
INTRODUCTION
Thalidomide and its immunomodulatory derivatives (IMiDs®) lenalidomide and pomalidomide have greatly improved the outcome of patients with multiple myeloma (MM), a malignant plasma cell (PC) disorder [1–3]. IMiDs directly kill multiple myeloma cells (MMCs) and also target the tumor environment by stimulating T cell function and inhibiting angiogenesis and tumor necrosis factor production by monocytes [4]. These activities are mediated through IMiD ability to promote binding of the Ikaros and Aiolos transcription factors to cereblon (CRBN), which forms an E3 ubiquitin ligase complex together with damaged DNA binding 1 (DDB1), cullin-4A (CUL4A) and regulator of cullins 1 (ROC1) [5, 6]. This results in the activation of CRBN E3 ligase activity, leading to Ikaros and Aiolos ubiquitination and proteasomal degradation [7–9]. As Aiolos and Ikaros repress interleukin-2 (IL-2) production by T cells [10], their proteasomal degradation induced by lenalidomide releases this inhibition and enhances T cell stimulation [9].
The killing of MMCs by lenalidomide is partially explained by the down-regulation of interferon regulatory factor 4 (IRF4), a transcription factor essential for MMC survival [11, 12]. Recent works have shown that lenalidomide-induced IRF4 inhibition in MMCs occurs downstream of Ikaros/Aiolos reduction and suggest that IRF4 is a transcriptional target of Ikaros and/or Aiolos [7, 8, 13].
IMiD effects on PCs, the healthy counterpart of MMCs, are unknown. Aiolos has a critical role in the generation of bone marrow (BM) PCs in mice [14], and IMiDs could target human PC generation as well. Indeed, Aiolos-deficient mice are unable to generate high affinity antibodies upon T cell-dependent antigen vaccination, whereas their ability to form germinal center and memory B cells (MBCs) is unaffected [14]. The generation of BM PCs requires the sequential activation, differentiation and trafficking of B cells, plasmablasts and PCs in different compartments (lymph nodes, lymph, blood and BM) [15]. Circulating PBs are short-lived cells that must find a niche in the BM or mucosal tissues to provide them with the factors required to survive and to fully differentiate into BM PCs (about 0.25% of human BM cells) [15].
We and others have shown that PC generation can be modeled using multi-step culture systems where various combinations of activation molecules and cytokines are successively used to reproduce the sequential cell differentiation occurring in the different organs/tissues in vivo [16–20]. In these culture models, MBCs differentiate into CD20low/-CD38- pre-plasmablasts (prePBs), CD20-CD38+CD138- PBs, CD20-CD38+CD138+ early PCs and long-lived PCs (LLPCs), which may survive and produce continuously high amounts of immunoglobulins (Igs) for months in vitro [21, 22]. The phenotype of in vitro-generated PBs and early PCs is similar to the phenotype of the few PBs detected in the peripheral blood (2 PBs/mm3) [19, 23]. Moreover, the molecular events occurring during differentiation of B cells into PCs are recapitulated in these in vitro differentiation models. In prePBs, which secrete Igs weakly, PC transcription factors (BLIMP1 and XBP1, mainly unspliced XBP1 mRNA) start to be expressed, while BCL6, PAX5 and other B cell transcription factors are progressively down-regulated. This change is more pronounced in early PCs (high Ig secretion) in which expression of PAX5 and BCL6 is inhibited and the ratio of spliced to unspliced XBP1 mRNA is increased [20].
Using this model, here we show that lenalidomide mainly targets the generation of highly proliferating prePBs, poorly proliferating early PCs and non-proliferating LLPCs. Conversely, lenalidomide does not affect much the generation of proliferating PBs and does not alter the long-term survival of LLPCs, once generated. Despite the different sensitivity of PBs and early PCs to lenalidomide, the expression of lkaros and Aiolos is comparably reduced in both cell types upon incubation with this drug.
RESULTS
Sequential generation of long-lived plasma cells
To investigate the effect of lenalidomide on the generation of human LLPCs from MBCs, we used an in vitro model that mimics the various steps associated with this process in lymph nodes, blood and BM [19, 20, 22]. In step 1 (four days of culture with soluble CD40 ligand (CD40L), phosphorothioate CpG oligodeoxynucleotides (ODN), IL-2, IL-10 and IL-15), purified MBCs are activated and induced to differentiate into highly proliferating CD20low/-CD38- prePBs that start to differentiate into CD20-CD38+ PBs [20]. In step 2, cells are cultured with IL-2, IL-10, IL-15 and IL-6, but without CD40L and ODN for three days (day 4 to 7) to promote differentiation into CD20-CD38+ PBs, which start to differentiate into poorly proliferating CD20-CD38+CD138+ early PCs. In step 3, cells are cultured in the presence of IL-6, IL-15 and interferon-alpha to complete PB maturation into CD20-CD38+CD138+ early PCs. In step 4 (addition of IL-6, APRIL and stromal cell-conditioned medium), early PCs finally differentiate into CD20-CD38+CD138+ non-cycling LLPCs and in step 5, newly generated LLPCs are allowed to survive and produce Igs continuously for months. Figure 1A-1B shows a schema of the culture model with the times of lenalidomide addition.
