or without early-onset or late-onset liver tumors (Figure 4 and Table 1). Whole exome sequences were screened for mutations in 10-18 mice of each genotype at different ages to identify unique, mutually exclusive, and co-occurring genes pertinent to HBsAg-induced HCC with or without PML deletion (PML-/-). Except for the mutated exonic sequences of PML in the PML-knockout mice, confirmed to match those previously reported [27], there were no differences in exonic DNA variants or polymorphisms at one month of age between different genotypes, indicating homogeneous genetic background at the baseline. Furthermore, no HBsAg transgene had integrated into the exomes (Supplementary file 1). The cumulative DNA variants, found in more than 50% of the wild-type mice that did not develop adenoma and HCC at two years of age, were regarded as age-related mutations (Figure 4A). By comparing the variants of genetically engineered mice with those of age-matched, wild-type mice, we removed the age effect on nonspecific gene mutations. At 6 months of age, the PML-/-, PML-/-HBsAgtg/0 and PML+/+HBsAgtg/0 mutation maps show low mutation rates (or cold areas) on chromosomes 15 and X; hypermutational regions (or kataegis) are visible on chromosomes 4-9, 11-14, and 17 (Figure 4B). We then identified the nonsynonymous mutation landscape in each genotype (Figure 4C).
Table 1: Common gene mutations compared between PML-/- and PML-/-HBsAgtg/0 and between HBsAgtg/0 and PML-/-HBsAgtg/0
(a) Co-occurring gene alterations in PML-/- and PML-/-HBsAgtg/0mice |
||||||
|---|---|---|---|---|---|---|
Cell growth & signal |
DNA repair |
Ubiquitination |
Membrane trafficking |
Lipid metabolism |
Cell adhesion |
Ribosomal biogenesis |
Ppp1r13l |
Pcna |
Usp2 |
Pld5 |
Ugt1a9 |
Itgad |
Rpl32 |
Fat2 |
Ercc1 |
Sae1 |
Vps52 |
Nr1d2 |
Itgb2l |
Mrpl55 |
Edaradd |
Fancc |
Ube4a |
Cpnel |
Glud1 |
Fat2 |
Rrs1 |
Fam59a |
Ybx1 |
Siah1a |
Gpi1 |
|||
Loxl4 |
Rest |
Rnf167 |
||||
Ptprg |
Purb |
Cbl |
||||
Pla2g4e |
Ncapd2 |
|||||
Ybx1 |
Cep164 |
|||||
Cdk6 |
Rrm2 |
|||||
Nop2 |
Smc5 |
|||||
Relb |
||||||
Cdc51 |
||||||
Lcmt2 |
||||||
Ccndbp1 |
||||||
Thap1 |
||||||
Gfra1 |
||||||
Jak3 |
||||||
(b) Co-occurring gene alterations in HBsAgtg/0and PML-/-HBsAgtg/0mice |
||||
|---|---|---|---|---|
Cell growth & signal |
ROS pathway |
Transcription & translation |
Ubiquitination |
Others |
Trib3 |
Duox2 |
Creb312 |
Fbxw9 |
Col6a3 |
Nanog |
Gstcd |
Nanog |
Cyld |
Ramp1 |
Cnksr1 |
Pon2 |
Pnrc1 |
Haus7 |
Sycp1 |
Larp7 |
Gtf3a |
Ccdc112 |
||
Disc1 |
Dlx5 |
Slc5a6 |
||
Map3K9 |
Ubtf |
|||
Tceb2 |
||||
Eif5 |
||||
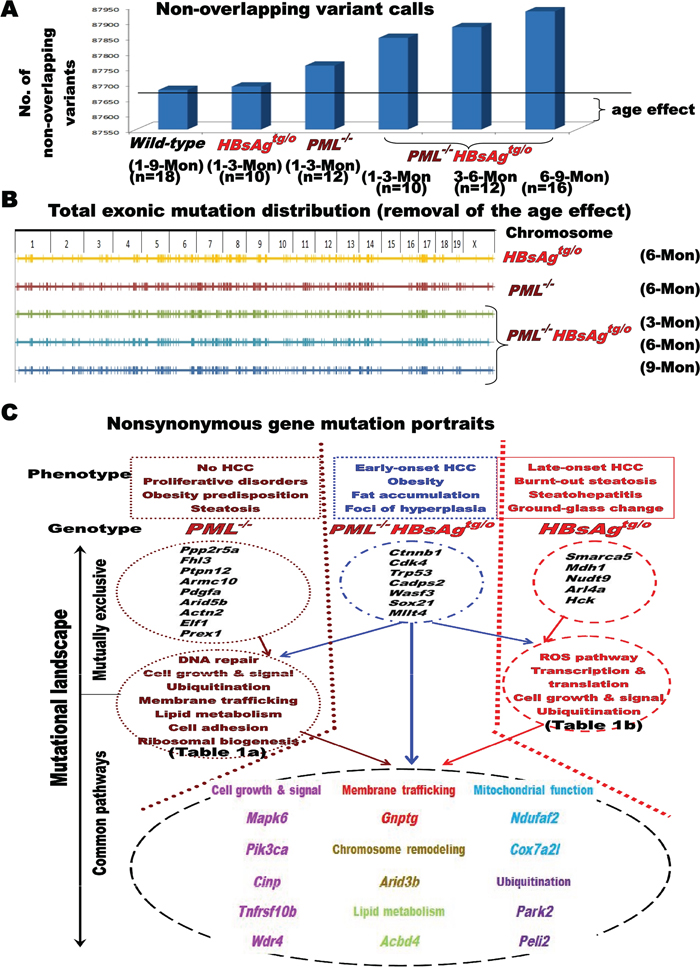
Figure 4: Impact of PML on the mutational landscape of HBsAg-related HCC. A. Effect of age on gene mutations in wild-type, PML+/+HBsAgtg/0 and PML-/-HBsAgtg/0 mice. B. Representative chromosome maps showing the effects of PML deletion and/or HBsAg expression on genomic stability after removing the age effect. C. Diagram illustrating mutually exclusive and co-occurring, nonsynonymous gene mutations in PML-/-, PML-/-HBsAgtg/0 and PML+/+HBsAgtg/0 mice, as well as their functions. Enriched genes were categorized by gene ontology analysis. Known and established gene alterations (>50% mutation rates) are listed and grouped for each genotype to facilitate correlations with the representative phenotype.
