
INTRODUCTION
Development of drug resistance is an important factor in the failure of anticancer therapeutic treatments [1]. Such resistance results from a variety of factors including individual variations in patients and somatic cell genetic differences in tumors. The ability to evade medicinal drugs is intrinsic to cancer cells. Reasons for acquisition of anticancer drug resistance include enhanced expression of transporters that increases anticancer drugs efflux, alterations in drug metabolism, mutations of drug targets and the activation of survival or inactivation of downstream death signaling pathways (Figure 1) [1]. Studies on cancer drug resistance mechanisms have yielded valuable information on how to circumvent resistance to improve cancer chemotherapy [1-3].
Reactive oxygen species (ROS) are chemical oxygen species with reactive properties, which comprise hydrogen peroxide (H2O2), the hydroxyl radical (•OH), superoxide(O2-) and singlet oxygen (1O2) [4]. Under physiological conditions, cells are capable of maintaining a balance between cellular oxidants and antioxidants, called redox homeostasis. Submicromolar levels of ROS act as second messengers to regulate cell proliferation, cell death, and other cellular processes [5]. Excessive levels of ROS induce oxidative stress that leads to various pathological states, including aging, neurological disorders, and cancer [6]. In general, most tumors exhibit higher levels of ROS than normal tissues, thus promoting tumor progression and development [5]. Moreover, oxidative stress controls the efficacy of cancer treatments in multiple ways, including chemosensitivity, apoptosis, angiogenesis, metastasis and inflammatory responses [6]. However, when ROS concentrations become extremely high, they lead to tumor cell death [7]. Thus, a variety of drugs with direct or indirect effects on ROS induction have been used for effective cancer therapies (Table 1). Nonetheless, some tumor cells can overcome drug-induced oxidative stress by enhancing their antioxidant systems, with the outcome that a new redox balance with a more higher ROS level is established, the process of ‘Redox Resetting’ (Figure 2). Such drug-induced redox resetting has recently been shown to result in drug resistance. For example, increased levels of reduced glutathione lead to elevated chemotherapeutic drug resistance in numerous cancers [8, 9].
Redox resetting has been implicated in drug resistance at multiple levels, including elevated drug efflux, altered drug metabolism and mutated drug targets [10, 11]. In addition, ROS-induced activation of survival signaling pathways and inactivation of downstream death signaling pathways can lead to drug resistance (Figure 1) [1, 12, 13]. Here, we focus on the effects of redox resetting on drug resistance mechanisms and on current research efforts to reveal the detailed mechanisms of resistance to cancer therapies.
Table 1: Roles of anticancer treatments in regulating ROS levels
Name |
Mechanism of action |
Effects on ROS |
Cancer types |
Refs |
Ionizing radiation |
Photons or particles affect chemical bonds and produce highly ROS, which cause damage to DNA and other cellular components |
Increases ROS production |
Different types of cancer |
[160] |
Methotrexate |
Triggers ROS associated cell apoptosis |
Increases ROS production |
Different types of cancer |
[161] |
Mitoxantrone |
Triggers cell membrane scrambling |
Significant increases of ROS formation |
Different types of cancer |
[162] |
Tamoxifen |
Promotes cancer cell senescence |
Promotes ROS generation |
Breast, colon cancer |
[163] |
Cisplatin |
Generation of nuclear DNA adducts |
Induces a mitochondrial-dependent ROS generation |
Different types of cancer |
[164] |
Paclitaxel (Taxol) |
Inhibitor of cell division |
Increases ROS production |
Different types of cancer |
[165] |
Adriamycin |
Reduces cell viability through initiating cell apoptosis and strong G2/M phase cell cycle arrest |
Increases ROS production |
Different types of cancer |
[166] |
Imatinib |
Protein tyrosine kinase inhibitor that induce apoptosis |
Increases ROS production |
Different types of cancer |
[167] |
Camptothecin |
Quinolone alkaloid that induces cytotoxicity |
Increases ROS production |
Different types of cancer |
[168] |
Flavopiridol |
Semisynthetic flavonoid that inhibits cyclin-dependent kinases |
Increases ROS production |
Leukemia |
[169] |
6-thioguanine |
UVA photosensitizer |
Increases ROS production |
Skin cancer |
[170] |
Procarbazine |
Isolated DNA could be degraded by procarbazine in the presence of oxygen |
Increases ROS production |
Lymphoma, primary brain cancers |
[171] |
NOV-002 |
Glutathione disulphide mimetic |
Alters intracellular GSSG/GSH ratio |
Lung, breast and ovarian cancer |
[172] |
Sulphasalazine |
Inhibitor of cysteine/glutamate transporter xCT |
Reduces intracellular transport of cysteine required for GSH synthesis |
Pancreatic and lung cancer |
|
L-asparaginase |
Depletes glutamine |
Reduces GSH |
Leukemia, pancreatic cancer |
|
Buthionine sulphoximine (BSO) |
Glutamate-cysteine ligase complex inhibitor |
Inhibits de novo GSH synthesis |
Ovarian and breast cancer, melanoma |
|
Carboplatin |
Induction of cell cycle arrest |
Induction of ROS owing to ER stress |
Different types of cancer |
[179] |
Gefitinib |
Selective epidermal growth factor receptor tyrosine kinase inhibitor |
Activates FOXO3a and in turn reduces ROS |
Different types of cancer |
[180] |
Irinotecan |
Topoisomerases inhibitor |
Causes oxidative stress |
Different types of cancer |
[181] |
Etoposide |
Selective Topo II α inhibitor |
Increases ROS production |
Neuroblastoma, breast cancer |
[182] |
Tunicamycin |
Glycosylation inhibitor that causes protein accumulation in the ER |
Triggers ER stress production |
Leukemia |
[183] |
Thapsigargin |
Sarco(endo)plasmic reticulum Ca2+ ATPase inhibitor that releases ER Ca2+ and stimulates Ca2+ influx |
Triggers ER stress production |
Leukemia |
[183] |
Chloroethylnitrosoureas |
Alkylating agent that causes DNA damage |
Increases ROS production |
Melanoma tumors |
[184] |
Temozolomide |
Alkylating agent |
Increases ROS production |
Brain cancer |
[185] |
Celecoxib |
Inhibits cyclooxygenase 2 (COX2) activity but it also induces ER stress by causing leakage of calcium from the ER into the cytosol |
Induction of ROS owing to ER stress |
Colorectal cancer, myeloma, Burkitt’s lymphoma and prostate cancer |
[186] |
Nelfinavir |
Originally developed as HIV protease inhibitor but it also induces ER stress by an unknown mechanism |
Induction of ROS owing to ER stress |
HPV-transformed cervical carcinoma, head and neck cancer, pancreatic cancer, melanoma and glioma |
[187] |
Bortezomib |
Proteasome inhibitor |
Induces ROS owing to ER stress |
Mantle cell lymphoma, multiple myeloma |
|
Anthracyclines (doxorubicin, daunorubicin or epirubicin) |
Insert into the DNA of replicating cells and inhibit topoisomerase II, which prevents DNA and RNA synthesis. |
Induce the generation of oxygen-derived free radicals through two main pathways: anon-enzymatic pathway that utilizes iron, and anenzymatic mechanism that involves the mitochondrial respiratory chain |
Different types of cancer |
[190] |
17-allylaminogeldanamycin (17-AAG) |
HSP90 inhibitor |
Decrease protein homeostasis during oxidative stress by disrupting HSP90–client protein complexes and promoting the degradation of the client proteins |
Breast cancer, non-small-cell lung cancer |
[191] |
Capecitabine |
Prodrug that is enzymatically converted to 5-fluorouracil (5-FU) in the body |
Decreases ROS production |
Colorectal, breast, gastric, and oesophageal cancer |
[192] |
5-fluorouracil (5-FU) |
Inhibits thymidylate synthetase and/or incorporates into RNA and DNA |
Induces intracellular increase inO2·- levels |
Colon cancer, rectum cancer, and head and neck cancer |
[88] |
Arsenic trioxide (As2O3) |
Reacts with cysteine residues on crucial proteins |
Inhibits mitochondrial respiratory function, thereby increasing free radical generation |
Leukemia, myeloma |
[193] |
2-methoxyestradiol(2-ME) |
Metabolite of estradiol-17β |
Induces free radicals and loss of mitochondrial membrane potential |
Prostate cancer, leukemia |
[194] |
N-(4 hydroxyphenyl)retinamide (4-HPR) |
Synthetic retinoid derivative |
Induces apoptosis through the production of ROS and mitochondrial disruption |
Prostate cancer, breast cancer, neuroblastoma |
[195] |
PARP inhibitors |
Inhibit the action of the enzyme PARP |
Reduce the capacity to repair ROS-induced DNA damage |
Breast cancer |
[196] |
Erastin |
Down regulates mitochondrial VDACs and cysteine redox shuttle |
Alters the mitochondrial membrane permeability and blocks GSH regeneration |
RASV12-expressing tumor cells |
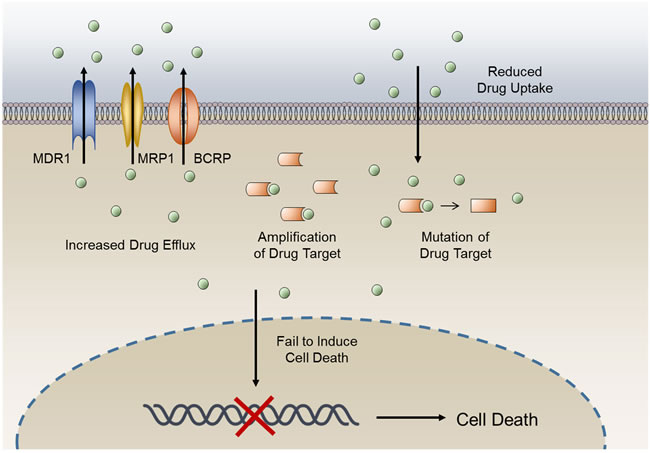
Figure 1: General mechanisms of cancer drug resistance. The anticancer activity of a drug can be limited by reduced drug influx or increased drug efflux, changes in expression levels of drug target, mutation of drug target, and a lack of cell death induction.