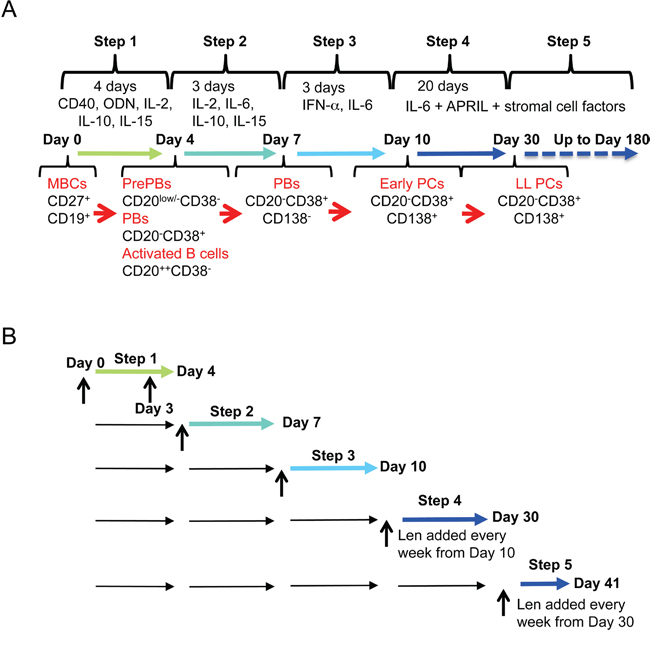
Figure 1: In vitro model to investigate lenalidomide effect during memory B cell differentiation into long-lived plasma cells. A. Using a five-step culture system, human memory B cells are induced to differentiate into long-lived plasma cells from day 0 to day 30. They can then be maintained in culture up to day 180. The cytokines used and the phenotype of the obtained cell populations at each step are indicated. B. The effect of lenalidomide on the different populations (from memory B cells to plasma cells) is investigated at the end of each differentiation step. Vertical arrows indicate when lenalidomide is added.
Lenalidomide impairs the generation of proliferating pre-plasmablasts mainly by reducing the number of cell divisions
Addition of lenalidomide at the start of step 1 (day 0 to 4; differentiation of MBCs mainly into CD20low/-CD38- prePBs and then CD20-CD38+ PBs) reduced the cell count (IC50 = 0.75 μM, a concentration in the range of those observed in patients treated with 25 mg lenalidomide daily) (Figure 2A) [24], but marginally reduced cell viability (Figure 2B). This effect was observed in the final day of step 1, when cells started cycling (Figure 2C). Moreover, 0.75 μM lenalidomide inhibited the generation of CD20low/-CD38- prePBs by 58% compared to control cells (DMSO alone) (Figures 3A-3C). As cell viability was not affected, we investigated whether this inhibition was due to a reduction in the number of cycling and dividing cells. Indeed, the percentage of prePBs in S phase was decreased by 42% (45% of control cells versus 26% of cells incubated with 0.75 μM lenalidomide were in S phase) and the fraction of prePBs in G1 phase was increased by 34% (Supplementary Figure S1A). The mean number of cell divisions in prePBs was decreased by 17% (from 3.5 to 2.9 divisions) (Figure 3D and Table 1). Detailed flow cytometry data of a representative experiment are shown in Supplementary Figure S2.
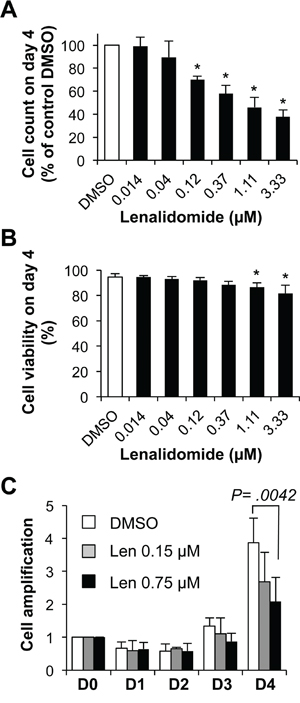
Figure 2: Effect of lenalidomide on cell growth in the first four days of in vitro memory B cell differentiation into plasma cells. 1.5 x 105 MBCs were activated with ODN and CD40L and cultured for four days with IL-2, IL-10, IL-15 and in the presence of increasing concentrations of lenalidomide or the largest DMSO concentration used to dilute lenalidomide (DMSO; control). A. Counts of cells recovered at the end of the four days of culture. B. Cell viability was assayed by trypan blue exclusion at day 4. C. Kinetics of cell amplification from D0 to D4, cells were cultured in the presence of control DMSO, 0.15 μM or 0.75 μM lenalidomide. Results are the mean value ± SD of four experiments. * P ≤ .05, compared with the control DMSO group using a paired t-test.
Table 1: Lenalidomide reduces the number of cell division at day 4
Mean cell division number |
|||
|---|---|---|---|
DMSO |
Lenalidomide |
Lenalidomide |
|
Total cells |
3.2 ± 0.4 |
2.9 ± 0.5* |
2.5 ± 0.7* |
Activated B cells |
2.0 ± 0.4 |
1.5 ± 0.5* |
1.4 ± 0.5* |
PrePBs |
3.5 ± 0.2 |
3.2 ± 0.2* |
2.9 ± 0.4* |
PBs |
4.1 ± 0.3 |
4.0 ± 0.4 |
3.5 ± 0.4 |
Mean number of cell divisions ± SD (three separate experiments) to generate activated B cells, prePBs, or PBs at day 4. Cells were labeled with CFSE at the start of the culture and the decrease in CFSE staining due to cell division was evaluated at day 4. * P ≤ .05 compared with the control group (DMSO) using a paired t-test.
Concomitantly with the generation of CD20low/-CD38- prePBs, MBC activation resulted also in the production of activated CD20highCD38- B cells (17% of all day-4 cells) (Figures 3A-3C) as previously reported [20]. In the presence of 0.75 μM lenalidomide, the count of activated B cells was reduced by 51% compared to control cells (Figure 3C), as a result of the decrease in the fraction of cycling and dividing cells (Supplementary Figure S1B, Figure 3D and Table 1).
