
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains a dismal disease with a median survival under six months and a five-year survival rate of 5% [1]. No effective therapies for locally advanced or metastatic tumors exist, demonstrating the need to define novel therapeutic strategies and markers, which predict responsiveness.
Phosphatidylinositol 3-kinases (PI3K) build a conserved group of lipid kinases, composed of catalytically and regulatory subunits, which phosphorylate the 3` hydroxyl group of phosphatidylinositols, whereby generating phophatidylinositol (3,4,5) trisphosphate (PI(3,4,5)P3) from PI(4,5)P2 at the inner plasma membrane [2]. Intracellular PIP3 levels are tightly regulated by the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN), which dephosphorylates PIP3 and therefore terminates PI3K signaling [3]. PIP3-induced proximity between the phosphoinositide-dependent protein kinase 1 (PDK1) and the AKT kinase allows PDK1 to phophorylate threonine 308 of AKT1 [2]. The PTEN-PI3K-AKT-mTOR pathway is frequently deregulated in PDAC. At the genetic level, mutations in the catalytical active class Ia PI3K subunit p110α [4](www.sanger.ac.uk/genetics/CGP/cosmic/) and the regulatory subunit p85α [5] as well as amplification of AKT2 [6] were described. Accordingly, mutant p110αH1047R can initiate and drive the carcinogenesis in the murine pancreas and the PI3K-PDK1 pathway is an essential node for KrasG12D-driven murine PDAC [7]. Furthermore, PTEN expression is lost or significantly reduced in human PDAC cell lines and tumor specimens [8] and surrogate markers of PI3K activity, like phosphorylation of AKT [7, 9, 10], were commonly detected in human and murine PDAC. Of note, AKT phosphorylation is inversely correlated with survival of PDAC patients [9, 11, 12]. Functionally, PI3K signaling is linked to processes like proliferation, therapeutic/apoptosis resistance, control of metabolism and angiogenesis in PDAC [13].
Due to the frequent activation of the PI3K pathway, PI3K inhibitors are promising therapeutic targets in solid tumors [14] including PDAC [13]. In agreement, the imidazoquinoline dual PI3K-mTOR inhibitor Bez235 revealed efficacy in in vivo models of PDAC [15, 16]. Furthermore, the pan-class I PI3K inhibitor GDC 0941 prevented tumor progression in an endogenous genetically defined mouse model and a humanized primary orthotopic xenotransplant model of PDAC [7]. Nevertheless, markers, which predict and modulate the response towards PI3K-mTOR inhibitors in PDAC are ill defined. In an attempt to unbiased define modulators of PI3K inhibitor sensitivity, we used a large murine PDAC cell line platform. We demonstrate here that Efemp1 as well as p27Kip1 axis controls responsiveness of PDAC cells towards Bez235.
Results
Murine PDAC cells are sensitive to the dual PI3K/mTor Inhibitor Bez235
To determine the sensitivity of murine KrasG12D-driven or p110αH1047R-driven PDAC cells towards the dual PI3K/mTor inhibitor Bez235, we treated 35 cell lines with Bez235 for 72 hours. Viability was measured using MTT assays and the IC50 values were calculated using a non-linear regression analysis [17]. IC50 values between 2.4 nmol/L for the most sensitive up to 30.8 nmol/L were determined (figure 1A). Statistics can be found in supplemental table 1. No statistically significant difference in the mean IC50 values of murine KrasG12D-driven (mean IC50 value: 9.85 +/- 1.15 nmol/L) and p110αH1047R-driven (mean IC50 value: 7.51 +/- 0.97 nmol/L) PDAC cells was detected (figure 1B), arguing that the PI3K pathway is equally important to maintain viability in both models investigated. Interestingly, cell lines isolated from metastases reveal significantly higher IC50 values (mean IC50 value: 12.15 +/- 1.97 nmol/l) compared to cell lines isolated from primary PDAC (mean IC50 value: 7.43 +/- 0.72 nmol/L) (figure 1C). In contrast to the high sensitivity of the murine PDAC cell lines towards Bez235, IC50 values for the mTOR inhibitor Rad001 are high ranging from 0.28 to 6.49 µmol/L (supplemental table 1), which might argue for a significant contribution of the PI3K inhibition for the Bez235 sensitivity.
To unbiased find genes differentially expressed in murine PDAC cells sensitive to Bez235, we used microarrays available from 28 murine PDAC cell lines. We defined two groups according to an Bez235 IC50 cutoff of 10 nmol/L that best separates the available 28 cell lines with high and low Bez235 IC50 values. The 50 most significant genes that are differentially expressed in cell lines with low and high Bez235 IC50 values are shown in figure 1D. The gene, which was statistically significantly differentially expressed between cells with a low and high IC50 value and revealed the greatest expression difference in both groups, was the EGF-containing fibulin-like extracellular matrix protein 1 (Efemp1/Fibulin3) gene (log fold-change -4.7, p-value 0.02) (figure 1D).