Loss of PML increased the mutation rates (> 50%) of the pathways, signaling molecules, or genes involved in DNA repair (Fancc, Ercc1, and Pcna), chromatin remodeling (Arid5b), cell growth (Jak3, Cdk6, and Pdgfa), lipid metabolism (Glud1 and Ugt1a family), ubiquitination (Ube4a, Usp21, and Siah1a), membrane trafficking (Pld5 and Cpne1), and ribosomal biogenesis (Rrs1). Based on our finding that PML-/- mice did not develop HCC, but did develop severe steatosis and proliferative disorders in multiple organs throughout their lifetime (supplementary Fig. S1), a predisposition for mutations in DNA repair genes did not appear to be a prerequisite for HCC initiation.
The knockdown of PML facilitated early mutations in HBsAg-transgenic liver cells. In the preneoplastic livers of PML-/-HBsAgtg/0 mice by 3 months of age, we observed an exceptionally high prevalence (>50%) of mutations in the cancer drivers Ctnnb1 (which encodes β-catenin of the Wnt signaling cascade), Trp53 (which encodes p53 and maintains genomic integrity), Cdk4 (which drives cell cycle progression) and Mllt4 (which contributes to chromatin remodeling). These genes are commonly mutated in human HCCs and other solid tumors [28][29].
The mutation rate was lower in PML+/+HBsAgtg/0 mice than in PML-/- and PML-/-HBsAgtg/0 mice. However, in the presence of PML, albeit at a slow rate after two years, the HBsAg transgene still progressively induced frequent mutations (>50%) in genes of oxidative stress pathways (Duox2, Gstcd, and Pon2), cell growth signaling (Nanog, Trib3, Larp7, Hck, and Disc1), transcriptional factors (Dlx5, Tceb2, and Ubtf), metabolism (Mdh1 and Nudt9), chromatin remodeling (Smarca5), and ubiquitination (Haus7, Fbxw9, and Cyld). The enrichment in oxidative stress-related gene mutations in HBsAg-transgenic mice corresponded to the long-term HBsAg-induced ER stress in the liver of human HBV carriers and subsequent HCC development very late in life [13][14].
The overall longitudinal mutational profiles of the life time in the livers of PML-/-, PML+/+HBsAgtg/0 and PML-/-HBsAgtg/0 mice revealed overlapping features, but also mutually exclusive portraits, which allowed us to correlate genotypes with phenotypic evolution. Many mutually exclusive mutations were previously shown to be involved in human cancer initiation and progression [28][29]. Here, Ctnnb1, Cdk4, Trp53 and Mllt4 were identified in early-onset HCCs of PML-/-HBsAgtg/0 mice, and Smarca5, Mdh1 and Nudt9 were identified in late-onset HCCs of PML+/+HBsAgtg/0 mice. Since the PML gene of PML+/+HBsAgtg/0 mice was not mutated during the whole HBsAg-induced pathogenesis, spatiotemporal correlations of the restoration of PML with HCC progression and burnt-out steatosis after decline of HBsAg implicated that the late-onset, aggressive, angiogenic HCC in PML+/+HBsAgtg/0 mice might be addicted to PML [30][31].