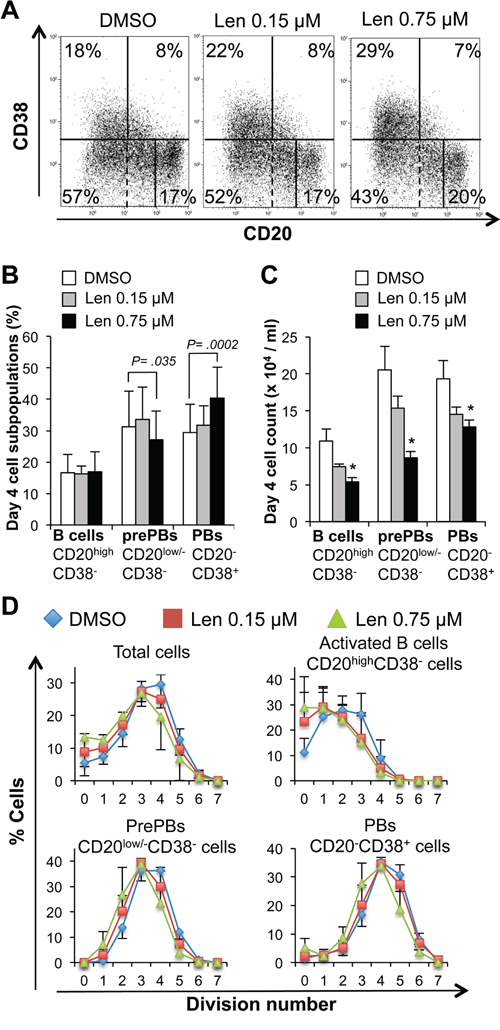
Figure 3: Lenalidomide reduces the generation of pre-plasmablasts in the first step of the B cell to plasma cell differentiation model. MBCs were activated with ODN and CD40L and cultured for four days with IL-2, IL-10, IL-15 and in the presence of increasing concentrations of lenalidomide or the largest DMSO concentration used to dilute lenalidomide (DMSO; control). The percentage and count of activated B cells (CD20highCD38-), prePBs (CD20low/-CD38-) and PBs (CD20-CD38+) were determined by FACS analysis at day 4. A. Dot plot of CD20 and CD38 expression in day-4 cells (one representative experiment out of seven). The numbers in the panels (gates) are the percentages of gated day-4 cells. Dashed lines indicated the limit of CD20 positivity determined using an isotype-matched control antibody. B and C. Mean percentage and count ± SD (seven separate experiments) of activated B cells, prePBs and PBs at day 4 following incubation with 0.15 μM or 0.75 μM lenalidomide. D. Mean number (three separate experiments) of cell divisions to generate activated B cells, prePBs, or PBs at day 4. Cells were labeled with CFSE at the start of the culture and the decrease in CFSE staining due to cell division was evaluated at day 4. * P ≤ .05, compared with the control DMSO group using a paired t-test.
During step 1, some CD20low/-CD38- prePBs further differentiated into CD20-CD38+ PBs (Figures 3A-3C). Since exposure to lenalidomide hindered the generation of prePBs and because PBs originate from this compartment,[20] PB count was also reduced in the treated cultures, compared with control (DMSO), at the end of step 1 (Figure 3C). However, the effect on PB count was about 2-fold lower than that on prePBs (34% versus 58% reduction, P ≤ .05). Similarly, the effect of 0.75 μM lenalidomide on the cell cycle was less strong in PBs than in prePBs: the PB fraction in S phase was reduced only by 17% compared with 42% in prePBs (P ≤ .05, Supplementary Figure 1A and 1C). Thus, lenalidomide mainly impairs prePB generation.
Lenalidomide did not significantly affect the frequency of IgM-, IgA- or IgG-producing activated B cells, prePBs and PBs (Supplementary Figure S3) or the expression of various membrane markers (CD19, CD24, CD30, CD45, CD126 and FCRL4). It only slightly increased CD31 expression in PBs (P = .009) (Supplementary Figure S4).
Lenalidomide strongly inhibits the generation of CD138+ early plasma cells, but not of plasmablasts
In step 2 (day 4 to 7), CD20low/-CD38- prePBs differentiate into CD20-CD38+ PBs and then into CD20-CD38+CD138+ early PCs. Addition of lenalidomide at the beginning of this step led to a concentration-dependent reduction of early PC number at day 7 (50% reduction with 0.15 μM lenalidomide) (Figure 4A-4B). Conversely, PB number was considerably less affected. It was significantly reduced (by 17%) only upon incubation with 1.33 μM lenalidomide, a concentration which is about 10-fold higher than the early PC 50% inhibitory concentration (IC50) (P = .02, Figure 4C). Lenalidomide did not inhibit PB cell cycle at day 7 (Supplementary Figure S1D). The count and viability of all cells present in the cultures at day 7 were decreased, respectively, by 21% and 16% upon incubation with 0.15 μM lenalidomide and increasing lenalidomide concentrations had a stronger effect (Figures 4D-4E).
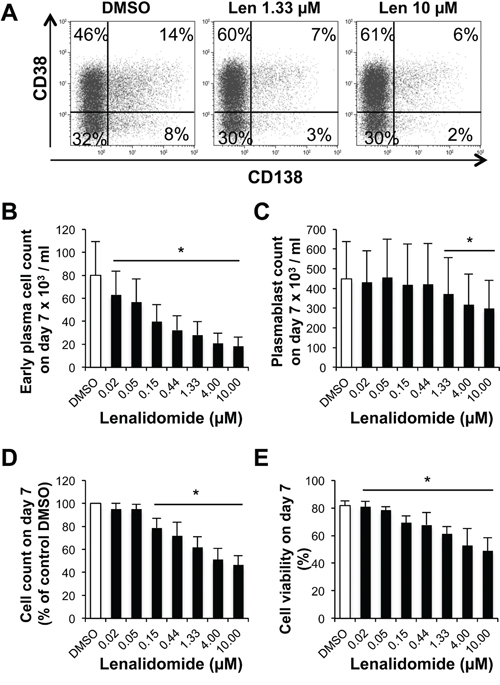
Figure 4: Lenalidomide preferentially targets the generation of CD138+ early plasma cells and poorly affects that of plasmablasts in the second step of in vitro B cell differentiation into plasma cells. Cells harvested at the end of step 1 (day-4 cells: mainly prePBs and PBs) were cultured with IL-2, IL-6, IL-10 and IL-15 in the presence of graded concentrations of lenalidomide or the largest DMSO concentration used to dilute lenalidomide (DMSO) for three days. The percentage and counts of PBs (CD20-CD38+CD138-) and early PCs (CD20-CD38+CD138+) were determined by FACS analysis at day 7. A. Dot plot of CD38 and CD138 expression in day-7 cells (one representative experiment out of four). The numbers in the panels (gates) are the percentage of gated day-7 cells. B and C. Mean count ± SD of early PCs and PBs at day 7 in the presence of increasing lenalidomide concentrations from day 4 to day 7 (four separate experiments). D and E. Mean count and viability ± SD of all day-7 cells in the presence of increasing lenalidomide concentrations from day 4 to day 7 (four separate experiments). * P ≤ .05, compared with the DMSO control group using a paired t-test.