INCREASED RATES OF DRUG EFFLUX
Drug export from cells is a primary cause of the cellular resistance to anticancer drugs and poses a significant threat to clinical tumor therapy. Several cell membrane transporter proteins have been implicated in drug resistance to commonly used chemotherapeutics by promoting drug efflux [1]. Among them, the ATP-binding cassette (ABC) transporter family is the most notable. There are 49 members of the ABC transporter family, but only multi-drug resistance protein 1 (MDR1), MDR-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP) have been studied extensively in relation to multidrug resistance (MDR) [10]. All three transporters have broad substrate specificity and promote the efflux of various hydrophobic cancer chemotherapeutics such as topoisomerase inhibitors, taxanes, and antimetabolites [14]. Here, we summarize the effects of redox reactions and redox signals on these three drug efflux transporters.
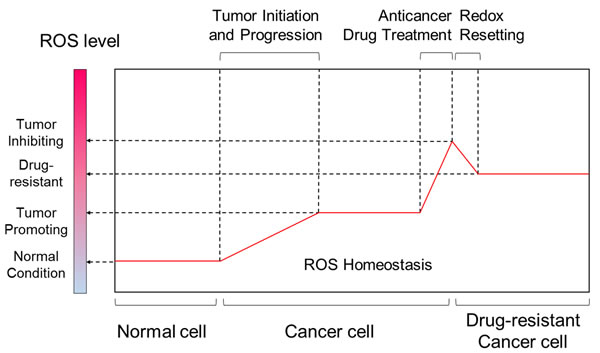
Figure 2: Comparisons of ROS level between different stages of tumor progression and tumor drug-resistance. While in normal cells ROS generation and antioxidants are in balance, increased ROS levels are hallmarks of cancer cells. Marked increase in ROS can be achieved by chemotherapeutic agents, resulting in irreparable cellular damages and cancer cell death. However, some cancer cells can develop drug resistance by redox resetting.
Redox reactions promote conformational changes of the transporters
All ABC transporters contain four domains - two nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs) (Figure 3) [15]. These four domains can be fused into multi-domain polypeptides in a variety of ways. The driving force for drug transport is achieved by a switch between two principal conformations of the NBD dimer [16]. The conformations of ABC transporters are maintained by multiple chemical interactions, including covalent bonds—the intra- and inter-molecular disulfide bond formed between reactive cysteine residues [17]. The cellular redox status has a great impact on reversible disulfide bond formation and is essential for proper protein folding as well as transporter functions.
The drug transport activity of human MDR1 is correlated with the redox states of its two cysteine residues (Cys431 and Cys1074). The ATP hydrolysis activity is strongly inhibited by the covalent reaction of either of these two cysteine residues with N-ethylmaleimide (NEM), a sulfhydryl blocker [18]. These two cysteine residues are present in NBD1 and NBD2 (Figure 3A), and are located very close to the bound nucleotide. The ready formation of the intramolecular disulfide between Cys431 and Cys1074 shows that the two nucleotide-binding sites of MDR1 are structurally very close and capable of intimate functional interactions, consistent with our current understanding of the catalytic mechanism [19].
MRP1 has a topological configuration similar to MDR1, whereas MRP1 has an additional membrane-spanning domain located at the N-terminus, called MSD0 [20]. The MSD0 functions as a plug that controls gating during drug transport (Figure 3B) [21]. Mutations at certain cysteine residues within MSD0 drastically reduce drug-transport activities [22, 23]. A previous study has identified that MSD0 is necessary for the dimerization of MRP1, which can be disrupted by treatment with dithiothreitol (DTT), a reducing agent [24, 25]. These data suggest that dimerization is formed through disulfide linkage between cysteine residues. Yang et al [23] investigated the roles of Cys7 and Cys32, which are located in the MSD0 domain, in MRP1 dimerization (Figure 3B). Mutations at Cys7 caused conformational changes and prevented dimerization in MRP1 [26]. In addition to dimerization, cancer cells activate antioxidant systems after treatment of ROS-inducing anticancer drugs, including enhanced expression of glutathione (GSH), which can form glutathione S-conjugated molecules to facilitate drug efflux by MRP1 [27].
In contrast to the molecular structures of MDR1 and MRP1, BCRP comprises six transmembrane domains and only one ATP-binding cassette, and is known as a ‘half-transporter’ [28]. Human BCRP exists in the plasma membrane as a homodimer due to disulfide-bonded cysteine residues (Figure 3C) [29]. Treatment with 2-mercaptoethanol (2-ME) reduces the BCRP from homodimer to monomer [30]. Three of the cysteine residues, Cys592, Cys 603, and Cys608 in BCRP are located on the extracellular face between TMD5 and TMD6 (Figure 3C) [31-33]. Cys592 and Cys608 are critical for protein stability by intramolecular disulfide bond formation. Mutations at these two cysteine residues result in protein misfolding and degradation, thereby increasing drug sensitivity because of inefficient drug elimination [31-33]. Cys603 is implicated in intermolecular disulfide bond formation, resulting in dimerization of BCRP (Figure 3C). Mutation at Cys603 prevents homodimerization [33]. However, functional analyses demonstrates that mutation at Cys603 do not change the transport activity of the drugs SN-38 and mitoxantrone, even though monomeric BCRP represents only a half-molecule of a functional ABC transporter [32]. Recently, Cys284, Cys374, and Cys438 are also reported to be involved in intramolecular disulfide bond formation and necessary for BCRP function [34].
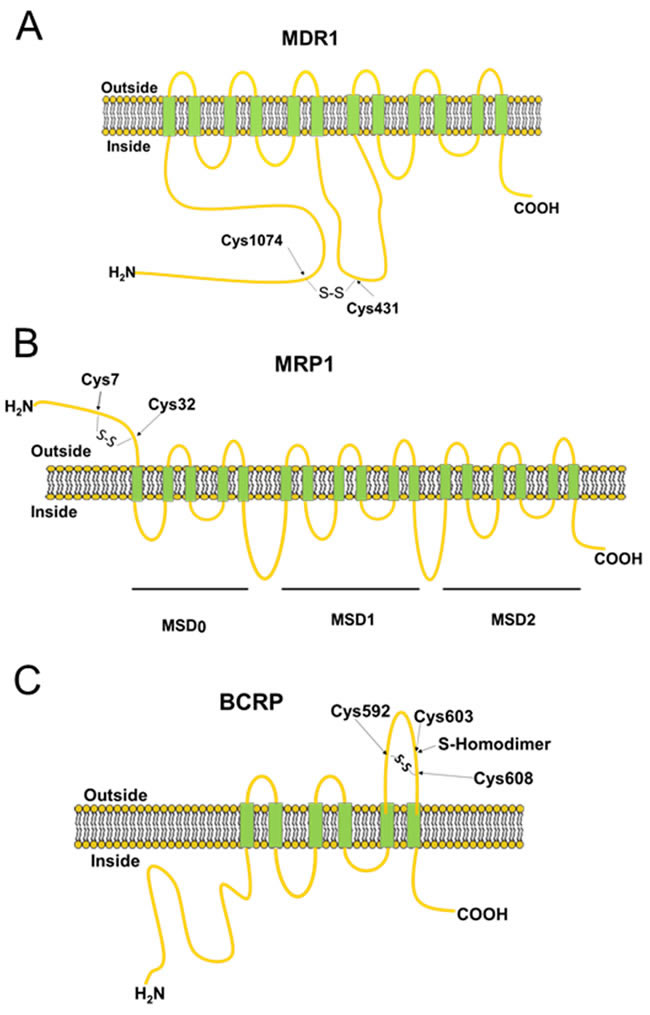
Figure 3: Schematic diagrams showing the structures of MDR1, MRP1 and BCRP. All ABC transporters contain transmembrane and membrane-spanning domains. The disulfide bonds between the cysteine residues identified in the figure are required for maintenance of protein stability and transporter function.
Redox determine transporter gene expression
Apart from the conformational changes of those drug efflux pumps mentioned above, redox-induced overexpression of efflux pumps provides alternative ‘gates’ by which drugs can be exported from cells. Overexpressed transporters have been frequently observed in many types of human malignancy, and correlated with reduced response to chemotherapeutic agents [35]. After treatment with anticancer drugs, redox signaling networks are activated to regulate these transporters expression in multiple layers, including transcriptional, translational, post-translational, and epigenetic levels.
Transcriptional regulation
Accumulating evidence shows that redox-sensing transcription factors take part in the transcriptional regulation of drug efflux transporters (Figure 4). Nuclear factor-erythroid 2 related factor 2 (NRF2), a redox-sensing transcription factor, can bind to antioxidant response element (ARE) and regulates a broad spectrum of genes involved in redox balancing, glutathione synthesis, and drug detoxification [36]. AREs are identified in the promoter region of efflux transporters, such as BCRP and MRPs [36]. In general, NRF2 is anchored in the cytoplasm by Kelch-like ECH-associated protein 1 (KEAP1), which facilitates NRF2 ubiquitination and proteasomal degradation. Cys273 and Cys288 of KEAP1 are the crucial target residues for oxidation. Redox modifications dissociate KEAP1 from NRF2, allowing the translocation of NRF2 to the nucleus, where it transactivates target gene expression (Figure 4) [37]. Recent studies showed that higher levels of NRF2 could promote tumorigenesis and contribute to chemoresistance, suggesting a “dark side” of the NRF2 pathway [38-43]. For example, the expression of NRF2 is increased during acquired resistance to tamoxifen and doxorubicin in breast and ovarian cancer cells [44, 45]. Nuclear accumulation of NRF2 can lead to enhanced expression of ARE-containing genes including drug efflux transporters, which facilitate the development of drug resistance [46]. In addition, overexpression of NRF2 causes enhanced resistance to chemotherapeutic agents, including cisplatin, doxorubicin and etoposide [40]. Higher expressions of NRF2 and its target genes are associated with taxol resistance and anchorage-independent growth in MCF-7 and MDA-MB-231 mammospheres compared to adherent cells [47]. Moreover, transport activities of several MRPs are activated by γ-glutamylcysteine synthetase (γ-GCS, the rate-limiting enzyme for GSH de novo biosynthesis), which can be induced by NRF2 [48].