Oncogenic addiction to PML in late-onset HBsAg-induced HCC progression in mice
As2O3 degrades PML in vivo and in vitro, mimicking PML loss in genetically engineered mice [32][33]. We thus treated one-year-old male and female PML+/+HBsAg, PML-/-HBsAgtg/0 and wild-type mice with intraperitoneal injections of As2O3 (3 mg/kg) or PBS (control) every two days for four weeks. Among the three genotypes of mice, only PML+/+HBsAg mice showed increased levels of PML while HBsAg decayed (Figure 5A). Although slight body weight loss was noted in mice treated with As2O3, we observed no differences in physical activity, eating, or physical appearance between mice treated with this dose of As2O3 and mice treated with PBS (placebo control) in groups of the same genotype. After four weeks of As2O3 treatment, we performed in-situ TUNEL analysis on excised mouse livers. We detected an induction of focal and confluent apoptosis in the PML+/+HBsAgtg/0 mice, but not wild-type or PML-/-HBsAgtg/0 mice (Figure 5B). Liver pathology was also examined in the livers excised at one month after the completion of As2O3 treatment (Figure 5C). The livers of wild-type mice treated with As2O3 showed glycogen accumulation (predominantly clear cells positive to PAS-staining) with no hepatocyte degeneration or necrosis. The continuing growth of early-onset, fat-like HCC tumors was not inhibited in PML-/-HBsAgtg/0 mice treated with As2O3, compared to those treated with PBS. In contrast, the severely dysplastic livers with microadenomas of PML+/+HBsAgtg/0 male mice treated with As2O3 exhibited confluent necrotic areas with selective loss of hepatocytes and preservation of endothelial and mesenchymal cells, resulting in sinusoidal reticular ectacia development with inflammatory cell infiltration. However, the mildly dysplastic livers of PML+/+HBsAgtg/0 female mice treated with As2O3 showed multifocal, small necrotic spots filled with inflammatory cell aggregates, similar to a clone-like clustering cell death. These differences were consistent with the finding that HBV-associated pathogenesis typically developed 3-5 months later in females than in males [23].
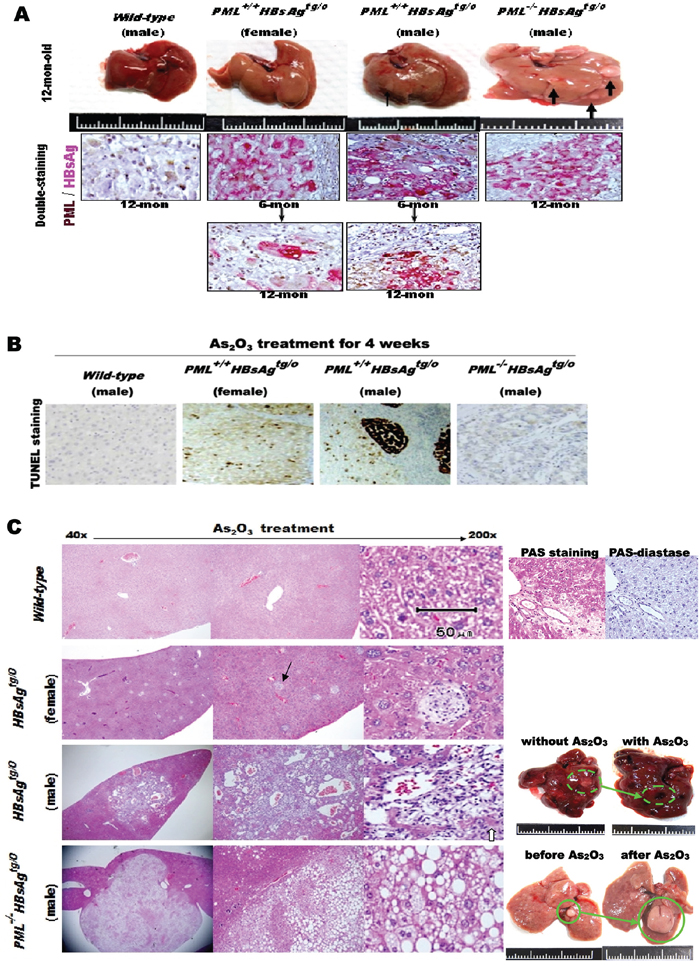
Figure 5: Effects of As2O3 on liver cell death in HBsAg-transgenic mice, with or without PML deletion. A. Macroscopic analysis of representative livers from one-year-old wild-type (n=16), PML+/+HBsAgtg/0 (n-36) and PML-/-HBsAgtg/0 (n=24) mice before As2O3 treatment (top row), showing pale hepatomegaly and multiple small HCCs (thick black arrows) in the PML-/-HBsAgtg/0 liver, and small adenoma (thin black arrow) in the PML+/+HBsAgtg/0 male liver. Double-staining immunohistochemistry of PML and HBsAg in mouse liver sections (lower panels), showing that cytoplasmic HBsAg expression (pink) in HBsAg-transgenic mice is gradually lost over time. Concomitantly, nuclear PML (brown) is restored in the HBsAg-lost liver cells of PML+/+HBsAgtg/0 mice. B. Representative TUNEL-stained sections from the livers of mice treated with As2O3 for 4 weeks. In PML+/+HBsAgtg/0 mice, apoptosis (dark brown) occurred focally in females and confluently in males C. Representative histological images of H&E stained liver sections from mice one month after completion of As2O3 treatment showing glycogen accumulation (purple-magenta) (PAS-diastase (an enzyme that breaks down glycogen) as a control) in wild-type mice and multiple holes (black arrow) in PML+/+HBsAgtg/0 mice, filled with inflammatory cell aggregates due to hepatocyte apoptosis in females, and large apoptotic areas with sinusoidal ectasia due to confluent hepatocyte death in males. Some residual hepatocytes (white arrow) remained within the sinusoidal ectasia. In addition to selective hepatocyte death in PML+/+HBsAgtg/0 mice, there were also vast differences in liver fat accumulation and tumor-onset timing in the livers of PML+/+HBsAgtg/0 and PML-/-HBsAgtg/0 mice at one year of age.