During step 3 (day 7 to day 10; maturation into CD20-CD38+CD138+ early PCs), incubation with lenalidomide reduced the generation of day-10 early PCs, with an IC50 of 0.76 μM (Figure 5A-5B). Conversely, it affected PB count only when used at a concentration higher than 4 μM (Figure 5C). The cell count and viability of all cells present in the culture at day 10 were decreased, respectively, by 27% and 28% with 0.76 μM lenalidomide and by 40% and 45% when lenalidomide concentration reached 10 μM (Figure 5D-5E). Thus, lenalidomide strongly inhibits the generation of CD138+ early PCs, but not of PBs in steps 2 and 3.
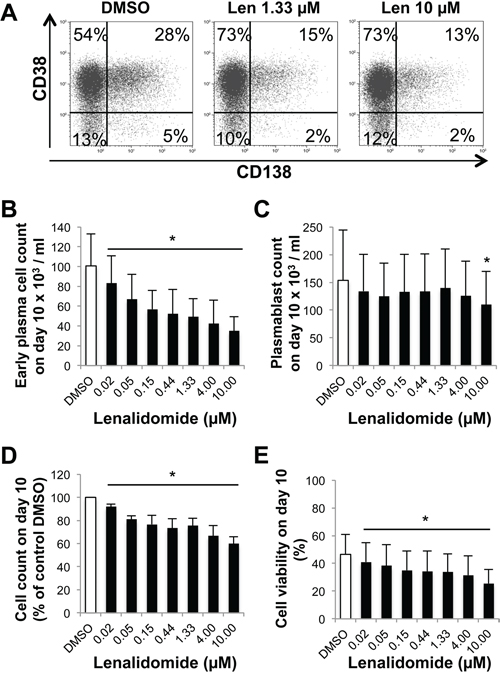
Figure 5: Lenalidomide preferentially targets the generation of CD138+ early plasma cells and poorly affects plasmablasts in the third step of the B cell to plasma cell differentiation model. Cells harvested at the end of step 2 (day-7 cells, mainly PBs and early PCs) were cultured with IL-6, IL-15 and IFN- α for three days (step 3) in the presence of increasing concentrations of lenalidomide or the largest DMSO concentration used to dilute lenalidomide (DMSO; control). The percentage and counts of PBs (CD20-CD38+CD138-) and early PCs (CD20-CD38+CD138+) were determined by FACS analysis at day 10. A. Dot plot of CD38 and CD138 expression in day-10 cells (one representative experiment out of four). The numbers in the panels (gates) are the percentage of gated day-10 cells. B and C. Mean count ± SD of early PCs or PBs at day 10 in the presence of increasing lenalidomide concentrations from day 7 to day 10 (four separate experiments). D and E. Mean count and viability ± SD of all day-10 cells in the presence of increasing lenalidomide concentrations from day 7 to day 10 (four separate experiments). * P ≤ .05, compared with the DMSO control group using a paired t-test.
Lenalidomide reduces the generation of long-lived plasma cells, but does not affect their survival once established
In step 4 (generation of CD20-CD38+CD138+ LLPCs), purified day-10 early PCs were cultured with IL-6, APRIL and stromal cell-conditioned medium (SC-CM) for 20 days. Lenalidomide was added at the beginning and then every week to ensure a continuous drug exposure according to the manufacturer’s recommendations. In this step, cells poorly cycle and only 20% survive and differentiate into LLPCs [22]. Lenalidomide reduced the number of LLPCs at day 30 with an IC50 below 1 μM (Figure 6A). During step 5, newly generated LLPCs may survive for months, if fresh IL-6, APRIL and SC-CM are added every week [22]. Incubation of LLPCs harvested at day 30 with up to 10 μM lenalidomide for four (day 34) or 11 days (day 41) did not significantly affect the survival of established LLPCs (Figure 6B). Lenalidomide also did not modify their ability to produce IgG and IgA (Supplementary Figure S5A- S5B). These LLPCs did not cycle without or with lenalidomide (Supplementary Figure S1E).