Forkhead box O (FOXO) proteins, a family of transcription factors, are deregulated in several cancers including prostate, breast, glioblastoma, rhabdomyosarcoma, and leukemia [49]. As inactivation of FOXOs has been determined to be a crucial step in carcinogenesis, increasing their activity could be a potential therapeutic strategy for cancer treatment [49, 50]. FOXOs are not only responsible for the initial therapeutic response to anticancer drugs, but also involved in the acquisition of drug resistance (Figure 4) [51, 52]. Under continuous stress induced by anticancer drugs, FOXOs can elicit the expression of relevant genes for drug efflux and antioxidant defense, such as MDR1, MRP2, Mn-SOD and catalase [50, 53-55]. For instance, FOXO3 and FOXO1 can induce MDR1 expression in adriamycin-resistant breast cancer cells and K562 leukemic cells [50, 54]. In addition, the promoter region of the human MRP2 gene contains four FOXO binding sites, and transcription of MRP2 gene is stimulated by overexpressed FOXO1 in MCF-7 cells [53]. FOXO1 expression is significantly upregulated in a paclitaxel resistant cells and further enhanced by exposure to paclitaxel [56]. Furthermore, FOXO1 overexpression has been frequently observed in cancer tissue samples obtained from chemoresistant patients [57]. Paradoxically, recent studies showed that FOXO3 expression levels were decreased in cisplatin-resistant cells [58], and FOXO3 knockdown increased cell proliferation and enhanced resistance to cisplatin [59].
Ataxia telangiectasia mutated (ATM) is a serine/threonine protein kinase that participates in activation of the DNA damage checkpoint, resulting in cell cycle arrest, DNA repair or apoptosis [60]. Recent studies have revealed a novel mechanism of ATM activation via direct oxidation [61, 62]. When ATM is activated by double-strand breaks (DSBs), the protein undergoes monomerization that requires free DNA ends and the Mre11-Rad50-Nbs1 (MRN) complex. By contrast, when ATM is activated by direct oxidation, oxidized ATM forms an active dimer covalently linked by intermolecular disulfide bonds [61]. Residue Cys2991 is crucial for ATM activation by oxidation. A C2991L mutant cannot be activated by H2O2 but can be normally activated by the MRN complex and DNA [61]. A recent study showed that both camptothecin and cisplatin treatment not only induced ATM activation, but also upregulated MDR-related genes BCRP and MRP2 expression in NCI-H446 cells. Moreover, cisplatin and camptothecin-induced BCRP and MRP2 upregulation can be suppressed by ATM inhibitors, indicating the role of ATM activation on MDR formation in lung cancer chemotherapy [63].
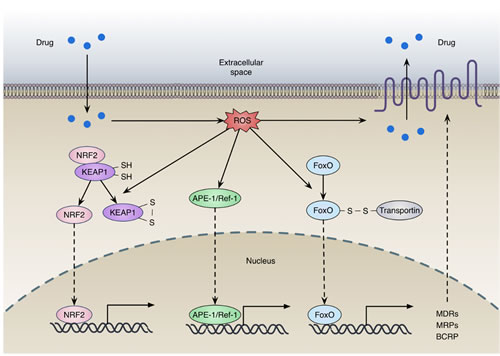
Figure 4: Redox regulation of drug efflux transporters expression. (a) Oxidation of KEAP1 dissociates NRF2 from the complex, allowing the translocation and activation of NRF2; (b) Oxidative stress promotes the translocation of APE-1, facilitating transcription of numerous gene including MDRs, MRPs and BCRP; (c) FOXO can be activated by interacting with transportin through disulfide linkage under oxidative stress. The activation of these transcription factors contributes to the expression of drug efflux transporters.
Post-translational regulation
MDR1 is a phosphorylation substrate for a number of protein kinases, including protein kinase C (PKC) and protein kinase A (PKA) [64]. PKA is shown to be activated by redox modifications through the formation of intramolecular disulfide bonds which cause a subcellular translocation, resulting in phosphorylation of established protein substrates [65]. PKC catalytic properties can be altered by redox mechanisms, which in turn influence the activity of MDR1 [66]. Activation of PKC has been reported to increase the phosphorylation of MDR1 in multidrug-resistant cells [67] and decrease drug accumulation and sensitivity [68]. Conversely, treatment with PKC inhibitors has been shown to decrease the phosphorylation of MDR1, resulting in attenuated drug efflux activity and MDR1 drug binding [69].
Epigenetic regulation
The promoter region of MDR1 is highly GC-rich and contains several CpG islands that are prone to be methylated for transcriptional silencing. Studies have demonstrates that the methylation status of the MDR1 promoter is correlated with MDR1 gene transcriptional activity [70-72]. The methylation is catalyzed by DNA methyltransferases (DNMTs) and use of S-adenosylmethionine (SAM) as a methyl donor. SAM is the first metabolite in the methionine cycle catalyzed by S-adenosylmethionine synthetase (also known as methionine adenosyltransferase, MAT). The activities of MATs are profoundly correlated with redox conditions, through the maintenance of a homotetrameric conformation [73]. The methionine cycle is the primary source of cysteine, a precursor of GSH in the transsulfuration pathway. Intracellular GSH levels are essential in the maintenance of methylated DNA. GSH depletion by hepatotoxin bromobenzene results in a reduction of intracellular methionine pools and genome-wide DNA hypomethylation [74].
ALTERED DRUG METABOLISM
Besides increased rates of drug efflux, altered drug metabolism is another important resistance mechanism, including drug inactivation or deficient drug activation. The redox resetting induced by anticancer drugs may hinder the therapeutic effects by such mechanisms. Antioxidant systems can directly inhibit the antitumor activity of some anticancer agents, such as paclitaxel [75], bortezomib [76] and radiation therapy [77]. For example, buthionine sulphoximine (BSO) significantly increases paclitaxel cytotoxicity through ROS accumulation [75]. Also, platinum drugs, which generate extremely high ROS levels, can be inactivated by GSH [78].
Alternatively, the cellular redox state is correlated with enzymic expression required for the conversion of antimetabolites, such as 5 fluorouracil (5-FU) and methotrexate, to their most active forms [79, 80]. Capecitabine is a fluoropyrimidine prodrug that is converted into 5-FU by thymidinephosphorylase [81]. The gene encoding thymidinephosphorylase can be inactivated by DNA methylation, thereby causing capecitabine resistance [82]. These epigenetic alterations have been shown to be induced by H2O2, where DNMT1 binds more tightly to chromatin after H2O2 treatment and then alters the methylation status of CpG regions [83]. As observed in the case of the topoisomerase inhibitor irinotecan, the inactivation by UDP glucuronosyl transferase 1 (UGT1A1) is induced by the redox-sensing NRF2-KEAP1 pathway [84]. Epigenetic silencing can also promote drug activity, and the expression of UGT1A1 is reduced by DNA methylation of the promoter. Therefore, in this case, promoter methylation promotes irinotecan activity [85, 86].
ALTERATIONS IN THE DRUG TARGETS
Drug response and resistance are also determined by alterations in the drug target, such as mutations or changes in expression level. The deregulated or prolonged production of cellular oxidants has been linked to mutations (induced by oxidant-mediated DNA damage), as well as modification of gene expression [87]. Thus, target alteration is more likely to happen with anticancer drugs that induce high ROS levels.
The fluoropyrimidine 5-FU is widely used in the treatment of a variety of cancers, including colorectal, breast, and aerodigestive tract cancer [88]. It is converted intracellularly to three active metabolites: fluorodeoxyuridinemonophosphate (FdUMP), fluorodeoxyuridinetriphosphate (FdUTP) and fluorouridine triphosphate(FUTP) (Figure 5).These active metabolites disrupt RNA synthesis and the function of thymidylate synthase (TYMS). TYMS plays a crucial role in catalyzing deoxyuridylate (dUMP) to thymidylate (dTMP), which provides the sole intracellular de novo source of dTMP [89]. Human TYMS protein can specifically bind to its own TYMS mRNA and functions as a translational repressor. The RNA binding activity is determined by its redox state. In the presence of reducing agents, the RNA binding activity of TYMS protein is significantly enhanced. In contrast, treatment of TYMS protein with the oxidizing agent diamide inhibits RNA binding [90]. These results demonstrate that the oxidation of TYMS, resulting in loss of translational repressor function, could lead to 5-FU resistance in cancer cells.
Drug target changes through epigenetic events have also been shown to be involved in resistance to cytotoxic chemotherapy drugs in a range of tumor cells. Hypermethylation of the DNA promoter regions of the drug targets results in cell resistance to anticancer drugs, such as cisplatin and carboplatin [91, 92]. In addition, methylation of genes involved in apoptosis, including the DNA mismatch repair (MMR) gene human mutL homolog 1 (hMLH1), can occur in drug-resistant tumor models. This has led to the concept that the use of a DNA demethylating agent such as 2′-deoxy-5-azacytidine in combination with anticancer drugs may reverse this resistance mechanism [93].
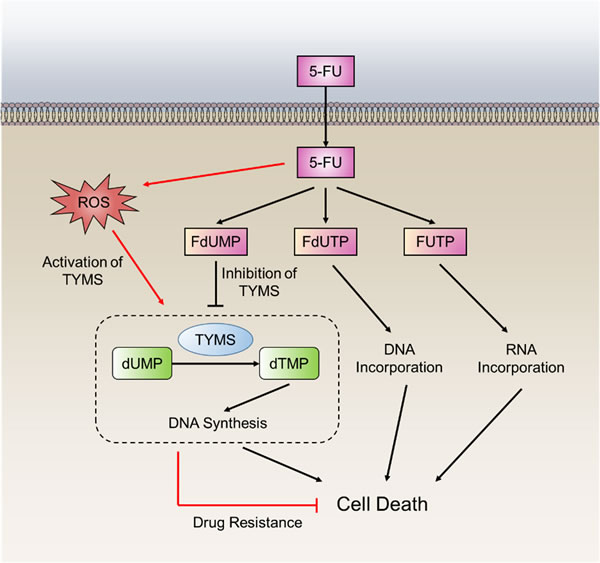
Figure 5: 5-FU resistance in cancer cells by TYMS oxidation. The fluoropyrimidines (5-FU) are broken down into three metabolites, fluorodeoxyuridine monophosphate (FdUMP), fluoro-deoxyuridine triphosphate (FdUTP) and fluorouridine triphosphate (FUTP). The principal mechanism of action of 5-FU is the inhibition of thymidylate synthase (TYMS), but alternative pharmacodynamic pathways acting through incorporation of drug metabolites into DNA and RNA. TYMS can also be activated through direct oxidation that leads to 5-FU resistance.