The extent of As2O3-induced cell death in one-year-old PML+/+HBsAgtg/0 mice reflected on decrease in the incidence of liver tumor development at 1.5 years of age (Figures 5C and 6A). The HBsAg transgene, but not the PML deletion, induced HCC and/or hepatocellular adenoma (HCA) and As2O3 treatment significantly increased hepatocyte death while decreasing HCC development only in PML+/+HBsAgtg/0 mice. However, the tumor-inhibitory effect of As2O3 did not dramatically improve survival at two years of age (Figure 6B). Survival rates for PML+/+HBsAgtg/0 mice with very late-onset angiogenic HCC responding to As2O3 were lower than for PML-/-HBsAgtg/0 mice with very early-onset adipose-like HCC resistant to As2O3, suggesting that HCC without PML might have a relatively indolent, low-grade character. Indeed, the survival rate of HBsAg-transgenic mice with HCC was inversely correlated with the severity of long-term HBsAg-induced liver injury, but did not correspond to the HCC size (Figure 6C).
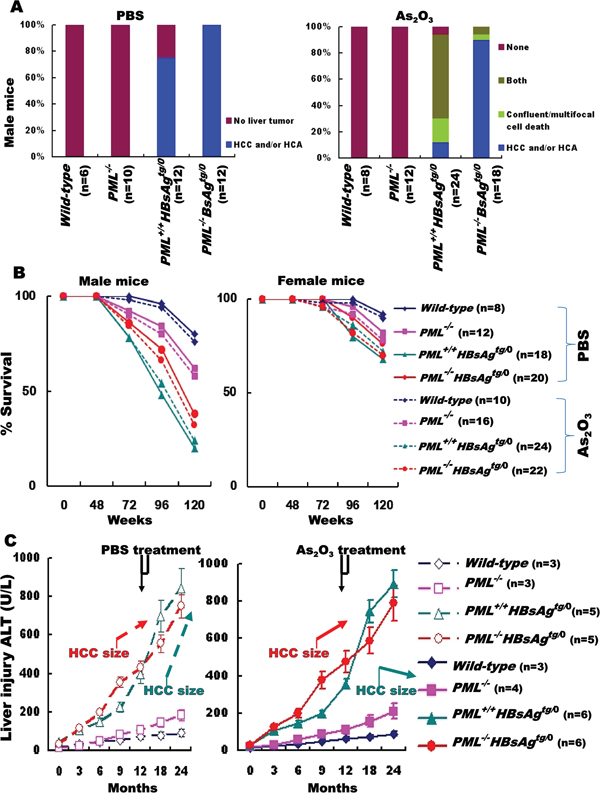
Figure 6: Outcomes of As2O3 treatment for HCC in HBsAg-transgenic mice with or without PML deletion. A. Summary of histopathology performed on livers from one-year-old male mice with different genotypes (wild-type, PML-/-, PML+/+HBsAgtg/0 and PML-/- HBsAgtg/0), one month after treatment with PBS (left) or As2O3 (right) B. Survival rates for male and female mice of various genotypes treated with (dashed lines) or without (solid lines) As2O3. C. Alanine aminotransferase (ALT) levels in surviving male mice (n=3-6) before, during and after PBS (left) or As2O3 (right) treatment (4-week window indicated at top).
DISCUSSION
Unlike 30% of the chronic HBV human carriers susceptible to infection-promoted immune-inflammatory processes of hepatitis, cirrhosis and/or carcinogenesis of the liver, the transgenic mice expressing the entire HBV genome, carrying the HBe or HBV-core protein, or producing infective HBV particles show no increased incidence of HCC and no pathological changes [34][35]. However, 80-100% of the HBsAg-transgenic and 40-80% of the HBx-transgenic mice under different promoter control developed oxidative stress and then a trabecular type of HCC in a male-predominant manner around 1.5-2 years of age, as observed in human HCCs [23][35]. Since HBsAg appears to be a dominant HBV onco-protein, the transgenic mice carrying HBsAg alone might provide insight into virus-host interactions and serve as animal models for evaluation of anti-HBV therapy [36]. In the present study, we found that the growth regulator PML, which can be upregulated by interferon or downregulated by As2O3 [33][37], acts as a physiological, reciprocal interaction partner of HBsAg. Also, PMLs spatiotemporally dose-dependent action affects the phenotypic evolution, tumorigenesis and lipid metabolism of HBsAg-transgenic mice.