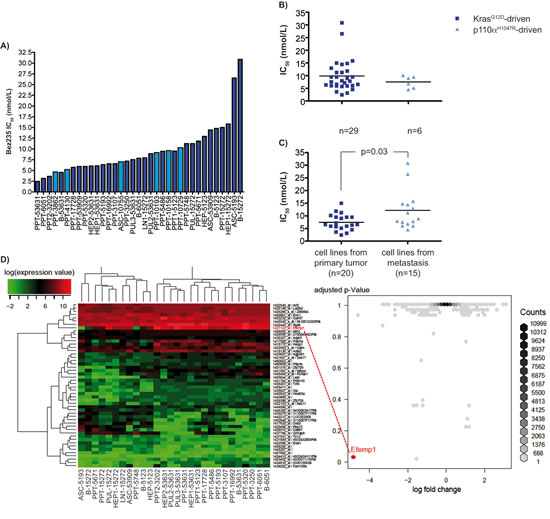
Figure 1: Bez235 IC50 values and differential expressed genes. A) IC50 values of murine PDAC cell lines. 29 murine KrasG12D-driven (dark blue) or 6 p110αH1047R-driven (light blue) PDAC cell lines were treated with different concentrations of Bez235 and viability was determined after 72 hours using MTT assay`s. IC50 values were calculated using a non-linear regression analysis. B) Comparison of the IC50 values of KrasG12D-driven (dark blue) (n=29) or p110αH1047R-driven (light blue) (n=6) PDAC cell lines. Shown is the mean IC50 value of both groups. The p value is indicated. C) Comparison of the IC50 values of cell lines derived from primary PDAC (dark blue) (n=20) or metastasis (light blue) (n=15). Shown is the mean IC50 of both groups. The p value is indicated. D) Differential expression analysis of transcriptome profiles of 18 murine PDAC cell lines with a Bez235 IC50 < 10 nM and 10 murine PDAC cell lines with a Bez235 IC50 > 10 nM. Volcano-plot (right) showing the fold-change and p-value for all probesets calculated by comparing the two groups of samples. Most probesets (dark-grey) show no change in expression levels but some show observable higher probability to be differentially expressed. Efemp1 (log fold-change -4.7, p-value 0.02) stands out from the rest showing high expression levels in Bez235 sensitive murine PDAC cells and low expression in Bez235 insensitive murine PDAC cells. The 50 probesets with the highest fold-change are shown in a heatmap (left).
Efemp1 expression correlates with Bez235 IC50 values
Efemp1 belongs to the Fibulin protein family of secreted glycoproteins, which are components of the extracellular matrix [18]. When array mRNA expression data of 28 cell lines for Efemp1 were correlated with Bez235 IC50 values, a Spearman correlation coefficient of r=0.86 (p<0.0001) was calculated (figure 2A). To corroborate the array expression data, we quantified Efemp1 mRNA expression in 35 murine PDAC cell lines using qPCR (figure 2B). Again, a significant correlation of Efemp1 with Bez235 IC50 values was evident (Spearman correlation coefficient of r=0.62; p<0.0001) (figure 2C), arguing that Efemp1 is a marker for Bez235 sensitivity.
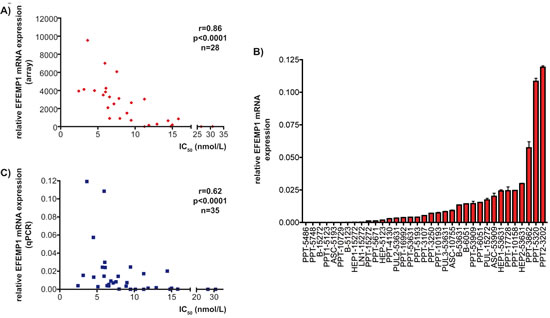
Figure 2: Efemp1 expression correlates with Bez235 IC50 values. A) Bez235 IC50 values of 28 murine PDAC cell lines were correlated with relative Efemp1 expression from the microarray dataset. Spearman r and statistics are indicated. B) Efemp1 mRNA expression of 35 murine PDAC cell lines was determined by qPCR using cyclophilin A mRNA as reference. C) Bez235 IC50 values of 35 murine PDAC cell lines were correlated with relative Efemp1 expression determined by qPCR. Spearman r and statistics are indicated.
Efemp1 increases sensitivity towards Bez235
To demonstrate a functional relevance of Efemp1 modulating Bez235 sensitivity we used gain- and loss-of-function studies. We stably transfected the murine ASC-5193 cell line (IC50 high) with an Efemp1 expression vector. Figure 3A, demonstrates increased expression of the Efemp1 mRNA in two clones compared to control transfected cells. We were not able to detect murine Efemp1 protein with the available Efemp1 antibodies. Bez235 leads to PI3K pathway inhibition, as measured by dephosphorylation of AKT and S6, irrespectively of the stable transfection of Efemp1 (figure 3B). However, as suggested by the correlation of Efemp1 expression with Bez235 IC50 values, elevation of Efemp1 expression increased the sensitivity of murine ASC-5193 towards Bez235 (figure 3C).
In addition to the gain-of-function approach, we used RNAi to demonstrate the effects of Efemp1 towards the Bez235 sensitivity in the murine PPT-53631 cells (IC50 low) as well as in human BxPc3 cells. Knockdown of Efemp1 in PPT-53631 and BxPc3 cells was demonstrated at the level of mRNA using qPCR in PPT-53631 (figure 3D, left graph) and BxPc3 cells (figure 3E, left graph). Reducing Efemp1 expression decreased Bez235 sensitivity in PPT-53631 (figure 3D, right graph) and BxPc3 cells (figure 3E, right graph). Together, these data demonstrate that Efemp1 modulates sensitivity towards Bez235 in cell-based PDAC models.