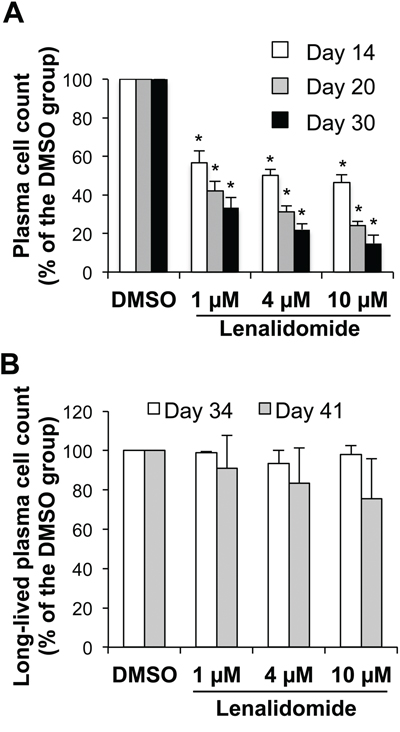
Figure 6: Lenalidomide targets the generation, but not the survival of long-lived plasma cells. Early PCs generated at the end of step 3 (day-10 cells) were FACS-sorted based on CD138 expression and lack of CD20 expression (> 95% purity). A. Purified early PCs were cultured for 4, 10 or 20 days with IL-6, APRIL and stromal cell-conditioned medium (SC-CM) in the presence of increasing concentrations of lenalidomide or the largest DMSO concentration used to dilute lenalidomide (DMSO). Fresh lenalidomide, DMSO, culture medium, cytokines and SC-CM were refreshed every week by replacing half of the culture medium. Counts of metabolically active LLPCs were assayed at day 14, 20 and 30 using a Cell Titer Glo Assay. Data are expressed as the percentage (mean ± SD; n= three separate experiments) of the values obtained in controls (DMSO). B. 30-day LLPCs were incubated with increasing lenalidomide concentrations or the largest DMSO concentration used to dilute lenalidomide (DMSO) for 4 or 11 days. Fresh lenalidomide, control DMSO, culture medium, cytokines and SC-CM were refreshed every week, by replacing half of the culture medium. Counts of metabolically active LLPCs were assayed at day 34 and 41. Data are expressed as the percentage (mean ± SD; n= three separate experiments) of the values obtained in controls (DMSO). * P ≤ .05, compared with the control group (DMSO) using a paired t-test.
Ikaros and Aiolos are expressed in plasmablasts and early plasma cells and their expression is inhibited by lenalidomide
Ikaros and Aiolos expression levels in FACS-sorted day-7 PBs, day-10 PBs and day-10 early PCs were reduced by 70-80% and 50-65%, respectively, following incubation with lenalidomide (Figure 7A-7D). Similar results were obtained in OMP2 myeloma cells (positive control) treated with lenalidomide (Figure 7A-7D). This indicates that the differential sensitivity of PBs and early PCs to lenalidomide is not due to a difference in expression/targeting of Ikaros and Aiolos. The different sensitivity to lenalidomide could not be explained by differences in CRBN gene expression because it was similarly expressed in the various cell populations generated during plasma cell differentiation (Supplementary Figure S6A). CRBN protein expression levels, normalized to actin content, also were comparable in PBs and early PCs, despite the 10-fold higher sensitivity to lenalidomide of early PCs (Supplementary Figure S6B).
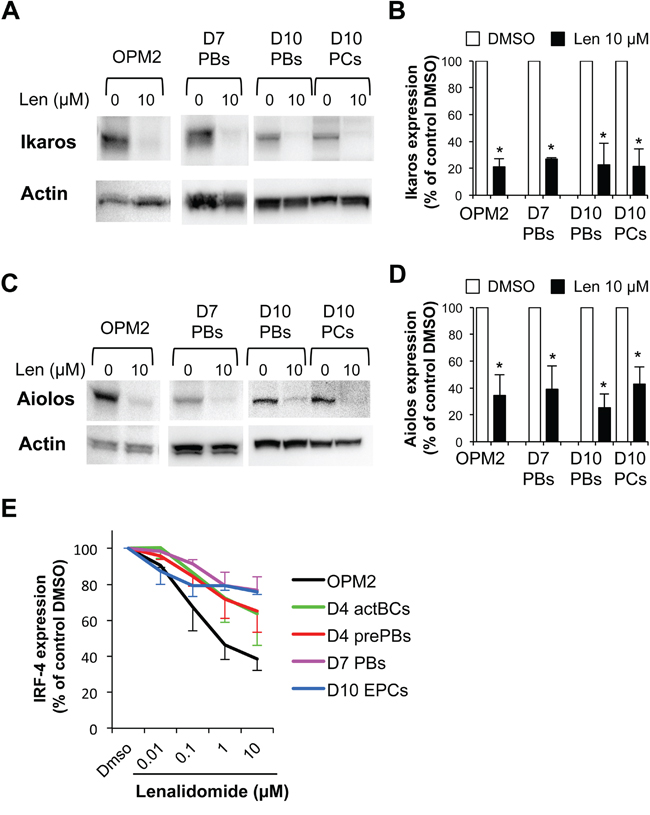
Figure 7: Ikaros, Aiolos and IRF4 are expressed in plasmablasts and early plasma cells and their expression is reduced by lenalidomide. PBs generated at step 2 (day-7 cells), PBs and early PCs generated at step 3 (day-10 cells) were incubated with 10 μM lenalidomide or control DMSO for the last 24 hours of culture and were FACS-sorted based on CD38 expression and lack of CD20 and CD138 expression19 (PBs, ≥ 95% purity), and on CD138 and CD38 expression and lack of CD20 expression (early PCs ≥ 95% purity). OPM2 myeloma cells were incubated with 10 μM lenalidomide or control DMSO for 24 hours. Cell lysates were separated by SDS-PAGE and Ikaros and Aiolos expression assessed by immunoblotting. β actin expression was used as loading control. Representative western blot of Ikaros A. and Aiolos C. expression (three separate experiments). Data in B and D. are the mean Ikaros and Aiolos expression in the three experiments, quantified by densitometry analysis and normalized to actin levels. * P ≤ .05, compared with the control group (DMSO) using a paired t-test. E. Lenalidomide reduces IRF4 expression. Cells were cultured in the presence of increasing concentrations of lenalidomide or DMSO (control). Lenalidomide was added for three or four days at the beginning of each step, as indicated in Supplementary Figure S1B. The human myeloma cell line OPM2 was cultured for 3 days. At the end of each step, IRF4 expression was measured by flow cytometry analysis. Data are expressed as the percentage (mean ± SD; 3 separate experiments) of the IRF4 staining index values obtained in the control group (DMSO).