PML was initially identified as a tumor suppressor that maintains genomic stability, activates the function of p53, inhibits the PI3K pathway [38] and is commonly degraded in virus infection and frequently lost in various types of cancer [15][16][39]. In contrast, PML has also been shown to behave as an oncogene or pro-survival factor that activates PPAR signaling and enhances fatty acid oxidation to maintain stem cell self-renewal and promote cancer cell survival [30][31][32][40][41][42][43]. In line with the two opposing activities of PML shown in chronic myelocytic leukemia and triple-negative breast carcinoma cells [15][26][40][41][42], we also found a dual role for PML in HBsAg-related pathogenesis. Loss of PML at the early phase predisposed to tumor development while presence of PML at the later phase enhanced metabolic reprogramming and tumor progression. Knockdown of PML increased genome instability and prompted HBsAg-transgenic mice to develop early-onset adipose-like HCCs. When HBsAg was lost and HBsAg-associated PML suppression was relieved, re-appearance of PML in later phase enhanced a metabolic shift from glycogen storage to lipolysis, which implicated increased energy availability for driving the angiogenic HCC progression. Thus, the reciprocal negative interaction observed with differential ratios between HBsAg and PML, which started with HBsAgextensive/PMLsuppression and progressed to HBsAglost/PMLre-appearance, might represent a spectrum of liver diseases with different biological consequences in hepatocarcinogenesis, mutation landscapes and metabolic reprogramming.
Our findings also indicated that parts of the early HBsAg effects on genomic instability and steatosis might be related to degradation of PML, and the late manifestation of burnt-out steatosis during HBsAg-related HCC progression might reflect restoration of PML from the HBsAg-associated degradation due to decline of HBsAg. Previous in vitro studies demonstrated that proteasome-mediated degradation of PML could be triggered by ectopically overexpressed SIAH-1/2 [20]. However, we found that strong expression of SIAH-1/2 in human HBV-related HCC specimens did not correlate with downregulation of PML (Figure 1), suggesting that PML is not a physiological substrate of SIAH-1/2 and the supposed negative interaction between PML and SIAH-1/2 may not account for the tumorigenic effect of HBsAg. Since the PML gene is rarely mutated in solid tumors, and it is not epigenetically silenced, whether the mechanism involved in HBsAg-induced PML degradation is associated with hepatocarcinogenesis remains to be determined [15][16]. Nevertheless, given that PML deficiency resulted in altered metabolism from a glycogenotic state to enhanced adipogenesis and even obesity in HBsAg-transgenic mice (Figure 2 and 5), PML could be the long-sought link between chronic HBV-infection and metabolic reprogramming toward HCC development [43][44][45]. However, it remains unclear how to reconcile PML’s tumor suppressive effects on cancer growth versus its oncogenic activities in enhancing lipid metabolism in different cellular and pathological contexts [13][40][41][42]. Interferon-mediated upregulation of PML transcription and As2O3-mediated PML protein degradation have both been shown to suppress growth and induce apoptosis in human HCC cell lines and animal HCC models [33][37]. However, treatment with either interferon or As2O3 was not effective in patients with advanced HCC in clinical trials [46][47]. This raises concern about the timing of PML-targeting treatments for HBV-related HCC. We showed that pharmacological inhibition of PML by treating mice with As2O3 only selectively killed the long-term HBsAg-transformed liver cells that had low HBsAg levels and rising PML levels. Our findings suggested that it might be counterproductive to use As2O3 prematurely for preventing early-onset HBV-related hepatocarcinogenesis, because in that state (HBsAghigh/PMLlow), PML functions as a tumor suppressor. However, As2O3 is effective when given in the late phase of HBV-related HCC progression (HBsAglow/PMLhigh), because it inhibits PML oncogenic pro-survival effects. On the contrary, interferon should be given early in the PML suppression phase.
Our observations might explain why both interferon and As2O3 treatment failed to show therapeutic benefits on unselected patients with HCC in clinical trials. Thus, our study emphasizes the need to profile HBsAg and PML expression in liver cells prior to embarking on therapy. Since the interaction of PML and HBsAg could be a predictive factor, reanalysis of the previous interferon and As2O3 trials is warranted in HBV-related HCC patients that are stratified by the HBsAg/PML expression ratio. Nevertheless, the corresponding survival gain might be lower than expected. Our study showed that As2O3 decreased the size and incidence of HBsAglow/PMLhigh HCCs but it did not stop the progression of HBsAg-induced liver injury that is also a determining survival factor. On the other hand, based on its effect on promoting tumor progression by enhancing fatty acid oxidation, PML could also be a prognostic factor of cancers that undergo metabolic reprogramming. Consistent with the clinical findings that loss or low levels of PML reflected less aggressive characters in cancer metabolism and better survival [40][41], PML-/-HBsAgtg/0 mice that developed very early-onset fat-accumulating HCC by one year of age lived as long as PML+/+HBsAgtg/0 mice that developed late-onset fat-burning HCC after 1.5 years (Figure 6C).