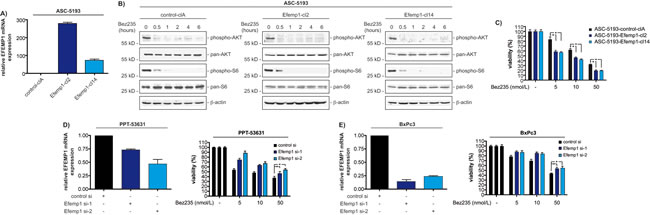
Figure 3: Efemp1 expression modulates Bez235 responsiveness. A) ASC-5193 cells were stably transfected with a control (control-clA) or an Efemp1 expression vector (Efemp1-cl2 and Efemp1-cl14). Efemp1 mRNA expression was determined by qPCR using cyclophilin A mRNA as reference. B) ASC-5193 control-clA, Efemp1-cl2 and Efemp1-cl14 cells were treated with 50 nmol/L Bez235 over time as indicated. Western blots determine phosphorylation of AKT (S473) and S6 (S235/236) as well as expression of pan-AKT and pan-S6. β-actin controls equal protein loading. C) ASC-5193 control-clA, Efemp1-cl2 and Efemp1-cl14 were treated with Bez235 for 72 hours as indicated. Afterwards viability was measured using MTT assays. D) Left graph: PPT-53631 cells were transfected with control siRNA or two specific Efemp1 siRNAs. After 72 hours relative Efemp1 mRNA expression was determined by qPCR using cyclophilin A mRNA as reference. Right graph: PPT-53631 cells were transfected with control siRNA or two specific Efemp1 siRNAs. After 24 hours cells were treated for 72 hours as indicated. Afterwards viability was measured using MTT assays. E) Left graph: BxPc3 cells were transfected with control siRNA or two specific Efemp1 siRNAs. After 72 hours relative Efemp1 mRNA expression was determined by qPCR using cyclophilin A mRNA as reference. Right graph: BxPc3 cells were transfected with control siRNA or two specific Efemp1 siRNAs. After 24 hours cells were treated for 72 hours as indicated. Afterwards viability was measured using MTT assays.
Efemp1 increases the expression of the cyclin-dependent kinase inhibitor p27Kip1
Since we have demonstrated that inhibition of the PI3K pathway in human PDAC models induces a cytostatic response with a G1-phase arrest in the cell cycle involving the cyclin-dependent kinase inhibitor p27Kip1 [19, 20] and p27Kip1 was recently shown to control sensitivity towards dual PI3K/mTOR inhibition [21], we investigated p27Kip1 in ASC-5193 overexpressing Efemp1. p27Kip1 protein levels were significantly elevated in stably Efemp1 transfected clones compared to the control clone (figure 4A). Increased p27Kip1 protein was distributed in the cytoplasm and the nucleus in the investigated ASC-5193 clones (figure 4A, right panel). Consistently, RNAi targeting Efemp1 leads to the downregulation of p27Kip1 protein expression in murine PPT-3202 and PPT-53631 cells (figure 4B). Knockdown in PPT2-3202 and PPT-53631 cells was controlled at the level of Efemp1 mRNA expression (data not shown).
To investigate how Efemp1 controls p27Kip1 protein expression, we measured p27Kip1 mRNA expression and protein turnover. A slight increase in p27Kip1 mRNA expression was observed in stably Efemp1 transfected ASC-5193 cells compared to controls (figure 4C). Protein turnover of p27Kip1 was not changed in ASC-5193 cells, irrespectively of Efemp1 expression (figure 4D). Consistently, the expression of a main regulator of p27Kip1 degradation in PDAC cells, the F-box protein S-phase kinase associated protein 2 (Skp2) [19], was not changed in the stable Efemp1 transfected ASC-5193 clones (figure 4E). To confirm the correlation of p27Kip1 and Bez235 responsiveness, we measured p27Kip1 protein expression in murine PDAC cell lines with different IC50 values. Consistent with the data from stable Efemp1 transfected ASC-5193 cells, p27Kip1 was higher expressed in murine PDAC cell lines with lower Bez235 IC50 values (figure 4F).
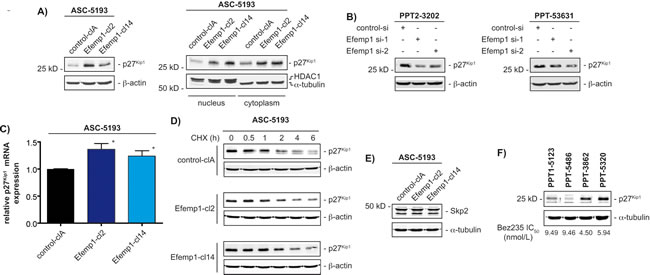
Figure 4: Efemp1 controls expression of p27Kip1. A) Left panel: Western blot detected expression of p27Kip1 in whole cell extracts of ASC-5193 cells stable transfected with a control (control-clA) or an Efemp1 expression vector (Efemp1-cl2 and Efemp1-cl14). β-actin controls equal protein loading. Right panel: Western blot of p27Kip1 expression in cytoplasmatic and nuclear extracts of ASC-5193 cells transfected with a control (control-clA) or an Efemp1 expression vector (Efemp1-cl2 and Efemp1-cl14). HDAC1 and α-tubulin controls cytoplasmatic and nuclear fractionation and loading. B) PPT2-3202 and PPT-53631 cells were transfected with a control siRNA or two specific Efemp1 siRNAs. After 48 hours western blot detected expression of p27Kip1. β-actin controls equal protein loading. C) ASC-5193 cells were stable transfected with a control (control-clA) or an Efemp1 expression vector (Efemp1-cl2 and Efemp1-cl14). p27Kip1 mRNA expression was determined by qPCR using cyclophilin A mRNA as reference. D) ASC-5193 control-clA, Efemp1-cl2 and Efemp1-cl14 cells were treated with cycloheximide (50 µg/ml) over time as indicated. Western blot detected expression of p27Kip1. β-actin controls equal protein loading. E) Skp2 western blot in ASC-5193 control-clA, Efemp1-cl2 and Efemp1-cl14 cells. β-actin controls equal protein loading. F) Western blot of p27Kip1 expression in murine PPT1-5123, PPT-5486, PPT-3862 and PPT-5320 PDAC cell lines. α-tubulin controls equal protein loading. Bez235 IC50 values for each cell line are depicted.