INEFFECTIVE INDUCTION OF CELL DEATH
Following the action of an activated drug on its cellular target, the therapeutic outcome is then determined by the next key process; the response of cancer cells to drug treatment. Generally, oxidative stress causes by anticancer drugs in turn leads to some cellular damage (e.g., DNA damage) that is tightly coupled to the induction of cell death. Nevertheless, some intrinsic redox adaptive responses can be triggered to enable the cancer cells to survive through inhibition of cell death and activation of cellular survival pathways, thus providing a mechanism of resistance to treatment with anticancer agents [7].
Deregulation of apoptosis
It is well known that resistance to apoptosis is a hallmark of cancer [94]. Thus, deregulation of apoptosis will protect cancer cells from cell death caused by drug-induced cellular damage. Cleavage of caspase-3 is known to play a central role in apoptosis. Substantial evidence reveals that the activity of caspase-3 is inhibited via redox modifications [95]. Caspase-3 has been found to be constitutively S-glutathionylated in human umbilical vein endothelial cells (HUVECs) [96]. Upon tumor necrosis factor α (TNFα) stimulation, de-glutathionylation of caspase-3 occurs mediated by glutaredoxin (Grx). Knockdown of Grx notably inhibit TNFα-induced cell death owing to attenuated caspase-3 cleavage, concomitant with enhanced caspase-3 S-glutathionylation [96]. Mutations of key S-glutathionylation sites of caspase-3 (C163S, C184S, and C220S) enhance cleavage compared with wild-type caspase-3 [97]. Furthermore, S-glutathionylated caspase-3 inhibits its cleavage by caspase-8 in vitro (Figure 6) [97]. In addition, caspase-3 can also be S-nitrosylated at Cys163 [98]. Upon the first apoptosis signal (Fas) ligation, de-nitrosylated caspase-3 leads to caspase-3 activation (Figure 6) [99]. Collectively, the higher ROS levels in drug-resistant cells may contribute to their escape from apoptosis by caspase-3 S-glutathionylation and S-nitrosylation.
Upon Fas ligand (FasL) binding, Fas interacts with Fas-associated protein with death domain (FADD) and procaspase 8 or 10, to form an active death inducing signaling complex (DISC) [100]. The FADD and procaspase-8 interaction can be inhibited by Flice inhibitory protein (FLIP) through competitive binding to FADD [100, 101]. Intriguingly, the activity of FLIP is shown to be enhanced by S-nitrosylation [102]. Loss of S-nitrosylation increases FLIP degradation, which in turn facilitates DISC complex formation, and results in activation of the downstream apoptosis cascade (Figure 6) [102]. FLIP have been shown to be involved in cisplatin-resistance to bladder cancer cells [103]. Also, fibroblast growth factor receptor 4 (FGFR4) has been indicated to be an inducer of chemoresistance in colorectal cancer through regulation of FLIP expression [104]. Thus, inhibition of FLIP may be a promising therapeutic strategy in numerous drug-resistant cancer scenarios [105]. Taken together, these studies described herein highlight that apoptosis is deregulated at multiple layers via redox-associated mechanisms.
Activation of autophagy
Autophagy plays paradoxical roles in acquired resistance to anticancer drugs. On one hand, cytotoxic drug treatment triggers persistent autophagy, which will produce excessive cellular damage and even lead to cell death, thus attenuating the drug resistance activity of cancer cells [106]. On the other hand, autophagy has a role in maintaining cancer cell survival during conditions of stress and might mediate resistance to anticancer therapies [107, 108]. For example, co-administration of cisplatin and an autophagy inhibitor chloroquine significantly suppress tumor survival whereas cisplatin monotherapy fails to show anticancer activity in nude mice xenografts using EC109/CDDP cells [109]. Another study demonstrated that in chronic lymphocytic leukemia (CLL), autophagy was induced by multiple stimuli and acted as a mechanism of resistance against flavopiridol, an endoplasmic reticulum (ER)-stress-mediating agent [110].
Redox resetting has been shown to regulate autophagy at multiple levels. To start with, high cellular ROS accumulation has been widely proven to induce autophagy [111]. In response to ER stress induced by tunicamycin, but not thapsigargin, NADPH oxidase 4 (Nox4)-mediated production of H2O2 leads to cytoprotective autophagy in HUVEC cells [112]. Treatment with either the antioxidant N-acetyl-L-cysteine (NAC) or catalase hinder the conversion of LC3-I to LC3-II which is a key step in autophagy induction, thereby decreasing the formation of LC3-II positive autophagosomes, and reducing starvation-induced protein degradation [113]. The activity of Atg4, which has been shown to be involved in the processing of LC3, has also been proved to be sensitive to H2O2 [113]. However, antioxidant activity is also essential for autophagy induction. For example, overexpression of catalase increases LC3-II levels in both HCT116 cells and H460 cells with low levels of endogenous catalase [114, 115]. Inhibition or knockdown of catalase attenuates LC3-II accumulation in HCT116, H1299 as well as WI38 cells [114, 115]. Due to the paradoxical roles autophagy plays in cancers, a better understanding of how redox regulation distinguishes between the survival-supporting and death-promoting roles of autophagy is necessary [116].
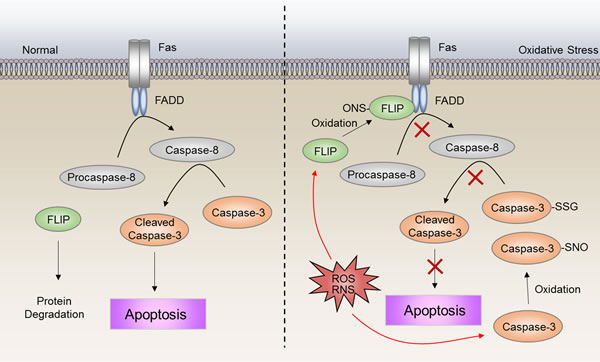
Figure 6: ROS-induced deregulation of apoptosis. S-nitrosylation of FLIP inhibits the interaction between procaspase-8 and FADD, leading to inactivation of caspase-8. S-glutathionylation and S-nitrosylation of caspase-3 inhibit cleavage to the active form. The high ROS level in drug-resistant cells may contribute to escape from apoptosis by S-nitrosylation of FLIP, as well as S-glutathionylation and S-nitrosylation of caspase-3.
DNA damage repair
The anticancer activity of most chemotherapy drugs relies on the induction of DNA damage in rapidly cycling tumor cells with inadequate DNA repair [117]. The cellular response to DNA damage is either repair or cell death. Therefore, the DNA damage repair capacity of cancer cells has a significant influence on the efficacy of DNA-damaging drugs.
The redox environment is capable of directly modulating DNA repair. One of the initial pieces of evidence is that both an increase in 8-oxoguanine (8-oxoG) and a reduction in DNA repair occurs in vitro following treatment with cadmium [118]. This is subsequently shown to be due to cysteine modification of 8-oxoguanine DNA glycosylase 1 (OGG1) [119]. Furthermore, an interaction between OGG1 and poly (ADP-ribose) polymerase 1 (PARP-1), a sensor of DNA damage involved in DNA repair, has recently been described [120]. This interaction is enhanced by oxidative stress and can stimulate PARP-1 activity [120]. Oxidative stress also causes the translocation of the Y-box binding protein (YB-1) to the nucleus, where it has a stable interaction with nei-like 2 protein (NEIL2) and increases NEIL2 activity in the base excision repair (BER) pathway [121]. A further example of redox regulation of DNA damage repair is the interaction between oxidized XRCC1 (x-ray cross-complementing group 1) and DNA polymerase b (Pol b) which is enhanced due to the formation of a disulfide bond [122].
Apurinic-apyrimidinic endonuclease 1 (APE-1) is a versatile protein that has both DNA repair and transcriptional regulatory functions by facilitating transcription factors binding to DNA [123, 124]. Overexpression of APE-1 has been found in several cancers and are correlated with the tumor radiosensitivity [125]. For example, APE-1 contributes to radioresistance [126] and alkylating agent resistance [127] in human glioma cells, as well as also promotes resistance to radiation combined with chemotherapy in medulloblastoma, primitive neuroectodermal tumors and pediatric ependymomas [128]. Knockdown of APE-1 dramatically sensitizes cancer cells to radiotherapy in pancreatic carcinoma [129]. APE-1 can be activated by nontoxic levels of ROS that promote translocation into the nucleus (Figure 4) [130]. ROS production following Ca2+ mobilization via purinergic receptors-induced extracellular ATP stimulation is responsible for the localization of APE-1 [131]. Furthermore APE-1 phosphorylation by PKC after an oxidative challenge has been shown to increase the activity of the APE-1 redox domain [132].
In addition, the activities of other DNA-repair proteins such as Ku, ATM, and human replication protein A (RPA) have been also reported to be altered by the redox resetting. In the nonhomologous end joining (NHEJ) double-strand DNA repair pathway, Ku DNA binding is lower in an oxidizing environment, although the mechanism is not clear [133]. Ku is a heterodimer that encircles broken DNA ends during repair and can recruit the DNA-PK catalytic subunit (DNA-PKcs) [134]. The duration of binding of Ku to the DNA is needed to improve recruitment of DNA-PKcs to the DNA-PK complex [135]. Ku is inactivated during oxidative stress in glucose-6-phosphate dehydrogenase (G6PD) null mutant Chinese hamster ovary cells [136]. ATM is subsequently activated through oxidation at specific cysteine residues [61]. Evidence also shows that ATM can promote an antioxidant response via regulation of the pentose phosphate pathway—one of the primary sources of NADPH [137]. An additional example of a DNA repair pathway protein involved in oxidative stress is human RPA. RPA is a DNA-binding protein involved in replication, repair, and recombination. In an oxidizing environment, the cysteines in the zinc-finger motif of the p70 subunit form disulfide bonds that impair its DNA binding [138].
It has been established that the detection of MMR is associated with resistance to many, but not all, DNA-damaging anticancer agents, such as monofunctional alkylating agents, cisplatin, and the antimetabolite 6-thioguanine [139]. Alkylating agents, including the chloroethylnitrosoureas (carmustine [BCNU], lomustine, and fotemustine), temozolomide, and procarbazine, are commonly used for the treatment of malignant brain tumors [140]. These agents add alkyl groups to DNA causing DNA damage and apoptosis [141]. Resistance to alkylating agents through direct DNA repair by O6-methylguanine methyltransferase (MGMT) remains a significant barrier to successful treatments of patients with malignant glioma [142, 143]. The relative expressions of MGMT in tumor cells may determine the response to alkylating agents [141]. Moreover, promoter methylation can silence MGMT expression in gliomas [144]. Recent studies showed that oxidative damage induced the formation of a large complex containing the DNMTs and polycomb repressive complex 4 (PRC4) members, which could lead to MGMT promoter methylation [145]. Early clinical studies showed that glioma patients with methylated MGMT promoters had a survival benefit treated with radiotherapy [146].