Resulting from the reciprocally dynamic interactions between HBsAg and PML, the dual activities of PML, the subsequent divergent mutations in growth signaling pathways, and the different phenotypic and metabolic evolutions driven by high and low HBsAg/PML ratios, inter-tumor and intra-tumor heterogeneity should occur with time. Based on this heterogeneity, single-target therapy for HBV-related HCC would be expected, at most, to produce modest response rates with short durations. These insights into the diverse, pleiotropic roles of PML at different stages of HBsAg-induced HCC development and progression could lead to novel therapeutic interventions. For example, combinatorial therapies could be developed to hit multiple targets, including HBsAg (with anti-sense approaches), PML-nuclear bodies (with As2O3 and retinoic acid applications), HBsAg-induced ER stress (with phenylbutyrate), PML-deficient pathways involved in DNA repair (with olaparib), and alterations in lipid metabolism (with bezafibrate). This strategy may lead to therapies with synthetic lethality in both early-onset and late-onset HBV-related HCC and therapies that prevent HBV-associated pathogenesis from liver injury and metabolic reprogramming.
MATERIALS AND METHODS
Ethics statement
This study strictly adhered to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animals were handled according to approved guidelines by the Institutional Animal Care and Use Committee. The animal protocols were approved by the Institutional Review Board of the Koo Foundation Sun Yat-Sen Cancer Center, Taipei, Taiwan. The mice were housed and maintained at the National Laboratory Animal Center in Taiwan, under institutionally approved conditions. The collection of tissue samples followed the recommendations given in the Declaration of Helsinki and its amendments.
Mice
PML-/- mice (129/SV-Pmltm/Ppp from the National Cancer Institute, USA) [26] were crossed with liver-specific HBsAg-transgenic mice (C57BL/6J-Tg(Alb1HBV)44Bri/J from Jackson Lab, USA) [23]. The resulting offspring were then crossed to produce different mouse genotypes, including wild-type, PML+/-, PML-/-, PML+/+HBsAgtg/0, PML+/-HBsAgtg/0, and PML-/-HBsAgtg/0, after several generations. These strains, with similar genetic backgrounds (129/SV x C57BL/6J), were confirmed by whole exome sequencing, PCR genotyping and immunohistochemistry. About 20-50 mice of each genotype were sacrificed at 1-24 months of age for macroscopic and microscopic analyses of the liver, pancreas, spleen, kidneys, brain, intestines, heart and lungs.
Previously, we found that there were no differences in the pathogenesis, incidence, timing, sex disparity and survival of HCC development between the parentalPML+/+HBsAg-transgenic C57BL/6J strain and the PML+/+HBsAg-transgenic mice derived from crossing C57BL/6J with 129/SV background [21]. Similar to the parental PML-knockout 129/SV strain, our genetically mixed PML-/- mice (129/SV x C57BL/6J) did not develop malignancy spontaneously but were prone to proliferative disorders.
Histopathology, in-situ TUNEL staining, and immunohistochemistry
Tissues were fixed in formalin, dehydrated, and embedded in paraffin. Deparaffinized sections (4-5 μm) were treated with 3% H2O2 to block endogenous peroxidase activity, then immersed in boiling 0.01% citric acid (pH 6.0) in a microwave oven for 15 min to enhance antigen retrieval. After cooling, some sections were rehydrated with PBS and stained with hematoxylin and eosin (H&E), the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) reagent (R&D systems), or periodic acid-schiff (PAS) or PAS-diastase (Sigma-Aldrich) for liver glycogen according to the manufacturer’s instructions. Other sections were treated with the following antibodies: anti-PML (Santa Cruz, 1:25 dilution), anti-HBsAg (Dako, 1;100 dilution), or antibodies raised against the E3 ligases, seven in absentia homolog-1 and -2 (Cell Signalling, 1:50 dilution). Immunohistochemistry was performed with the ABC Kit (Dako). An isotype antibody was also used as a control. Immunohistochemical analyses of archival paraffin blocks derived from patients was approved by the Institutional Review Board of the Koo Foundation Sun Yat-Sen Cancer Center, Taipei, Taiwan.
Drug treatment
As2O3 (arsenic trioxide) directly binds PML, which promotes covalent modification of PML by the small ubiquitin-like modifier (SUMO). PML SUMOlyation is followed by ubiquitylation and proteasome-mediated degradation [33]. To test the effect of pharmacological PML inhibition, As2O3 or an equal volume of PBS was injected intraperitoneally into treatment or control mice, respectively. Symptoms of toxicity were evaluated once each week, including changes in skin pigmentation, hair loss, physical activity, food consumption, lethargy and body weight. One month after completing the drug treatment, mouse livers were grossly examined and sectioned for pathology. In addition, approximately 60-100 μl of blood from surviving male mice (n=3-6) were collected from the tail vein. Alanine aminotransferase (ALT), an indicator of the severity of liver injury, was measured in the Clinical Laboratory Medicine at the Koo Foundation Sun Yat-Sen Cancer Center using standard procedures.