p27Kip1 haploinsufficiency accelerates cancer development in a murine KrasG12D-driven PDAC model
To demonstrate the modulatory effect of p27Kip1 towards the PI3K inhibitor response at the genetic level, we attempted to generate p27Kip1+/- and p27Kip1-/- murine PDAC cell lines. We crossed the p27Kip1 knockout mouse line [22] into the Ptf1aCre/+;LSL-KrasG12D/+ (KC mice thereafter) mouse model of PDAC [23]. Kaplan-Meier analysis demonstrated that p27Kip1 haploinsufficiency accelerates cancer development in the investigated model (figure 5A). PDACs developing in KCp27Kip1+/- (figure 5B, II) mice are differentiated ductal adenocarcinomas as observed in KC mice (figure 5B, I). Furthermore, there is a tendency of an increased macroscopic metastasis rate into lymph nodes, liver and lungs in KCp27Kip1+/- mice (KC, n=43, 39.1% macroscopic metastasis; KCp27Kip1+/-, n=13, 69.2% macroscopic metastasis; Fisher`s exact test p=0.067). Although KCp27Kip1-/- mice die with a median survival of 179 days (figure 5A), no invasive cancers were detected (figure 5B, III). Histological, pancreata of KCp27Kip1-/- mice show desmoplastic reaction with numerous PanIN lesions (figure 5B, III). Pancreata of KCp27Kip1-/- mice are massively enlarged (figure 5B, IV and 5C), leading to obstruction of the surrounding GI-tract and subsequent death. Furthermore, cell lines isolated from KCp27Kip1-/- tumors undergo a senescence crisis in vitro (figure 5B, V).
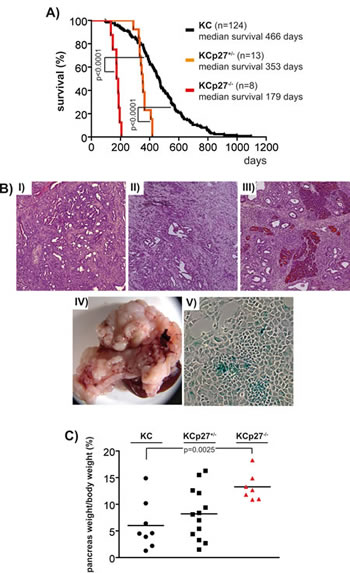
Figure 5: p27Kip1 haploinsufficiency accelerates KrasG12D-dependent cancer development in the murine pancreas. A) Kaplan-Meier survival curves of the indicated genotypes. p values of the log-rank test are indicated. B) Microscopie of the PDAC of I) KC mice and II) KCp27Kip1+/- mice and of the pancreas with typical pre-malignant changes observed in III) KCp27Kip1+/- mice. (original magnification 50x) IV) Macroscopic appearance of the pancreas of KCp27Kip1-/- mice. V) Microscopic images of epithelial cells isolated from KCp27Kip1-/- mice in culture stained for SA-β-galactosidase. (original magnification 100x). C) Pancreas weight/body weight % of the indicated genotypes. p value of the Student`s t-test is indicated.
p27Kip1 expression levels determine Bez235 sensitivity
Expression levels of p27Kip1 were downregulated at protein (figure 6A) and mRNA (figure 6B) level in cell lines from KCp27Kip1+/- mice compared to KC cell lines. p27Kip1 protein and mRNA expression was absent in cell lines from KCp27Kip1-/- mice (figure 6A and 6B). To prove the contribution of p27Kip1 towards the control of Bez235 sensitivity, IC50 values of four cell lines from KCp27Kip1+/- as well as cell lines from KCp27Kip1-/- tumors were determined and compared to the IC50 values of p27Kip1-proficient PDAC cell lines (n=35). As shown in figure 6C, lowering p27Kip1 expression increased IC50 values in a gene dose-dependent manner.
To further establish the role of p27Kip1 as a regulator of Bez235 sensitivity, we reduced the expression of p27Kip1 by increasing Skp2 expression in the murine PPT-6554 cell line and the human MiaPaCa2 cell line. Increasing the expression of Skp2 leads to a significant downregulation of p27Kip1 in PPT-6554 (figure 6D, left panel) and MiaPaCa2 cells (figure 6E, left panel). Lowering p27Kip1 by Skp2 decreases sensitivity of PPT-6554 (figure 6D, right graph) and MiaPaCa2 (figure 6E, right graph) cells for Bez235, further supporting the notion that p27Kip1 is an important regulator of Bez235 sensitivity in cell-based PDAC models.
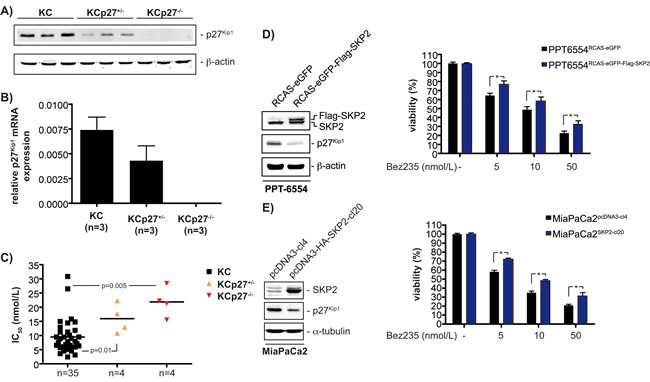
Figure 6: p27Kip1 controls responsiveness of PDAC cells towards Bez235. A) Western blot detects expression of p27Kip1 in the murine cell lines with the indicated genotype. β-actin controls equal protein loading. B) p27Kip1 mRNA expression in cell lines with the indicated genotype was determined by qPCR using cyclophilin A mRNA as reference. C) IC50 values of four KCp27Kip1+/- and four KCp27Kip1-/- cell lines were determined and compared to the IC50 values of 35 murine PDAC cell lines. The p value is indicated. D) Left panel: Murine PPT-6554 cells were transduced with RCAS-Flag-Skp2-EGFP virus or the RCAS-EGFP control retrovirus. Western blot detected expression of Skp2 and p27Kip1. β-actin controls equal protein loading. Right graph: Murine PPT-6554 cells transduced with RCAS-Flag-Skp2-EGFP virus or the RCAS-EGFP control retrovirus were treated with Bez235 as indicated or were left as vehicle treated controls. After 72 hours viability was determined with MTT assays. E) Left panel: Western blot of Skp2 and p27Kip1 expression in MiaPaCa2 cells stably transfected with a control (pcDNA3-cl4) or a HA-tagged Skp2 expression vector (pcDNA3-HA-Skp2-cl20). α-tubulin controls equal protein loading. Right graph: MiaPaCa2 cells stably transfected with a control (pcDNA3-cl4) or a HA-tagged Skp2 expression vector (pcDNA3-HA-Skp2-cl20) were treated with Bez235 as indicated or were left as vehicle treated controls. After 72 hours viability was determined with MTT assays.