TARGETING REDOX ALTERATIONS IN CANCER THERAPY
In general, cancer cells exhibit higher levels of ROS than normal cells that facilitate tumorigenesis and tumor progression. Therefore, the treatment of antioxidants can suppress cancer initiation or progression. A number of studies suggest that antioxidants could diminish cancer initiation by suppressing DNA damage and genomic instability. For example, ATM-deficiency-accelerated transgenic murine lymphomagenesis is suppressible by NAC [147]. Another study has also claimed that NAC slowed tumor progression in a p53-dependent mouse lymphomagenesis model, seemingly by reducing genomic instability [148]. Furthermore, a recent study has also observed that NAC and vitamin C have significant antitumorigenic effects in vivo. But in stark contrast to earlier studies, they found that the effect of NAC and vitamin C highly relied on hypoxia-inducible factor 1 (HIF-1) rather than on reduction of genomic instability in a MYC-dependent lymphoma model [149]. However, other studies showed that supplementation with vitamin E significantly increased the risk of prostate cancer [150] and taking b-carotene, vitamin A or E supplements increased the incidence of lung cancer [151]. A recent study showing that NAC promotes melanoma progression supports these findings [152]. Likewise, a study showed that NAC increased melanoma metastasis in vivo through the small guanosine triphosphatase (GTPase) RHOA activation [153].
Based on the intrinsic oxidative stress of cancer cells, further ROS inductions have been shown to be efficient in preferentially killing malignant cells, with some showing promise in clinical studies. However, upregulated antioxidant capacity has been found in some cancer cells, especially those in advanced stages. This redox adaptation enables the cancer cells to survive under increased oxidative stress, and provides a mechanism of drug resistance. For example, elevated levels and activity of catalase are found in multidrug resistant HL-60 leukaemia cells [154]. Upregulation of HMOX1, SOD1 and GSH are found to be associated with arsenic trioxide resistance [155]. Also, several studies suggest that the resistance to doxorubicin, paclitaxel or platinum-based drugs, which induce intracellular ROS production, is correlated with increased antioxidant capacity [156, 157].
For those cancer cells that have adapted to higher level of oxidative stress by increasing their antioxidant capacity, simply ROS-generating agents treatment might not be effective. It is possible to combine ROS-generating drugs with compounds that restrain the cellular antioxidant capacity. For example, a combination of arsenic trioxide and 2-Me, a SOD inhibitor, shows significantly enhanced cytotoxic activity in primary chronic lymphocytic leukaemia (CLL) cells [158]. A combination of arsenic trioxide and ascorbic acid, mediating GSH depletion, are also reported to be effective for relapsed or refractory multiple myeloma in a clinical study [159].
CONCLUSIONS
Due to significant advances in the research arena in the last few decades, cancer drug resistance is now realized to be more complicated than originally conceived. The emergence of drug resistance is the result of dynamic battles between cancer cells and chemotherapeutic agents. Although new anticancer drugs will continue to be developed, it is anticipated that novel drug-resistance mechanisms will follow. Therefore, deciphering the intrinsic mechanisms of drug resistance induction may be an effective strategy to solve this significant problem in cancer therapy.
Redox resetting, which usually occurs in anticancer drug treatment, is a protective response from tumor cells that can buffer drug-induced stresses and damage by rebuilding redox homeostasis and activating multiple redox signaling pathways, thereby leading to drug resistance. The versatility of redox signaling is such that it can affect almost every cell and involve multiple signaling processes. Thus it is anticipated that redox signaling will continue to be an important stimulator in the development of drug resistance with new therapeutic agents. Therefore, it is essential to reveal and understand the molecular mechanisms underlying redox resetting-induced drug resistance.
Despite the universality of redox resetting in chemotherapeutic treatments, different agents for distinct cancer types, patients with individual variations, and genetic heterogeneity in tumors may lead to inequable situations. Therefore, only when we have screened and identified enough usable drug-resistant biomarkers related to redox resetting, will it be feasible to overcome drug resistance by monitoring and regulating the process of redox resetting. The use of modern genomic, proteomic and other omics techniques has dramatically promoted the ability to identify novel genes and signaling networks involved in tumor responsiveness to drug treatment. Moreover, high-throughput techniques combined with bioinformatics approaches has allowed the identification of genotypes and molecular signatures also aided the interrogation of clinical samples, facilitating the prediction of drug responses to certain drugs. These will provide abundant data that can be used to identify potential predictive biomarkers for patient stratification.
Abbreviations
ROS : Reactive oxygen species
ABC : the ATP-binding cassette
MDR : multidrug resistance
MRP : MDR-associated protein
BCRP : breast cancer resistance protein
NRF2: Nuclear factor-erythroid 2 related factor 2
ARE : antioxidant response element
FOXO : Forkhead box O
ATM: Ataxia telangiectasia mutated
DNMTs : DNA methyltransferases
5-FU: 5 fluorouracil
APE-1: Apurinic-apyrimidinic endonuclease 1
ACKNOWLEDGMENTS
This work was supported by grants from the National 973 Basic Research Program of China (2013CB911300), the Chinese NSFC (81225015, 81430071, 81401951, 81502131), Sichuan Science-Technology Innovative Research Team for Young Scientist (2013TD0001), and Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJ1400207, KJ1500332).
CONFLICTS OF INTEREST
There is no any competing interest.
Author contributions
All authors contributed to prepare, review, and write the manuscript.
REFERENCES
1. Holohan C, Van Schaeybroeck S, Longley DB and Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013; 13:714-726.
2. Kuczynski EA, Sargent DJ, Grothey A and Kerbel RS. Drug rechallenge and treatment beyond progression—implications for drug resistance. Nature reviews Clinical oncology. 2013; 10:571-587.
3. Bock C and Lengauer T. Managing drug resistance in cancer: lessons from HIV therapy. Nature reviews Cancer. 2012; 12:494-501.
4. Tanaka H, Matsumura I, Ezoe S, Satoh Y, Sakamaki T, Albanese C, Machii T, Pestell RG and Kanakura Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Molecular cell. 2002; 9:1017-1029.
5. Gorrini C, Harris IS and Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature reviews Drug discovery. 2013; 12:931-947.
6. Nathan C and Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nature reviews Immunology. 2013; 13:349-361.
7. Trachootham D, Alexandre J and Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature reviews Drug discovery. 2009; 8:579-591.
8. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM and Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxidative medicine and cellular longevity. 2013; 2013:972913.
9. Tew KD, O’Brien M, Laing NM and Shen H. Coordinate changes in expression of protective genes in drug-resistant cells. Chemico-biological interactions. 1998; 111-112:199-211.
10. Gottesman MM, Fojo T and Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature reviews Cancer. 2002; 2:48-58.
11. Borst P and Elferink RO. Mammalian ABC transporters in health and disease. Annual review of biochemistry. 2002; 71:537-592.
12. Debatin KM and Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004; 23:2950-2966.
13. Lowe SW, Cepero E and Evan G. Intrinsic tumour suppression. Nature. 2004; 432:307-315.
14. Wong K, Ma J, Rothnie A, Biggin PC and Kerr ID. Towards understanding promiscuity in multidrug efflux pumps. Trends in biochemical sciences. 2014; 39:8-16.
15. Higgins CF, Hiles ID, Salmond GP, Gill DR, Downie JA, Evans IJ, Holland IB, Gray L, Buckel SD, Bell AW, Hermodsend MA. A family of related ATP-binding subunits coupled to many distinct biological processes in bacteria. Nature. 1986; 323:448-450.
16. Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007; 446:749-757.
17. Hohl M, Briand C, Grutter MG and Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nature structural & molecular biology. 2012; 19:395-402.
18. Loo TW and Clarke DM. Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. The Journal of biological chemistry. 1995; 270:22957-22961.
19. Jin MS, Oldham ML, Zhang Q and Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012; 490:566-569.
20. Cole SP. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annual review of pharmacology and toxicology. 2014; 54:95-117.
21. Chen Q, Yang Y, Li L and Zhang JT. The amino terminus of the human multidrug resistance transporter ABCC1 has a U-shaped folding with a gating function. The Journal of biological chemistry. 2006; 281:31152-31163.
22. Leslie EM, Letourneau IJ, Deeley RG and Cole SP. Functional and structural consequences of cysteine substitutions in the NH2 proximal region of the human multidrug resistance protein 1 (MRP1/ABCC1). Biochemistry. 2003; 42:5214-5224.
23. Yang Y, Chen Q and Zhang JT. Structural and functional consequences of mutating cysteine residues in the amino terminus of human multidrug resistance-associated protein 1. The Journal of biological chemistry. 2002; 277:44268-44277.
24. Yang Y, Liu Y, Dong Z, Xu J, Peng H, Liu Z and Zhang JT. Regulation of function by dimerization through the amino-terminal membrane-spanning domain of human ABCC1/MRP1. The Journal of biological chemistry. 2007; 282:8821-8830.
25. Yang Y, Li Z, Mo W, Ambadipudi R, Arnold RJ, Hrncirova P, Novotny MV, Georges E and Zhang JT. Human ABCC1 interacts and colocalizes with ATP synthase alpha, revealed by interactive proteomics analysis. Journal of proteome research. 2012; 11:1364-1372.
26. Maiti AK. Genetic determinants of oxidative stress-mediated sensitization of drug-resistant cancer cells. International journal of cancer. 2012; 130:1-9.
27. Krause MS, Oliveira LP, Jr., Silveira EM, Vianna DR, Rossato JS, Almeida BS, Rodrigues MF, Fernandes AJ, Costa JA, Curi R and de Bittencourt PI, Jr. MRP1/GS-X pump ATPase expression: is this the explanation for the cytoprotection of the heart against oxidative stress-induced redox imbalance in comparison to skeletal muscle cells? Cell biochemistry and function. 2007; 25:23-32.
28. Mo W and Zhang JT. Human ABCG2: structure, function, and its role in multidrug resistance. International journal of biochemistry and molecular biology. 2012; 3:1-27.