Whole exome library preparation and sequencing
Flash-frozen livers were digested overnight with proteinase K (Bioline) and genomic DNA was extracted with a DNeasy Tissue kit (Qiagen). Genomic DNA integrity was assessed by electrophoresis on 1% agarose gels. DNA quantity and quality were examined by analyzing samples on a High Sensitivity DNA Bioanalyzer chip (Agilent Technologies, Santa Clara, CA). Double-stranded DNA concentrations were determined with a Qubit Hi-sensitivity DNA assay kit (Life Technologies). Exome enrichment was performed with the Agilent Sure Select XT Mouse All Exon kit. According to the manufacturer’s recommended protocol, exomes were quantitated and qualified with Qubit and Bioanalyzer chip assays. The enriched libraries were then subjected to sequencing with the Illumina GAIIx or the Illumina HiSeq2500 (Illumina), according to the manufacturer’s protocols. To check the read quality after sequencing, nucleotides with low quality scores (<13) were first trimmed from the sequence reads. Overall, DNA sequencing generated a mean target coverage of 164×, and a mean of 95.5% of the target sequence was covered to a depth of at least 10×. Both coverage and depth were similar between cohorts. The analysis followed the best practices recommended by the Broad Institute. Read sequences were aligned to a mouse reference genome. The exome single nucleotide polymorphism (SNP) calls were produced with the GATK SNP calling pipeline [48]. Variant annotation was conducted with snpEff to add information, such as the name of the gene that harbored the variant, the effect of the mutation (missense variant, synonymous variant, etc.), and information from the SNP database, dbSNP [49][50].
Abbreviations
HBV: hepatitis B virus; HBsAg: HBV surface antigen; HCC: hepatocellular carcinoma; PML: Promyelocytic Leukemia; ER: endoplasmic reticulum; HBsAgtg/o: HBsAg-transgenic mouse; ALT: alanine aminotransferase.
CONFLICTS OF INTEREST
Nothing to report.
GRANT SUPPORT
This work was supported by Ministry of Science and Technology, R.O.C. grants 100-2320-B-368-001, 101-2320-B-368-001-MY3 and 104-2320-B-368-001 (to YC).
REFERENCES
1. Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001; 2: 533-543.
2. Bosch FX, Ribes J, Diaz M, Cléries R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology. 2004; 127: S5–S16.
3. Beasley RP, Lin CC, Hwang LY, Chien CS. Hepatocellular carcinoma and hepatitis B virus: A prospective study of 22,707 men in Taiwan. Lancet. 1981; 2: 1129-1133.
4. Howard CR. The structure of hepatitis B envelope and molecular variants of hepatitis B virus. J Viral Hepat. 1995; 2: 165-70.
5. Moolla N, Kew M, Arbuthnot P. Regulatory elements of hepatitis B virus transcription. J Viral Hepat. 2002; 9: 323-331.
6. Beck J, Nassal M. Hepatitis B virus replication. World J Gastroenterol. 2007; 13: 48-64.
7. Chu CJ, Hussain M, Lok AS. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology. 2002; 36: 1408-1415.
8. Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, Ooi LL, Lee CG. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013; 34: 787-798.
9. Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology. 1989; 9: 452-456.
10. Liaw YF, Sheen IS, Chen TJ, Chu CM, Pao CC. Incidence, determinants and significance of delayed clearance of serum HBsAg in chronic hepatitis B virus infection: a prospective study. Hepatology. 1991; 13: 627-631.
11. Jefferies C, Wynne C, Higgs R. Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat Rev Immunol. 2011; 11: 617-625.
12. Nissar AU, Tasduq SA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic Res. 2015; 30: 1-32.
13. Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet. 2014; 5: 112-119.
14. Churin Y, Roderfeld M, Roeb E. Hepatitis B virus large surface protein: function and fame. Hepatobiliary Surg Nutr. 2015; 4: 1-10.
15. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, Verbel DA, Cordon-Cardo C, Pandolfi PP. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst. 2004; 96: 269-279.
16. Rabellino A, Scaglioni PP. PML Degradation: Multiple Ways to Eliminate PML. Front Oncol. 2013; 3: 60-70.
17. Son SH, Yu E, Choi EK, Lee H, Choi J. Promyelocytic leukemia protein-induced growth suppression and cell death in liver cancer cells. Cancer Gene Ther. 2005; 12: 1-11.
18. Brauckhoff A, Malz M, Tschaharganeh D, Malek N, Weber A, Riener MO, Soll C, Samarin J, Bissinger M, Schmidt J, Longerich T, Ehemann V, Schirmacher P, Breuhahn K. Nuclear expression of the ubiquitin ligase seven in absentia homolog (SIAH)-1 induces proliferation and migration of liver cancer cells. J Hepatol. 2011; 55: 1049-1057.
19. Malz M, Aulmann A, Samarin J, Bissinger M, Longerich T, Schmitt S, Schirmacher P, Breuhahn K. Nuclear accumulation of seven in absentia homologue-2 supports motility and proliferation of liver cancer cells. Int J Cancer. 2012; 131: 2016-2026.
20. Fanelli M, Fantozzi A, De Luca P, Caprodossi S, Matsuzawa S, Lazar MA, Pelicci PG, Minucci S. The coiled-coil domain is the structural determinant for mammalian homologues of Drosophila Sina-mediated degradation of promyelocytic leukemia protein and other tripartite motif proteins by the proteasome. J Biol Chem. 2004; 279: 5374-5379.
21. Chung YL, Wu ML. Promyelocytic leukaemia protein links DNA damage response and repair to hepatitis B virus-related hepatocarcinogenesis. J Pathol. 2013; 230: 377-387.