Discussion
A goal of personalized medicine is to stratify patients to maximize therapeutic responses to specific targeted therapies. Therefore, there is the need to gain insights into the molecular mode of action of targeted therapies and to define markers and modulators of responsiveness. Here we used a large murine PDAC cell line platform to unbiased find modulators of the response towards the dual PI3K/mTOR inhibitor Bez235. By the use of gain- and loss-of-function experiments, we demonstrate that Efemp1 and p27Kip1 are involved in the control of Bez235 sensitivity of PDAC cells in this preclinical model.
In several tumor entities predictive markers for responsiveness towards PI3K-AKT pathway inhibition were described, whereat genetic lesions in the components of the PI3K-AKT pathway were found positively correlated with PI3K inhibitor sensitivity. Consistently, p110α mutation and/or loss of PTEN expression may characterize sensitive cancer cells [24-29]. Although a low number of patients were included, a recent clinical trial reported increased response rates towards PI3K-AKT-mTOR inhibitors, when patients with gynecologic malignancies were stratified with regard to the presence of a p110α mutation [30]. Consistent, an increased response rate towards PI3K-AKT-mTOR inhibitors in p110α mutated cancers was recently confirmed in a retrospective study [31]. In addition to genetic changes, surrogate marker of PI3K pathway activation, like phosphorylation of AKT, denote increased flux through the signaling pathway and therefore may indicate PI3K inhibitor sensitivity in some cancer models investigated [32, 33]. However, discrepant results for genetic markers as well as pathway activation markers to predict PI3K inhibitor sensitivity were reported. For example, no correlation of the PTEN status with the responsiveness of breast cancer cells towards GDC 0941 was detected [26] and neither p110 nor PTEN mutations were correlated with PI3K inhibitor sensitivity in the JFCR39 human cancer cell line panel [33]. Furthermore, no correlation between basal activity of PI3K-AKT signaling and the potency of Bez235 to induce dephosphorylation of AKT was observed [24] and the extend of inhibition of the PI3K/AKT signaling pathway at the biochemical level induced by PI3K inhibitors does not predict the biological response [34]. These results argue that tumor-type specificities contribute to the discrepant results with respect to markers predicting the response towards PI3K inhibitors. This notion is now also corroborated by our results. Concordantly, mutations in the oncogenes KRAS or BRAF are reported to confer resistance towards PI3K pathway inhibitors [24, 26, 33, 35, 36]. Consistent, murine p110αH1047R-driven lung cancers respond to Bez235 therapy, whereas murine KrasG12D-driven lung cancers do not [37]. In contrast our preclinical murine PDAC cell line platform demonstrates that murine PDAC cells are equally sensitive towards Bez235, irrespectively whether they are initiated and driven by p110αH1047R or KrasG12D oncogenes. These data argue that I) the PI3K pathway is an important node and target for therapeutic intervention in PDAC and II) that each tissue has its unique and specific signaling requirements for tumor maintenance. Furthermore, tumor entity specific markers might be necessary to predict sensitivity towards PI3K inhibition.
The combined partial/complete response rate of p110α mutated cancers towards PI3K-AKT-mTOR inhibitors is only 18% to 30% [30, 31], arguing that further markers predicting responsiveness towards these targeted therapies are needed. We detected the Efemp1 gene differentially expressed in murine PDAC cells with low and high sensitivity for Bez235. Efemp1 belongs to the fibulin protein family, glycoproteins of the extracellular matrix consisting out of Fibulin 1 to 7 [18]. Efemp1 can conduct pro-tumorigenic as well as tumor-suppressive functions dependent on the cancer type. In PDAC, Efemp1 mRNA was upregulated in 13 of 15 investigated cancer specimens [38]. Efemp1 mRNA expression was variable increased ranging from 1.5 to over 10 fold induction in cancers compared to normal tissues [38]. This is consistent with our expression analysis in the murine PDAC cell line platform, which also demonstrates a variable expression of the Efemp1 mRNA. The variable expression of Efemp1 might argue that high Efemp1 expression characterizes a subtype of PDAC. Since our gain- and loss-of-function studies shows that high Efemp1 expression increases the responsiveness towards Bez235, this PDAC subtype might benefit from PI3K inhibitors.
Considering recent observations that Efemp1 can easily be detected in serum of patients with pleural mesothelioma and that high serum Efemp1 levels discriminates asbestos-exposed persons without mesothelioma from asbestos-exposed persons with mesothelioma [39], points to an avenue to include Efemp1 serum levels as a biomarker in trials with PI3K-mTOR inhibitors in PDAC. However, high Efemp1 serum levels were not detected in breast cancer, prostate cancer, ovarian cancer, lung cancer (with effusion) or glioblastoma [39]. Therefore, the definite prove that a PDAC subgroup with high Efemp1 serum levels exists and responds to PI3K-mTOR inhibitors, awaits further clinical investigations.