29. Wakabayashi K, Tamura A, Saito H, Onishi Y and Ishikawa T. Human ABC transporter ABCG2 in xenobiotic protection and redox biology. Drug metabolism reviews. 2006; 38:371-391.
30. Mitomo H, Kato R, Ito A, Kasamatsu S, Ikegami Y, Kii I, Kudo A, Kobatake E, Sumino Y and Ishikawa T. A functional study on polymorphism of the ATP-binding cassette transporter ABCG2: critical role of arginine-482 in methotrexate transport. The Biochemical journal. 2003; 373:767-774.
31. Henriksen U, Fog JU, Litman T and Gether U. Identification of intra- and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. The Journal of biological chemistry. 2005; 280:36926-36934.
32. Kage K, Fujita T and Sugimoto Y. Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer science. 2005; 96:866-872.
33. Wakabayashi K, Nakagawa H, Adachi T, Kii I, Kobatake E, Kudo A and Ishikawa T. Identification of cysteine residues critically involved in homodimer formation and protein expression of human ATP-binding cassette transporter ABCG2: a new approach using the flp recombinase system. Journal of experimental therapeutics & oncology. 2006; 5:205-222.
34. Liu Y, Yang Y, Qi J, Peng H and Zhang JT. Effect of cysteine mutagenesis on the function and disulfide bond formation of human ABCG2. The Journal of pharmacology and experimental therapeutics. 2008; 326:33-40.
35. Thomas H and Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer control. 2003; 10:159-165.
36. Singh A, Wu H, Zhang P, Happel C, Ma J and Biswal S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Molecular cancer therapeutics. 2010; 9:2365-2376.
37. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M and Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:11908-11913.
38. Hayes JD and McMahon M. The double-edged sword of Nrf2: subversion of redox homeostasis during the evolution of cancer. Molecular cell. 2006; 21:732-734.
39. Lau A, Villeneuve NF, Sun Z, Wong PK and Zhang DD. Dual roles of Nrf2 in cancer. Pharmacological research. 2008; 58:262-270.
40. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK and Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008; 29:1235-1243.
41. Kensler TW and Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010; 31:90-99.
42. Ganan-Gomez I, Wei Y, Yang H, Boyano-Adanez MC and Garcia-Manero G. Oncogenic functions of the transcription factor Nrf2. Free radical biology & medicine. 2013; 65:750-764.
43. Sporn MB and Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nature reviews Cancer. 2012; 12:564-571.
44. Kim SK, Yang JW, Kim MR, Roh SH, Kim HG, Lee KY, Jeong HG and Kang KW. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free radical biology & medicine. 2008; 45:537-546.
45. Kaspar JW, Niture SK and Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free radical biology & medicine. 2009; 47:1304-1309.
46. Meijerman I, Beijnen JH and Schellens JH. Combined action and regulation of phase II enzymes and multidrug resistance proteins in multidrug resistance in cancer. Cancer treatment reviews. 2008; 34:505-520.
47. Wu T, Harder BG, Wong PK, Lang JE and Zhang DD. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Molecular carcinogenesis. 2014.
48. Glasauer A and Chandel NS. Targeting antioxidants for cancer therapy. Biochemical pharmacology. 2014; 92:90-101.
49. Myatt SS and Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nature reviews Cancer. 2007; 7:847-859.
50. Yang JY and Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clinical cancer research. 2009; 15:752-757.
51. Zhang Y, Gan B, Liu D and Paik JH. FoxO family members in cancer. Cancer biology & therapy. 2011; 12:253-259.
52. Gomes AR, Zhao F and Lam EW. Role and regulation of the forkhead transcription factors FOXO3a and FOXM1 in carcinogenesis and drug resistance. Chinese journal of cancer. 2013; 32:365-370.
53. Choi HK, Cho KB, Phuong NT, Han CY, Han HK, Hien TT, Choi HS and Kang KW. SIRT1-mediated FoxO1 deacetylation is essential for multidrug resistance-associated protein 2 expression in tamoxifen-resistant breast cancer cells. Molecular pharmaceutics. 2013; 10:2517-2527.
54. Han CY, Cho KB, Choi HS, Han HK and Kang KW. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis. 2008; 29:1837-1844.
55. Putker M, Madl T, Vos HR, de Ruiter H, Visscher M, van den Berg MC, Kaplan M, Korswagen HC, Boelens R, Vermeulen M, Burgering BM and Dansen TB. Redox-dependent control of FOXO/DAF-16 by transportin-1. Molecular cell. 2013; 49:730-742.
56. Wang J, Yang H, Li W, Xu H, Yang X and Gan L. Thioredoxin 1 upregulates FOXO1 transcriptional activity in drug resistance in ovarian cancer cells. Biochimica et biophysica acta. 2015; 1852:395-405.
57. Goto T and Takano M. Transcriptional role of FOXO1 in drug resistance through antioxidant defense systems. Advances in experimental medicine and biology. 2009; 665:171-179.
58. Tezil T, Bodur C, Kutuk O and Basaga H. IKK-beta mediates chemoresistance by sequestering FOXO3; a critical factor for cell survival and death. Cellular signalling. 2012; 24:1361-1368.
59. Shiota M, Yokomizo A, Kashiwagi E, Tada Y, Inokuchi J, Tatsugami K, Kuroiwa K, Uchiumi T, Seki N and Naito S. Foxo3a expression and acetylation regulate cancer cell growth and sensitivity to cisplatin. Cancer science. 2010; 101:1177-1185.
60. Shiloh Y and Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews Molecular cell biology. 2013; 14:197-210.
61. Guo Z, Kozlov S, Lavin MF, Person MD and Paull TT. ATM activation by oxidative stress. Science. 2010; 330:517-521.
62. Guo Z, Deshpande R and Paull TT. ATM activation in the presence of oxidative stress. Cell cycle. 2010; 9:4805-4811.
63. Ke SZ, Ni XY, Zhang YH, Wang YN, Wu B and Gao FG. Camptothecin and cisplatin upregulate ABCG2 and MRP2 expression by activating the ATM/NF-kappaB pathway in lung cancer cells. International journal of oncology. 2013; 42:1289-1296.
64. Mellado W and Horwitz SB. Phosphorylation of the multidrug resistance associated glycoprotein. Biochemistry. 1987; 26:6900-6904.
65. Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC and Eaton P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. The Journal of biological chemistry. 2006; 281:21827-21836.
66. Giorgi C, Agnoletto C, Baldini C, Bononi A, Bonora M, Marchi S, Missiroli S, Patergnani S, Poletti F, Rimessi A, Zavan B and Pinton P. Redox control of protein kinase C: cell- and disease-specific aspects. Antioxidants & redox signaling. 2010; 13:1051-1085.
67. Blobe GC, Sachs CW, Khan WA, Fabbro D, Stabel S, Wetsel WC, Obeid LM, Fine RL and Hannun YA. Selective regulation of expression of protein kinase C (PKC) isoenzymes in multidrug-resistant MCF-7 cells. Functional significance of enhanced expression of PKC alpha. The Journal of biological chemistry. 1993; 268:658-664.
68. Bates SE, Lee JS, Dickstein B, Spolyar M and Fojo AT. Differential modulation of P-glycoprotein transport by protein kinase inhibition. Biochemistry. 1993; 32:9156-9164.
69. Vanoye CG, Castro AF, Pourcher T, Reuss L and Altenberg GA. Phosphorylation of P-glycoprotein by PKA and PKC modulates swelling-activated Cl- currents. The American journal of physiology. 1999; 276:C370-378.
70. Chen M, Xue X, Wang F, An Y, Tang D, Xu Y, Wang H, Yuan Z, Gao W, Wei J, Zhang J and Miao Y. Expression and promoter methylation analysis of ATP-binding cassette genes in pancreatic cancer. Oncology reports. 2012; 27:265-269.
71. Baker EK and El-Osta A. MDR1, chemotherapy and chromatin remodeling. Cancer biology & therapy. 2004; 3:819-824.
72. Baker EK, Johnstone RW, Zalcberg JR and El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene. 2005; 24:8061-8075.
73. Elremaly W, Rouleau T and Lavoie JC. Inhibition of hepatic methionine adenosyltransferase by peroxides contaminating parenteral nutrition leads to a lower level of glutathione in newborn Guinea pigs. Free radical biology & medicine. 2012; 53:2250-2255.
74. Lertratanangkoon K, Wu CJ, Savaraj N and Thomas ML. Alterations of DNA methylation by glutathione depletion. Cancer letters. 1997; 120:149-156.
75. Alexandre J, Batteux F, Nicco C, Chereau C, Laurent A, Guillevin L, Weill B and Goldwasser F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. International journal of cancer. 2006; 119:41-48.
76. Llobet D, Eritja N, Encinas M, Sorolla A, Yeramian A, Schoenenberger JA, Llombart-Cussac A, Marti RM, Matias-Guiu X and Dolcet X. Antioxidants block proteasome inhibitor function in endometrial carcinoma cells. Anti-cancer drugs. 2008; 19:115-124.
77. Bairati I, Meyer F, Gelinas M, Fortin A, Nabid A, Brochet F, Mercier JP, Tetu B, Harel F, Abdous B, Vigneault E, Vass S, Del Vecchio P and Roy J. Randomized trial of antioxidant vitamins to prevent acute adverse effects of radiation therapy in head and neck cancer patients. Journal of clinical oncology. 2005; 23:5805-5813.
78. Meijer C, Mulder NH, Timmer-Bosscha H, Sluiter WJ, Meersma GJ and de Vries EG. Relationship of cellular glutathione to the cytotoxicity and resistance of seven platinum compounds. Cancer research. 1992; 52:6885-6889.
79. Schwartz PM, Moir RD, Hyde CM, Turek PJ and Handschumacher RE. Role of uridine phosphorylase in the anabolism of 5-fluorouracil. Biochemical pharmacology. 1985; 34:3585-3589.
80. Houghton JA and Houghton PJ. Elucidation of pathways of 5-fluorouracil metabolism in xenografts of human colorectal adenocarcinoma. European journal of cancer & clinical oncology. 1983; 19:807-815.
81. Malet-Martino M and Martino R. Clinical studies of three oral prodrugs of 5-fluorouracil (capecitabine, UFT, S-1): a review. The oncologist. 2002; 7:288-323.
82. Kosuri KV, Wu X, Wang L, Villalona-Calero MA and Otterson GA. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochemical and biophysical research communications. 2010; 391:1465-1470.