Lenalidomide reduces IRF4 expression in activated B cell to plasma cell populations
Lenalidomide reduced in a dose-dependent manner IRF4 expression (measured by flow cytometry) in OPM2 myeloma cells (60% reduction with 10 μg/ml lenalidomide, Figure 7E and Supplementary Figure S7). It also significantly (P ≤ .05) reduced IRF4 expression in a dose-dependent manner in activated B cells, prePBs, PBs and early PCs (Figure 7E and Supplementary Figure S7). This effect was already significant with 0.1 μM lenalidomide and with 10 μM lenalidomide, IRF4 decrease varied from 23% in PBs to 36% in activated BCs. IRF4 reduction was comparable in PBs and early PCs with 1 μM and 10 μM lenalidomide (23% and 24% respectively). Interestingly, IRF4 downregulation was more pronounced in early PCs than in PBs with low doses of lenalidomide (0.01 μM and 0.1 μM) (Figure 7E).
Treatment with lenalidomide does not affect the count of normal bone marrow plasma cells in vivo
Twelve allografted patients with MM received 25 mg lenalidomide daily for 3 to 18 months following the detection of residual MMCs. Patients with increasing counts of malignant PCs during lenalidomide treatment were not considered in order to eliminate a possible competition between malignant and normal PCs for the same niche. BM MMCs and normal PCs were monitored regularly during the treatment. Although the number of BM PCs in each patient fluctuated over time, possibly due to BM sample variation, the mean count during the lenalidomide treatment did not differ from that before treatment (Figure 8).
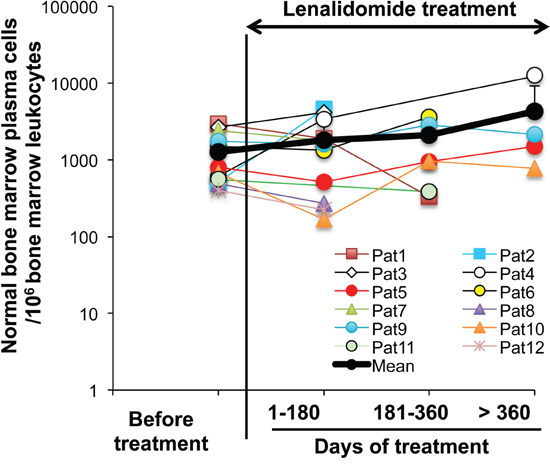
Figure 8: Lenalidomide treatment does not affect the counts of normal bone marrow plasma cells in allografted patients with multiple myeloma. Twelve patients with MM who received an allogeneic hematopoietic stem cell transplant were treated with 25 mg lenalidomide daily for 3-18 months. Tumor and normal PCs were regularly monitored. All the patients had stable counts of tumor PCs during lenalidomide treatment. Normal PCs were identified as CD38high, CD19+, CD27+, CD56-, CD117-, CD200- and cytoplasmic Kappa or Lambda Ig light chain+ cells using multicolor cytometry. Data are expressed as the normal PC counts/106 leukocytes in the BM of each patient, harvested before and at different times during lenalidomide treatment. The black bold line represents the mean value of the patients’ PC counts.
DISCUSSION
The main results of this study can be summarized in four points:
Lenalidomide reduces the generation of highly proliferating pre-plasmablasts
We and others have shown that the differentiation of MBCs into prePBs and then PBs is a process requiring several cell divisions [17]. In agreement, here we found that prePBs are actively cycling (45% of prePBs in S phase). Lenalidomide impaired prePB formation by reducing the percentage of prePBs in S phase and increasing the fraction in G1 without affecting their viability. This is in line with the ability of lenalidomide to induce the cell cycle inhibitors p21WAF1 and p27KIP1 in tumor cell lines, particularly by promoting demethylation of the p21WAF1 gene promoter [25].
Lenalidomide poorly affects PB formation but dramatically inhibits the generation of early PCs
Lenalidomide reduced the early PC count with an estimated 50% inhibitory concentration of 0.76 μM, similar to the concentration that inhibits prePB formation. Conversely, lenalidomide began to affect PB generation only at 10-fold higher concentrations and did not impair their cell cycle. Early PCs are more differentiated than PBs. They are poorly cycling cells that express more cytoplasmic Igs, lack membrane Igs (PBs still express a low amount of membrane Igs) and strongly express CD27 and CD138 (syndecan-1) [19]. The primary targets of lenalidomide are the Ikaros and Aiolos transcription factors [7-9, 13]. However, the 10-fold difference in sensitivity of proliferating PBs and poorly proliferating early PCs to lenalidomide is not explained by differences in Ikaros and Aiolos expressions in these two cell types. Alternatively, lenalidomide could degrade more efficiently Ikaros or Aiolos in early PCs than in PBs. This was not the case when these cells were incubated with 10 μM lenalidomide (60% to 80% reduction in both cell types). However, we cannot exclude that these transcription factors might be degraded more efficiently in early PCs than in PBs when lower lenalidomide concentrations are used. We could not fully investigate this point because of the difficulty in obtaining enough cells to assess protein expression by western blotting at different time points and upon incubation with various lenalidomide concentrations. Moreover, the anti-Ikaros and-Aiolos antibodies, which are reported to be efficient for protein quantification by FACS, worked with myeloma cells in our hands, but not with the in vitro generated cell populations, probably due to the weak expression of these proteins in these cells. Nevertheless, this point should be further investigated when more efficient antibodies become available.