Mechanistically, Efemp1 was shown to promote metastasis, cell cycle progression and apoptosis resistance in a human PDAC model [38]. To induce changes in cellular behavior of tumor cells, Efemp1 acts in a para- and autocrine fashion by modulating signaling pathways like the EGF receptor pathway [40] or the NOTCH pathway [41]. We detected that the cyclin-dependent kinase inhibitor p27Kip1 is connected to Efemp1 expression. Loss of p27Kip1 expression, which occurs in 50-70% of PDACs, is a marker for poor prognosis of the disease [42-44]. Consistent, we observed acceleration of the KrasG12D-driven carcinogenesis in the pancreas of p27Kip1+/- mice. Furthermore, we provide genetic evidence that higher p27Kip1 expression increases the sensitivity of PDAC cells for Bez235, which might characterize p27Kip1 as a further predictive marker for PI3K-mTOR inhibitor sensitivity in PDAC. Interestingly, p27Kip1 was shown to be predictive for dual PI3K-mTOR inhibitor sensitivity in a cell-based model of pituitary adenomas from rats with multiple endocrine neoplasia-like syndrom [21] and p27Kip1 expression correlated with rapalog sensitivity in the NCI-60 cell line platform [45].
Whereas increasing Efemp1 expression induces p27Kip1 expression, and vice versa, reducing Efemp1 expression reduces p27Kip1 expression in the investigated murine cell lines, it is currently unknown whether p27Kip1 is the sole effector directed by Efemp1 to modulate PI3K-mTOR inhibitor sensitivity. Since the aim of the current study was to unbiased define markers for PI3K-mTOR inhibitor sensitivity in PDAC models, clarification of this topic awaits further experiments beyond the scope of the current manuscript.
Although we provide some evidence that Efemp1 and p27Kip1 are also relevant in human PDAC models to control the sensitivity towards the dual PI3K-mTOR inhibitor, sufficient validation of both markers depends on the availability of a large low-passaged human PDAC cell line platform currently unavailable in our laboratory. Furthermore, additional in vivo studies, beyond the scope of the current manuscript, are needed to additional define the predictive value of Efemp1 and p27Kip1 for PI3K-mTOR inhibitor sensitivity in pre-clinical settings.
In conclusion, we have defined the Efemp1-p27Kip1 axis as predictive for the sensitivity of PDAC cells for the dual PI3K-mTOR inhibitor Bez235 in a murine pre-clinical model. These observations might help to better stratify clinical trials with PI3K-mTOR inhibitors in PDAC.
Material and Methods
Compounds
Are described in supplementary methods.
Cell lines, generation and culture of murine PDAC cells
Primary dispersed murine pancreatic cancer cells were established from genetically engineered KrasG12D- or p110αH1047R -based mouse models of PDAC and cultivated as described [46, 47]. A description of the used murine cell lines can be found in supplemental table 2. Human PDAC cells (MiaPaCa2 and BxPc3) were cultured as described [46].
Mouse strains
Mouse strains are described in supplementary methods. The strains were interbred to obtain mice with the respective genotypes as described [48]. All animal studies were conducted in compliance with European guidelines for the care and use of laboratory animals and were approved by the local authorities.
Histochemistry and SA-β-galactosidase staining
For histopathological analysis, murine specimens were fixed in 4% formaldehyde, embedded in paraffin and sectioned (3 µm thick). Tumors were stained with haematoxylin and eosin as described [48, 49]. SA-β-galactosidase staining is described in supplementary methods.
Total cell lysates, Nuclear extracts, Western blot and Viability assays
Whole cell lysates and nuclear extracts were prepared and western blots were carried out as recently described [46, 47, 50, 51]. Used antibodies are described in supplementary methods. Western blots were performed using Odyssey Infrared Imaging System (LI-COR Biosciences) as described [47]. Viability of the cells was measured using MTT-assays as described [46].
RNA interference
siRNAs were transfected with polyethylenimine (Sigma-Aldrich) at a final concentration of 50 nM as described [52]. siRNAs were purchased from Eurofins (Ebersberg, Germany) and sequences are depicted in supplementary methods.
Quantitative Reverse-Transcriptase PCR
Total RNA was isolated from pancreatic carcinoma cell lines using the RNeasy kit (Qiagen) following the manufacturer`s instructions. Quantitative mRNA analyses were performed as previously described using real-time PCR analysis (TaqMan, PE StepOnePlus, Real time PCR System, Applied Biosystems) [19]. Primers are depicted in supplementary methods.
Gene Expression profiling and bioinformatics
Total RNA from murine PDAC cell lines was prepared using the RNeasy kit (Qiagen). Labeled cRNA was synthesized, hybridized onto GeneChip Mouse Genome 430 2.0 arrays (Affymetrix) [17]. Microarray analysis is described in supplementary methods. Microarray data were submitted to the GEO repository (Accession: GSE40609).
Stable transfection, RCAS virus construction and RCAS virus transduction
Are described in supplementary methods.
Statistical methods
A two-tailed Student`s t-test was used to test statistical significance. * denotes a p-value of at least <0.05. p-values were calculated with GraphPad Prism4 software. IC50 values were calculated with GraphPad Prism4 using a non-linear regression model. Kaplan-Meier survival curves were compared by log-rank test. All data were obtained from three independent experiments performed in triplicate, and the results are presented as mean and standard error of the mean (S.E.M).
Acknowledgments
We thank Birgit Kohnke-Ertel for excellent technical support. We thank the Novartis AG for the synthesis and providing of Bez235. Furthermore, we thank all colleagues providing mouse lines and plasmids. This work was supported by the DFG (SFB824/C9 to D.S. and G.S. and SFB824/Z2 to I.E.), Novartis-Stiftung für therapeutische Forschung (to G.S. and D.S.) and the Rudolf-Bartling Stiftung (Projekt IV/108 to G.S.).