22. Chung YL. Defective DNA damage response and repair in liver cells expressing hepatitis B virus surface antigen. FASEB J. 2013; 27: 2316-2327.
23. Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989; 59: 1145-1156.
24. Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011; 8: 275-284.
25. Crawford DR, Ostrowski S, Vakharia D, Ilic Z, Sell S. Separate origins of hepatitis B virus surface antigen-negative foci and hepatocellular carcinomas in transgenic HBsAg (alb/psx) mice. Am J Pathol. 2006; 169:223-232.
26. Nagai S, Davoodi N, Gasser SM. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011; 21: 474-485.
27. Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F, Pandolfi PP. Role of PML in cell growth and the retinoic acid pathway. Science. 1998; 279: 1547-1551.
28. Villanueva A, Llovet JM. Liver cancer in 2013: Mutational landscape of HCC--the end of the beginning. Nat Rev Clin Oncol. 2014; 11: 73-74.
29. Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud AL, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc JF, Mazzaferro V, Calvo F, Villanueva A, Nault JC, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015; 47: 505-511.
30. Martin-Martin N, Sutherland JD, Carracedo A. PML: Not all about Tumor Suppression. Front Oncol. 2013; 3: 200-203.
31. Mazza M, Pelicci PG. Is PML a Tumor Suppressor? Front Oncol. 2013; 3: 174-182.
32. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008; 453: 1072-1078.
33. Lallemand-Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, Zhou J, Zhu J, Raught B, de Thé H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol. 2008; 10: 547-555.
34. Zheng Y, Chen WL, Louie SG, Yen TS, Ou JH. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology. 2007; 4:16-21.
35. Teng YC, Shen ZQ, Kao CH, Tsai TF. Hepatocellular carcinoma mouse models: Hepatitis B virus-associated hepatocarcinogenesis and haploinsufficient tumor suppressor genes. World J Gastroenterol. 2016; 22:300-325.
36. Ringelhan M, O'Connor T, Protzer U, Heikenwalder M. The direct and indirect roles of HBV in liver cancer: prospective markers for HCC screening and potential therapeutic targets. J Pathol. 2015; 235:355-367.
37. Herzer K, Hofmann TG, Teufel A, Schimanski CC, Moehler M, Kanzler S, Schulze-Bergkamen H, Galle PR. IFN-alpha-induced apoptosis in hepatocellular carcinoma involves promyelocytic leukemia protein and TRAIL independently of p53. Cancer Res. 2009; 69: 855-862.
38. Ivanschitz L, De Thé H, Le Bras M. PML, SUMOylation, and Senescence. Front Oncol. 2013; 3: 171-178.
39. Regad T, Chelbi-Alix MK. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene. 2001; 20:7274-7286.
40. Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, Adams AC, Sundvall M, Song SJ, Ito K, Finley LS, Egia A, Libermann T, Gerhart-Hines Z, Puigserver P, Haigis MC, Maratos-Flier E, Richardson AL, Schafer ZT, Pandolfi PP. A metabolic prosurvival role for PML in breast cancer. J Clin Invest. 2012; 122: 3088-3100.
41. Lallemand-Breitenbach V, de Thé H. Hematopoietic stem cells burn fat to prevent exhaustion. Cell Stem Cell. 2012; 11: 447-449.
42. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, Pandolfi PP. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012; 18: 1350-1358.
43. Kim MK, Yang S, Lee KH, Um JH, Liu M, Kang H, Park SJ, Chung JH. Promyelocytic leukemia inhibits adipogenesis, and loss of promyelocytic leukemia results in fat accumulation in mice. Am J Physiol Endocrinol Metab. 2011; 301: E1130-E1142.
44. Yoshioka Y, Hashimoto E, Yatsuji S, Kaneda H, Taniai M, Tokushige K, Shiratori K. Nonalcoholic steatohepatitis: cirrhosis, hepatocellular carcinoma, and burnt-out NASH. J Gastroenterol. 2004; 39: 1215-1218.
45. Budhu A1, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL, Fan J, Sun HC, Tang ZY, Wang XW. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013; 144: 1066-1075.
46. Wu J, Henderson C, Feun L, Van Veldhuizen P, Gold P, Zheng H, Ryan T, Blaszkowsky LS, Chen H, Costa M, Rosenzweig B, Nierodzik M, Hochster H, Muggia F, Abbadessa G, Lewis J, Zhu AX. Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2010; 28: 670-676.
47. Zhang CH, Xu GL, Jia WD, Li JS, Ma JL, Ge YS. Effects of interferon treatment on development and progression of hepatocellular carcinoma in patients with chronic virus infection: a meta-analysis of randomized controlled trials. Int J Cancer. 2011; 129: 1254-1264.
48. Zhu P, He L, Li Y, Huang W, Xi F, Lin L, Zhi Q, Zhang W, Tang YT, Geng C, Lu Z, Xu X. OTG-snpcaller: an optimized pipeline based on TMAP and GATK for SNP calling from ion torrent data. PLoS One. 2014; 9: e97507
49. Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front Genet. 2012; 3: 35-43.
50. Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999; 9: 677-679.