In myeloma cells, IRF4 is a major transcription factor for initiating plasma cell differentiation and Ikaros and Aiolos control IRF4 gene expression [7, 8, 13]. Lenalidomide also negatively modulates IRF4 expression as a consequence of the degradation of Ikaros and Aiolos [7, 8, 13]. In agreement, we found that lenalidomide degrades in a dose-dependent manner IRF4 in activated B cells, prePBs, PBs and early PCs. IRF4 degradation is much more pronounced in early PCs than in PBs treated with low doses of lenalidomide (i.e. 0,01 μM and 0,1 μM) but is comparable with higher doses (1 and 10 μM). At day-10 of culture, there is a significant decrease of cell viability affecting mainly early PCs [19]. IRF4 expression is critical for PC survival [11, 12], so we can hypothesize that early PCs surviving with high doses of lenalidomide are those selected according to high IRF4 expression. This could explain the absence of significant difference in IRF4 expression between early PC and PB at 10 μM lenalidomide concentration. Lenalidomide-induced IRF4 degradation in normal PBs and PCs is less pronounced than in OPM2 myeloma cells, possibly because IRF4 expression is deregulated in myeloma cells through continuous triggering by aberrant MYC expression [11, 12].
Lenalidomide efficiently targets the generation of long-lived PCs from early PCs in vitro
Lenalidomide inhibits the generation of LLPCs starting from purified early PCs with an IC50 lower than 1 μM, a concentration in the range of those inhibiting prePB or early PC generation. The ability to generate LLPCs in vitro was recently documented by Cocco et al. and by our group [21, 22]. Our results fit well with the phenotype of Aiolos-deficient mice [14]. Indeed, they cannot generate PCs that produce Igs for a prolonged time [14]. As lenalidomide specifically targets Ikaros and Aiolos, these in vivo [14] and in vitro (our study) findings suggest that Aiolos and/or Ikaros are critical for LLPC generation from early PCs. In the current study, we could not determine Ikaros and Aiolos expression in LLPCs because of the too limited number of available LLPCs for western blot analysis. Indeed, only a few LLPCs can be generated due to the high cell loss occurring during the early PC and LLPC generation steps (80% of differentiating cells will die) [19, 22]. Moreover, although Ikaros and Aiolos immunodetection by FACS was previously reported in myeloma cell lines [13], we did not obtain specific labeling in PBs or early PCs and therefore could not use this approach in LLPCs (results not shown).
Once LLPCs are generated, lenalidomide does not affect their long-term survival and Ig production
The in vitro generation of LLPCs from early PCs is a highly selective process, but once LLPCs are generated, they can efficiently survive for months in vitro [21, 22]. The current data indicate that Ikaros and Aiolos are not essential for LLPC survival in our in vitro differentiation system. Moreover, LLPC survival might not be affected by lenalidomide also in vivo. Indeed, in patients with MM who received a stem cell transplant, lenalidomide treatment for 3 to 18 months following detection of residual tumor cells did not affect the mean count of normal BM PCs, identified by flow cytometry. These data suggest that Ikaros and/or Aiolos are dispensable for the long-term survival of human LLPCs in vitro and in vivo. It should be interesting to investigate the changes in MBCs, prePBs, PBs and PCs in patients treated with lenalidomide.
In conclusion, this study shows that lenalidomide affects minimally the formation of human PBs, specifically targets the generation of prePBs from MBCs, of early PCs from PBs and of LLPCs from early PCs, and does not alter the survival of LLPCs once generated, in agreement with the lack of effect on normal BM PCs in patients treated with lenalidomide. As lenalidomide specifically targets Ikaros and Aiolos proteins, these data suggest a major role of these transcription factors in regulating the generation of early PCs and LLPCs in humans.
The specific targeting of plasma cell precursors by lenalidomide should prompt to investigate whether resistance of plasma cell tumors to lenalidomide is associated with the selection of plasmablast or mature plasma cell subclones. Multiple myeloma is a heterogeneous tumor the growth of which is supported by MM progenitors that differentiate into mature MMCs [26]. The phenotype of MM progenitors that generate tumors in immune-compromised mice is controversial, particularly their expression of CD138 [26, 27], the marker that discriminates early PCs (sensitive to lenalidomide) from PBs (poorly sensitive) in the current report. A recent study has shown that the proteasome inhibitor bortezomib kills preferentially mature and plasmablastic MMCs and that bortezomib resistance is associated with the emergence of pre-plasmablastic MMCs [28] (Figure 9B). The current data suggest that the MMC phenotype in patients resistant to lenalidomide should be investigated to determine whether resistance is associated with the emergence of poorly proliferating long-lived plasma cells or of CD38+CD138- plasmablasts. In this case, the clinical benefit of lenalidomide and bortezomib association could be explained by the ability of this drug combination to kill pre-plasmablastic up to mature MMCs (Figure 9A-9B).
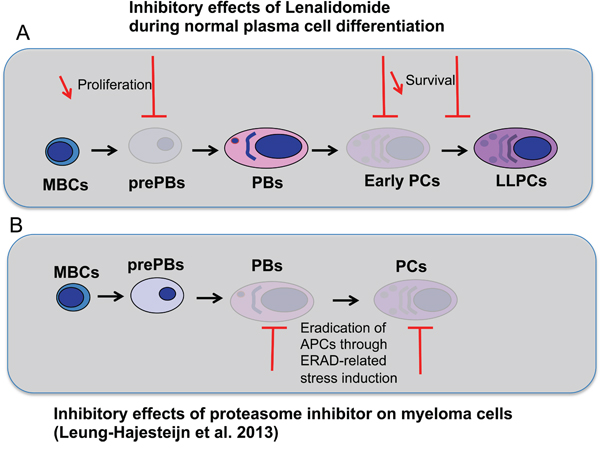
Figure 9: Effects of lenalidomide and proteasome inhibitor on the different stages of plasma cell differentiation. A. During plasma cell differentiation, lenalidomide inhibits the generation of prePBs, early PCs and LLPCs but has a moderate inhibitory effect on PBs and LLPCs once generated. B. As shown by Leung-Hagesteijn et al. in multiple myeloma [28], antibody-producing cells (APCs), PBs and PCs, are sensitive to proteasome inhibitor because of their high level of Ig synthesis. Proteasome inhibitor treatment induces endoplasmic reticulum-associated protein degradation (ERAD)-related stress.