83. O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, Casero RA, Sears CL and Baylin SB. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer cell. 2011; 20:606-619.
84. Yueh MF and Tukey RH. Nrf2-Keap1 signaling pathway regulates human UGT1A1 expression in vitro and in transgenic UGT1 mice. The Journal of biological chemistry. 2007; 282:8749-8758.
85. Belanger AS, Tojcic J, Harvey M and Guillemette C. Regulation of UGT1A1 and HNF1 transcription factor gene expression by DNA methylation in colon cancer cells. BMC molecular biology. 2010; 11:9.
86. Toffoli G, Cecchin E, Gasparini G, D’Andrea M, Azzarello G, Basso U, Mini E, Pessa S, De Mattia E, Lo Re G, Buonadonna A, Nobili S, De Paoli P and Innocenti F. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. Journal of clinical oncology. 2010; 28:866-871.
87. Klaunig JE and Kamendulis LM. The role of oxidative stress in carcinogenesis. Annual review of pharmacology and toxicology. 2004; 44:239-267.
88. Longley DB, Harkin DP and Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nature reviews Cancer. 2003; 3:330-338.
89. Field MS, Kamynina E, Watkins D, Rosenblatt DS and Stover PJ. Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112:400-405.
90. Lin X, Liu J, Maley F and Chu E. Role of cysteine amino acid residues on the RNA binding activity of human thymidylate synthase. Nucleic acids research. 2003; 31:4882-4887.
91. Chang X, Monitto CL, Demokan S, Kim MS, Chang SS, Zhong X, Califano JA and Sidransky D. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer research. 2010; 70:2870-2879.
92. Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Critical reviews in oncology/hematology. 2007; 63:12-31.
93. Plumb JA, Strathdee G, Sludden J, Kaye SB and Brown R. Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer research. 2000; 60:6039-6044.
94. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
95. Baker A, Santos BD and Powis G. Redox control of caspase-3 activity by thioredoxin and other reduced proteins. Biochemical and biophysical research communications. 2000; 268:78-81.
96. Anathy V, Roberson EC, Guala AS, Godburn KE, Budd RC and Janssen-Heininger YM. Redox-based regulation of apoptosis: S-glutathionylation as a regulatory mechanism to control cell death. Antioxidants & redox signaling. 2012; 16:496-505.
97. Pan S and Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circulation research. 2007; 100:213-219.
98. Mitchell DA and Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nature chemical biology. 2005; 1:154-158.
99. Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ and Stamler JS. Fas-induced caspase denitrosylation. Science. 1999; 284:651-654.
100. Hengartner MO. The biochemistry of apoptosis. Nature. 2000; 407:770-776.
101. Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, Owen LB, Pope RM, Tschopp J, Wajant H, Wallach D, Wiltrout RH, Zornig M and Lynch DH. The CD95 receptor: apoptosis revisited. Cell. 2007; 129:447-450.
102. Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH and Rojanasakul Y. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. Journal of immunology. 2008; 180:3072-3080.
103. Lee S, Yoon CY, Byun SS, Lee E and Lee SE. The role of c-FLIP in cisplatin resistance of human bladder cancer cells. The Journal of urology. 2013; 189:2327-2334.
104. Turkington RC, Longley DB, Allen WL, Stevenson L, McLaughlin K, Dunne PD, Blayney JK, Salto-Tellez M, Van Schaeybroeck S and Johnston PG. Fibroblast growth factor receptor 4 (FGFR4): a targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014; 5:e1046.
105. Wilson TR, McEwan M, McLaughlin K, Le Clorennec C, Allen WL, Fennell DA, Johnston PG and Longley DB. Combined inhibition of FLIP and XIAP induces Bax-independent apoptosis in type II colorectal cancer cells. Oncogene. 2009; 28:63-72.
106. Kopetz S, Lesslie DP, Dallas NA, Park SI, Johnson M, Parikh NU, Kim MP, Abbruzzese JL, Ellis LM, Chandra J and Gallick GE. Synergistic activity of the SRC family kinase inhibitor dasatinib and oxaliplatin in colon carcinoma cells is mediated by oxidative stress. Cancer research. 2009; 69:3842-3849.
107. Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Loze MT, Tang D and Cao L. DAMP-mediated autophagy contributes to drug resistance. Autophagy. 2011; 7:112-114.
108. Friesen C, Fulda S and Debatin KM. Induction of CD95 ligand and apoptosis by doxorubicin is modulated by the redox state in chemosensitive- and drug-resistant tumor cells. Cell death and differentiation. 1999; 6:471-480.
109. Hayes JD and Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Critical reviews in biochemistry and molecular biology. 1995; 30:445-600.
110. T Mahoney E, Byrd JC and Johnson AJ. Autophagy and ER stress play an essential role in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Autophagy. 2013; 9:434-435.
111. Scherz-Shouval R and Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends in biochemical sciences. 2011; 36:30-38.
112. Wu RF, Ma Z, Liu Z and Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Molecular and cellular biology. 2010; 30:3553-3568.
113. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L and Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. The EMBO journal. 2007; 26:1749-1760.
114. Oh SH, Kim YS, Lim SC, Hou YF, Chang IY and You HJ. Dihydrocapsaicin (DHC), a saturated structural analog of capsaicin, induces autophagy in human cancer cells in a catalase-regulated manner. Autophagy. 2008; 4:1009-1019.
115. Choi CH, Jung YK and Oh SH. Selective induction of catalase-mediated autophagy by dihydrocapsaicin in lung cell lines. Free radical biology & medicine. 2010; 49:245-257.
116. Kim Y and Sun H. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging cell. 2007; 6:489-503.
117. Bouwman P and Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature reviews Cancer. 2012; 12:587-598.
118. Dally H and Hartwig A. Induction and repair inhibition of oxidative DNA damage by nickel(II) and cadmium(II) in mammalian cells. Carcinogenesis. 1997; 18:1021-1026.
119. Bravard A, Vacher M, Gouget B, Coutant A, de Boisferon FH, Marsin S, Chevillard S and Radicella JP. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Molecular and cellular biology. 2006; 26:7430-7436.
120. Noren Hooten N, Kompaniez K, Barnes J, Lohani A and Evans MK. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). The Journal of biological chemistry. 2011; 286:44679-44690.
121. Das S, Chattopadhyay R, Bhakat KK, Boldogh I, Kohno K, Prasad R, Wilson SH and Hazra TK. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. The Journal of biological chemistry. 2007; 282:28474-28484.
122. Cuneo MJ and London RE. Oxidation state of the XRCC1 N-terminal domain regulates DNA polymerase beta binding affinity. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:6805-6810.
123. Kelley MR, Georgiadis MM and Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Current molecular pharmacology. 2012; 5:36-53.
124. Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, 2nd, Borch RF, Qiao X, Georgiadis MM and Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxidants & redox signaling. 2008; 10:1853-1867.
125. Evans AR, Limp-Foster M and Kelley MR. Going APE over ref-1. Mutation research. 2000; 461:83-108.
126. Naidu MD, Mason JM, Pica RV, Fung H and Pena LA. Radiation resistance in glioma cells determined by DNA damage repair activity of Ape1/Ref-1. Journal of radiation research. 2010; 51:393-404.
127. Silber JR, Bobola MS, Blank A, Schoeler KD, Haroldson PD, Huynh MB and Kolstoe DD. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clinical cancer research. 2002; 8:3008-3018.
128. Bobola MS, Jankowski PP, Gross ME, Schwartz J, Finn LS, Blank A, Ellenbogen RG and Silber JR. Apurinic/apyrimidinic endonuclease is inversely associated with response to radiotherapy in pediatric ependymoma. International journal of cancer. 2011; 129:2370-2379.
129. Chen S, Xiong G, Wu S and Mo J. Downregulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 enhances the sensitivity of human pancreatic cancer cells to radiotherapy in vitro. Cancer biotherapy & radiopharmaceuticals. 2013; 28:169-176.
130. Ramana CV, Boldogh I, Izumi T and Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:5061-5066.
131. Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, Kelley MR, Quadrifoglio F and Tell G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic acids research. 2005; 33:4379-4394.
132. Hsieh MM, Hegde V, Kelley MR and Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic acids research. 2001; 29:3116-3122.
133. Bennett SM, Neher TM, Shatilla A and Turchi JJ. Molecular analysis of Ku redox regulation. BMC molecular biology. 2009; 10:86.
134. Falck J, Coates J and Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005; 434:605-611.
135. Andrews BJ, Lehman JA and Turchi JJ. Kinetic analysis of the Ku-DNA binding activity reveals a redox-dependent alteration in protein structure that stimulates dissociation of the Ku-DNA complex. The Journal of biological chemistry. 2006; 281:13596-13603.
136. Ayene IS, Stamato TD, Mauldin SK, Biaglow JE, Tuttle SW, Jenkins SF and Koch CJ. Mutation in the glucose-6-phosphate dehydrogenase gene leads to inactivation of Ku DNA end binding during oxidative stress. The Journal of biological chemistry. 2002; 277:9929-9935.
137. Cosentino C, Grieco D and Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. The EMBO journal. 2011; 30:546-555.
138. Men L, Roginskaya M, Zou Y and Wang Y. Redox-dependent formation of disulfide bonds in human replication protein A. Rapid communications in mass spectrometry : RCM. 2007; 21:2743-2749.
139. Penney RB and Roy D. Thioredoxin-mediated redox regulation of resistance to endocrine therapy in breast cancer. Biochimica et biophysica acta. 2013; 1836:60-79.
140. Silber JR, Blank A, Bobola MS, Ghatan S, Kolstoe DD and Berger MS. O6-methylguanine-DNA methyltransferase-deficient phenotype in human gliomas: frequency and time to tumor progression after alkylating agent-based chemotherapy. Clinical cancer research. 1999; 5:807-814.
141. Fu D, Calvo JA and Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nature reviews Cancer. 2012; 12:104-120.
142. Haar CP, Hebbar P, Wallace GCt, Das A, Vandergrift WA, 3rd, Smith JA, Giglio P, Patel SJ, Ray SK and Banik NL. Drug resistance in glioblastoma: a mini review. Neurochemical research. 2012; 37:1192-1200.
143. Liu L and Gerson SL. Targeted modulation of MGMT: clinical implications. Clinical cancer research. 2006; 12:328-331.
144. Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS and Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nature reviews Cancer. 2014; 14:747-753.