Disclosure of Potential Conflicts of Interest:
There are no conflicts of interest.
Reference
1. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010; 362(17):1605-1617.
2. Engelman JA, Luo J and Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006; 7(8):606-619.
3. Salmena L, Carracedo A and Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008; 133(3):403-414.
4. Jimeno A, Tan AC, Coffa J, Rajeshkumar NV, Kulesza P, Rubio-Viqueira B, Wheelhouse J, Diosdado B, Messersmith WA, Iacobuzio-Donahue C, Maitra A, Varella-Garcia M, Hirsch FR, Meijer GA and Hidalgo M. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer Res. 2008; 68(8):2841-2849.
5. Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, Dela Vega T, Kenski DM, Bowman KK, Lorenzo M, Li H, Wu J, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009; 16(6):463-474.
6. Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK and Testa JR. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996; 93(8):3636-3641.
7. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, Michalski CW, Schlitter AM, Esposito I, Kind AJ, Rad L, Schnieke A, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013; in press.
8. Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL and Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004; 23(53):8571-8580.
9. Yamamoto S, Tomita Y, Hoshida Y, Morooka T, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H, Nakamori S, Monden M and Aozasa K. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2004; 10(8):2846-2850.
10. Schlieman MG, Fahy BN, Ramsamooj R, Beckett L and Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003; 89(11):2110-2115.
11. Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, Enders GH, Zhang R, Sansom OJ and Adams PD. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011; 42(1):36-49.
12. Nitsche C, Edderkaoui M, Moore RM, Eibl G, Kasahara N, Treger J, Grippo PJ, Mayerle J, Lerch MM and Gukovskaya AS. The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology. 2012; 142(2):377-387 e371-375.
13. Sun C, Rosendahl AH, Andersson R, Wu D and Wang X. The role of phosphatidylinositol 3-kinase signaling pathways in pancreatic cancer. Pancreatology. 2011; 11(2):252-260.
14. McCubrey JA, Steelman LS, Chappell WH, Sun L, Davis NM, Abrams SL, Franklin RA, Cocco L, Evangelisti C, Chiarini F, Martelli AM, Libra M, Candido S, Ligresti G, Malaponte G, Mazzarino MC, et al. Advances in Targeting Signal Transduction Pathways. Oncotarget. 2013.
15. Cao P, Maira SM, Garcia-Echeverria C and Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. Br J Cancer. 2009; 100(8):1267-1276.
16. Venkannagari S, Fiskus W, Peth K, Atadja P, Hidalgo M, Maitra A and Bhalla KN. Superior efficacy of co-treatment with dual PI3K/mTOR inhibitor NVP-BEZ235 and pan-histone deacetylase inhibitor against human pancreatic cancer. Oncotarget. 2012; 3(11):1416-1427.
17. Fritsche P, Seidler B, Schüler S, Schnieke A, Göttlicher M, Schmid RM, Saur D and Schneider G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009; 58(10):1399-1409.
18. de Vega S, Iwamoto T and Yamada Y. Fibulins: multiple roles in matrix structures and tissue functions. Cell Mol Life Sci. 2009; 66(11-12):1890-1902.
19. Reichert M, Saur D, Hamacher R, Schmid RM and Schneider G. Phosphoinositide-3-kinase signaling controls S-phase kinase-associated protein 2 transcription via E2F1 in pancreatic ductal adenocarcinoma cells. Cancer Res. 2007; 67(9):4149-4156.
20. Schild C, Wirth M, Reichert M, Schmid RM, Saur D and Schneider G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol Carcinog. 2009; 48(12):1149-1158.
21. Lee M, Theodoropoulou M, Graw J, Roncaroli F, Zatelli MC and Pellegata NS. Levels of p27 sensitize to dual PI3K/mTOR inhibition. Mol Cancer Ther. 2011; 10(8):1450-1459.
22. Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K and Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996; 85(5):733-744.
23. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003; 4(6):437-450.
24. Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J and Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008; 68(19):8022-8030.
25. Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, Wang S, Garcia-Echeverria C and Maira SM. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci U S A. 2009; 106(52):22299-22304.
26. O’Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Punnoose EA, Guan J, Berry L, Prior WW, Amler LC, Belvin M, Friedman LS and Lackner MR. Predictive biomarkers of sensitivity to the phosphatidylinositol 3’ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010; 16(14):3670-3683.
27. Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O and Aoki Y. The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011; 17(10):3272-3281.
28. Shoji K, Oda K, Kashiyama T, Ikeda Y, Nakagawa S, Sone K, Miyamoto Y, Hiraike H, Tanikawa M, Miyasaka A, Koso T, Matsumoto Y, Wada-Hiraike O, Kawana K, Kuramoto H, McCormick F, et al. Genotype-dependent efficacy of a dual PI3K/mTOR inhibitor, NVP-BEZ235, and an mTOR inhibitor, RAD001, in endometrial carcinomas. PLoS One. 2012; 7(5):e37431.
29. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, Basecke J, Mijatovic S, Maksimovic-Ivanic D, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012; 3(10):1068-1111.
30. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH and Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30(8):777-782.
31. Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, Lee JJ, Luthra R, Hong DS and Kurzrock R. PIK3CA Mutations in Advanced Cancers: Characteristics and Outcomes. Oncotarget. 2012; 3(12):1566-1575.
32. Boyd ZS, Wu QJ, O’Brien C, Spoerke J, Savage H, Fielder PJ, Amler L, Yan Y and Lackner MR. Proteomic analysis of breast cancer molecular subtypes and biomarkers of response to targeted kinase inhibitors using reverse-phase protein microarrays. Mol Cancer Ther. 2008; 7(12):3695-3706.