145. Ahuja N, Easwaran H and Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. The Journal of clinical investigation. 2014; 124:56-63.
146. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC and Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine. 2005; 352:997-1003.
147. Reliene R and Schiestl RH. Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA repair. 2006; 5:852-859.
148. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE and Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nature medicine. 2005; 11:1306-1313.
149. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, Lee LA, Semenza GL and Dang CV. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer cell. 2007; 12:230-238.
150. Klein EA, Thompson IM, Jr., Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, Klotz L, Parsons JK, Chin JL, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Jama. 2011; 306:1549-1556.
151. Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL, Valanis B, Williams JH, Barnhart S and Hammar S. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. The New England journal of medicine. 1996; 334:1150-1155.
152. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ and Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015; 527:186-191.
153. Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyurek LM, Lindahl P, Nilsson J and Bergo MO. Antioxidants can increase melanoma metastasis in mice. Science translational medicine. 2015; 7:308re308.
154. Lenehan PF, Gutierrez PL, Wagner JL, Milak N, Fisher GR and Ross DD. Resistance to oxidants associated with elevated catalase activity in HL-60 leukemia cells that overexpress multidrug-resistance protein does not contribute to the resistance to daunorubicin manifested by these cells. Cancer chemotherapy and pharmacology. 1995; 35:377-386.
155. Zhou P, Kalakonda N and Comenzo RL. Changes in gene expression profiles of multiple myeloma cells induced by arsenic trioxide (ATO): possible mechanisms to explain ATO resistance in vivo. British journal of haematology. 2005; 128:636-644.
156. Ramanathan B, Jan KY, Chen CH, Hour TC, Yu HJ and Pu YS. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer research. 2005; 65:8455-8460.
157. Hoshida Y, Moriyama M, Otsuka M, Kato N, Taniguchi H, Shiratori Y, Seki N and Omata M. Gene expressions associated with chemosensitivity in human hepatoma cells. Hepato-gastroenterology. 2007; 54:489-492.
158. Zhou Y, Hileman EO, Plunkett W, Keating MJ and Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003; 101:4098-4104.
159. Bahlis NJ, McCafferty-Grad J, Jordan-McMurry I, Neil J, Reis I, Kharfan-Dabaja M, Eckman J, Goodman M, Fernandez HF, Boise LH and Lee KP. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clinical cancer research. 2002; 8:3658-3668.
160. Yoshida T, Goto S, Kawakatsu M, Urata Y and Li TS. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free radical research. 2012; 46:147-153.
161. Chen P, Wang H, Duan Z, Zou JX, Chen H, He W and Wang J. Estrogen-related receptor alpha confers methotrexate resistance via attenuation of reactive oxygen species production and P53 mediated apoptosis in osteosarcoma cells. BioMed research international. 2014; 2014:616025.
162. Arnold M, Bissinger R and Lang F. Mitoxantrone-induced suicidal erythrocyte death. Cellular physiology and biochemistry. 2014; 34:1756-1767.
163. Lee YH, Kang BS and Bae YS. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life sciences. 2014; 97:116-122.
164. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS and Doetsch PW. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PloS one. 2013; 8:e81162.
165. Liu W, Gu J, Qi J, Zeng XN, Ji J, Chen ZZ and Sun XL. Lentinan exerts synergistic apoptotic effects with paclitaxel in A549 cells via activating ROS-TXNIP-NLRP3 inflammasome. Journal of cellular and molecular medicine. 2015; 19:1949-55.
166. Fan C, Zheng W, Fu X, Li X, Wong YS and Chen T. Strategy to enhance the therapeutic effect of doxorubicin in human hepatocellular carcinoma by selenocystine, a synergistic agent that regulates the ROS-mediated signaling. Oncotarget. 2014; 5:2853-2863. doi: 10.18632/oncotarget.1854.
167. Tibullo D, Barbagallo I, Giallongo C, La Cava P, Parrinello N, Vanella L, Stagno F, Palumbo GA, Li Volti G and Di Raimondo F. Nuclear translocation of heme oxygenase-1 confers resistance to imatinib in chronic myeloid leukemia cells. Current pharmaceutical design. 2013; 19:2765-2770.
168. Jayasooriya RG, Choi YH, Hyun JW and Kim GY. Camptothecin sensitizes human hepatoma Hep3B cells to TRAIL-mediated apoptosis via ROS-dependent death receptor 5 upregulation with the involvement of MAPKs. Environmental toxicology and pharmacology. 2014; 38:959-967.
169. Ge Y, Byun JS, De Luca P, Gueron G, Yabe IM, Sadiq-Ali SG, Figg WD, Quintero J, Haggerty CM, Li QQ, De Siervi A and Gardner K. Combinatorial antileukemic disruption of oxidative homeostasis and mitochondrial stability by the redox reactive thalidomide 2-(2,4-difluoro-phenyl)-4,5,6,7-tetrafluoro-1H-isoindole-1,3(2H)-dione (CPS49) and flavopiridol. Molecular pharmacology. 2008; 74:872-883.
170. Gueranger Q, Li F, Peacock M, Larnicol-Fery A, Brem R, Macpherson P, Egly JM and Karran P. Protein oxidation and DNA repair inhibition by 6-thioguanine and UVA radiation. The Journal of investigative dermatology. 2014; 134:1408-1417.
171. Wang J and Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer biology & therapy. 2008; 7:1875-1884.
172. Montero AJ, Diaz-Montero CM, Deutsch YE, Hurley J, Koniaris LG, Rumboldt T, Yasir S, Jorda M, Garret-Mayer E, Avisar E, Slingerland J, Silva O, Welsh C, Schuhwerk K, Seo P, Pegram MD, et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast cancer research and treatment. 2012; 132:215-223.
173. Lo M, Ling V, Low C, Wang YZ and Gout PW. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Current oncology. 2010; 17:9-16.
174. Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, Lam S, Gout PW and Wang YZ. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer chemotherapy and pharmacology. 2009; 64:463-472.
175. Dufour E, Gay F, Aguera K, Scoazec JY, Horand F, Lorenzi PL and Godfrin Y. Pancreatic tumor sensitivity to plasma L-asparagine starvation. Pancreas. 2012; 41:940-948.
176. Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A, Goekbuget N, Schrappe M and Pui CH. L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer. 2011; 117:238-249.
177. O’Dwyer PJ, Hamilton TC, LaCreta FP, Gallo JM, Kilpatrick D, Halbherr T, Brennan J, Bookman MA, Hoffman J, Young RC, Comis RL and Ozols RF. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. Journal of clinical oncology. 1996; 14:249-256.
178. Lewis-Wambi JS, Kim HR, Wambi C, Patel R, Pyle JR, Klein-Szanto AJ and Jordan VC. Buthionine sulfoximine sensitizes antihormone-resistant human breast cancer cells to estrogen-induced apoptosis. Breast cancer research : BCR. 2008; 10:R104.
179. Brozovic A, Vukovic L, Polancac DS, Arany I, Koberle B, Fritz G, Fiket Z, Majhen D, Ambriovic-Ristov A and Osmak M. Endoplasmic reticulum stress is involved in the response of human laryngeal carcinoma cells to Carboplatin but is absent in Carboplatin-resistant cells. PloS one. 2013; 8:e76397.
180. Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS, Du Y, Ko HW, Herbst R and Hung MC. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clinical cancer research. 2013; 19:845-854.
181. Halliwell B. Oxidative stress and cancer: have we moved forward? The Biochemical journal. 2007; 401:1-11.
182. Takeda S, Nishimura H, Koyachi K, Matsumoto K, Yoshida K, Okamoto Y, Amamoto T, Shindo M and Aramaki H. (-)-Xanthatin induces the prolonged expression of c-Fos through an N-acetyl-L-cysteine (NAC)-sensitive mechanism in human breast cancer MDA-MB-231 cells. The Journal of toxicological sciences. 2013; 38:547-557.
183. Zhang Y, Soboloff J, Zhu Z and Berger SA. Inhibition of Ca2+ influx is required for mitochondrial reactive oxygen species-induced endoplasmic reticulum Ca2+ depletion and cell death in leukemia cells. Molecular pharmacology. 2006; 70:1424-1434.
184. Demidem A, Morvan D and Madelmont JC. Bystander effects are induced by CENU treatment and associated with altered protein secretory activity of treated tumor cells: a relay for chemotherapy? International journal of cancer. 2006; 119:992-1004.
185. Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH, Chen TH and Shih CM. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PloS one. 2012; 7:e38706.
186. Zhu J, Song X, Lin HP, Young DC, Yan S, Marquez VE and Chen CS. Using cyclooxygenase-2 inhibitors as molecular platforms to develop a new class of apoptosis-inducing agents. Journal of the National Cancer Institute. 2002; 94:1745-1757.
187. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M, Hollander MC, Kawabata S, Tsokos M, Figg WD, Steeg PS and Dennis PA. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clinical cancer research. 2007; 13:5183-5194.
188. Chen Z, Pittman EF, Romaguera J, Fayad L, Wang M, Neelapu SS, McLaughlin P, Kwak L and McCarty N. Nuclear translocation of B-cell-specific transcription factor, BACH2, modulates ROS mediated cytotoxic responses in mantle cell lymphoma. PloS one. 2013; 8:e69126.
189. Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R and Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clinical cancer research. 2007; 13:5291-5294.
190. Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R and Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacological reports : PR. 2009; 61:154-171.
191. Neckers L and Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clinical cancer research. 2012; 18:64-76.
192. Lee RA, Kim HA, Kang BY and Kim KH. Hemoglobin induces colon cancer cell proliferation by release of reactive oxygen species. World journal of gastroenterology. 2006; 12:5644-5650.
193. Miller WH, Jr., Schipper HM, Lee JS, Singer J and Waxman S. Mechanisms of action of arsenic trioxide. Cancer research. 2002; 62:3893-3903.
194. Zhang Q, Ma Y, Cheng YF, Li WJ, Zhang Z and Chen SY. Involvement of reactive oxygen species in 2-methoxyestradiol-induced apoptosis in human neuroblastoma cells. Cancer letters. 2011; 313:201-210.
195. Lai WL and Wong NS. ROS mediates 4HPR-induced posttranscriptional expression of the Gadd153 gene. Free radical biology & medicine. 2005; 38:1585-1593.
196. Luo X and Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes & development. 2012; 26:417-432.
197. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd and Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012; 149:1060-1072.
198. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S and Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007; 447:864-868.