33. Dan S, Okamura M, Seki M, Yamazaki K, Sugita H, Okui M, Mukai Y, Nishimura H, Asaka R, Nomura K, Ishikawa Y and Yamori T. Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res. 2010; 70(12):4982-4994.
34. Raynaud FI, Eccles SA, Patel S, Alix S, Box G, Chuckowree I, Folkes A, Gowan S, De Haven Brandon A, Di Stefano F, Hayes A, Henley AT, Lensun L, Pergl-Wilson G, Robson A, Saghir N, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009; 8(7):1725-1738.
35. Ihle NT, Lemos R, Jr., Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL and Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009; 69(1):143-150.
36. Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, Heynck S, Stuckrath I, Weiss J, Fischer F, Michel K, Goel A, Regales L, Politi KA, Perera S, Getlik M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009; 106(43):18351-18356.
37. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008; 14(12):1351-1356.
38. Seeliger H, Camaj P, Ischenko I, Kleespies A, De Toni EN, Thieme SE, Blum H, Assmann G, Jauch KW and Bruns CJ. EFEMP1 expression promotes in vivo tumor growth in human pancreatic adenocarcinoma. Mol Cancer Res. 2009; 7(2):189-198.
39. Pass HI, Levin SM, Harbut MR, Melamed J, Chiriboga L, Donington J, Huflejt M, Carbone M, Chia D, Goodglick L, Goodman GE, Thornquist MD, Liu G, de Perrot M, Tsao MS and Goparaju C. Fibulin-3 as a blood and effusion biomarker for pleural mesothelioma. N Engl J Med. 2012; 367(15):1417-1427.
40. Camaj P, Seeliger H, Ischenko I, Krebs S, Blum H, De Toni EN, Faktorova D, Jauch KW and Bruns CJ. EFEMP1 binds the EGF receptor and activates MAPK and Akt pathways in pancreatic carcinoma cells. Biol Chem. 2009; 390(12):1293-1302.
41. Hu B, Nandhu MS, Sim H, Agudelo-Garcia PA, Saldivar JC, Dolan CE, Mora ME, Nuovo GJ, Cole SE and Viapiano MS. Fibulin-3 Promotes Glioma Growth and Resistance through a Novel Paracrine Regulation of Notch Signaling. Cancer Res. 2012; 72(15):3873-3885.
42. Lu CD, Morita S, Ishibashi T, Hara H, Isozaki H and Tanigawa N. Loss of p27Kip1 expression independently predicts poor prognosis for patients with resectable pancreatic adenocarcinoma. Cancer. 1999; 85(6):1250-1260.
43. Juuti A, Nordling S, Louhimo J, Lundin J, von Boguslawski K and Haglund C. Loss of p27 expression is associated with poor prognosis in stage I-II pancreatic cancer. Oncology. 2003; 65(4):371-377.
44. Fukumoto A, Ikeda N, Sho M, Tomoda K, Kanehiro H, Hisanaga M, Tsurui Y, Tsutsumi M, Kato JY and Nakajima Y. Prognostic significance of localized p27Kip1 and potential role of Jab1/CSN5 in pancreatic cancer. Oncol Rep. 2004; 11(2):277-284.
45. Chen G, Yang N, Wang X, Zheng SY, Chen Y, Tong LJ, Li YX, Meng LH and Ding J. Identification of p27/KIP1 expression level as a candidate biomarker of response to rapalogs therapy in human cancer. J Mol Med (Berl). 2010; 88(9):941-952.
46. Conradt L, Godl K, Schaab C, Tebbe A, Eser S, Diersch S, Michalski CW, Kleeff J, Schnieke A, Schmid RM, Saur D and Schneider G. Disclosure of erlotinib as a multikinase inhibitor in pancreatic ductal adenocarcinoma. Neoplasia. 2011; 13(11):1026-1034.
47. Schneider G, Henrich A, Greiner G, Wolf V, Lovas A, Wieczorek M, Wagner T, Reichardt S, von Werder A, Schmid RM, Weih F, Heinzel T, Saur D and Krämer OH. Cross talk between stimulated NF-kappaB and the tumor suppressor p53. Oncogene. 2010; 29(19):2795-2806.
48. Eser S, Messer M, Eser P, von Werder A, Seidler B, Bajbouj M, Vogelmann R, Meining A, von Burstin J, Algul H, Pagel P, Schnieke AE, Esposito I, Schmid RM, Schneider G and Saur D. In vivo diagnosis of murine pancreatic intraepithelial neoplasia and early-stage pancreatic cancer by molecular imaging. Proc Natl Acad Sci U S A. 2011; 108(24):9945-9950.
49. Seidler B, Schmidt A, Mayr U, Nakhai H, Schmid RM, Schneider G and Saur D. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc Natl Acad Sci U S A. 2008; 105(29):10137-10142.
50. Labisso WL, Wirth M, Stojanovic N, Stauber RH, Schnieke A, Schmid RM, Krämer OH, Saur D and Schneider G. MYC directs transcription of MCL1 and eIF4E genes to control sensitivity of gastric cancer cells toward HDAC inhibitors. Cell Cycle. 2012; 11(8):1593-1602.
51. Stauber RH, Knauer SK, Habtemichael N, Bier C, Unruhe B, Weisheit S, Spange S, Nonnenmacher F, Fetz V, Ginter T, Reichardt S, Liebmann C, Schneider G and Krämer OH. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget. 2012; 3(1):31-43.
52. Wirth M, Fritsche P, Stojanovic N, Brandl M, Jaeckel S, Schmid RM, Saur D and Schneider G. A simple and cost-effective method to transfect small interfering RNAs into pancreatic cancer cell lines using polyethylenimine. Pancreas. 2011; 40(1):144-150.