MATERIALS AND METHODS
Patients
This study was performed under approval of the Montpellier University Hospital Centre for Biological Resources (DC-2008-417). It included patients with multiple myeloma (MM) who received an allogeneic hematopoietic stem cell transplant after reduced intensity conditioning (fludarabine, intra-venous busulfan ± thymoglobulin) at the Department of Hematology, Montpellier University Hospital, France. They all signed a written informed consent according to the Helsinki Declaration and EBMT good clinical practice guidelines. Residual tumor disease was monitored using flow cytometry as indicated [29]. In the case of detection of ≥ 103 MMCs/L, patients (n=12) received 25 mg lenalidomide daily for 3-18 months with repeated monitoring of MMCs and normal PCs.
Reagents
Human recombinant interleukin (IL)-2 was purchased from R&D Systems (Minneapolis, MN, USA), interferon-alpha-2b (IFN- α, IntronA) from Merck Canada Inc. (Kirckland, Canada), IL-6, IL-10 and IL-15 from PeproTech (Rocky Hill, NJ, USA). Mouse monoclonal antibodies (mAbs) conjugated to allophycocyanin (APC), fluorescein isothiocyanate (FITC), peridinin chlorophyll protein-cyanin 5.5 (PerCP-Cy5.5), phycoerythrin (PE) or Pacific BlueTM (PB), and specific for human CD19 (clone HIB19), CD24 (clone ML5), CD27 (clone M-T271), CD30 (clone BerH8), CD31 (clone WM59), CD38 (clone HIT2), CD56 (cloneB159), CD117 (clone 104D2), CD138 (clone RF8B2), IgG (clone G18-145), IgM (clone G20-127), lambda Ig light chain (clone JDC-12), kappa Ig light chain (clone TB28-2) and Ki67 (clone B56) were purchased from BD Biosciences (Le Pont De Claix, France); for CD200 (clone OX104) from eBiosciences (San Diego, CA, USA); for CD20 (clone B9E9), CD45 (clone J33), CD126 (clone M91) and CD138 (clone B-A38) from Beckman Coulter (Fullerton, CA, USA); for IRF4 (clone IRF4.3E4) and FCRL4 (clone 580810) from BioLegend (San Diego, CA, USA); for IgA, IgM, and IgG (polyclonal goat Abs) from Southern Biotech (Birmingham, AL, USA).
Cell samples
Peripheral blood cells from healthy volunteers were purchased from the French Blood Center (Toulouse, France) and CD19+CD27+ MBCs were purified (≥ 95% purity) as described [19].
Cell cultures
MBCs were differentiated using a previously described five-step culture method [19, 20, 22]. All cultures were performed in Iscove modified Dulbecco medium (Invitrogen, Carlsbad, CA, USA) and 10% FCS. In step 1 (day 0 to day 4), 1.5 x 105/ml purified peripheral blood MBCs were seeded in 6-well culture plates and were activated for 4 days by 10 μg/ml of phosphorothioate CpG oligodeoxynucleotides (ODN) 2006 (Sigma-Aldrich, St Louis, MO, USA), 50 ng/ml histidine tagged soluble CD40 ligand (CD40L) and 5 μg/ml of an anti-poly-histidine mAb (R&D Systems) in the presence of 20 U/ml IL-2, 50 ng/ml IL-10 and10 ng/ml IL-15. In step 2 (day 4 to day 7), PBs were generated by removing ODN and CD40L and changing the cytokine cocktail (20 U/ml IL-2, 50 ng/ml IL-6, 50 ng/ml IL-10 and 10 ng/ml IL-15). In step 3 (day 7 to day 10), PBs were differentiated into early PCs by adding 50 ng/ml IL-6, 10 ng/ml IL-15 and 500 U/ml IFN- α. In step 4 (day 10 to day 30), early PCs were FACS sorted based on CD138 expression and lack of CD20 expression (> 95% purity) and differentiated into LLPCs by adding 10 ng/ml IL-6, 100 ng/ml APRIL and 50% of stromal cell-conditioned medium (SC-CM). Fresh culture medium, growth factors and SC-CM were added once per week and cultures were maintained until day 30. In step 5 (day 30 to day 41), LLPCs were allowed to survive and produce continuously Igs in the presence of 10 ng/ml IL-6, 100 ng/ml APRIL and 35% of SC-CM. The SC-CM was obtained by culturing confluent monolayers of Resto-6 SC cells for 5 days. The culture supernatant was filtered with 0.2 μM filters and frozen. The Resto-6 cell line was obtained from lymph node-derived SCs, as previously described [30].
Various lenalidomide concentrations (Celgene Corporation, Summit, NJ, USA) were added at the start of each step and its effects evaluated by analyzing cell counts and phenotype at the end of each step. Supplementary Figure S1A- S1B shows a schema of the culture model with the times of lenalidomide addition.
Cell viability and cell growth assays
Cell concentration and viability were assessed with the trypan blue dye exclusion test. The number of metabolically active cells was determined by intracellular ATP quantitation with the Cell Titer Glo Luminescent Assay (Promega Corporation, Madison, WI, USA).
Cell cycle and immunophenotypic analysis
Cycling cells were identified by DAPI staining (Sigma-Aldrich) and cells in the S phase by incubation with bromodeoxyuridine (BrdU) for 1 hour followed by labeling with an anti-BrdU antibody (APC BrdU flow kit, BD Biosciences) according to the manufacturer’s instructions. For immunophenotypic analysis, cells were stained with a combination of 4 to 7 mAbs conjugated to different fluorochromes, as indicated [22]. The Cytofix/Cytoperm kit (BD Biosciences) was used for intracellular staining of IgM, IgA, IgG, IRF-4 or Ki67 antigen [19]. Flow cytometry analysis was performed with a FACSAria cytometer using FACSDiva 6.1 (Becton Dickinson, San Jose, CA, USA) and with a Cyan ADP cytometer driven by the Summit software (Beckman Coulter). The Kaluza software (Beckman Coulter) was used for data analysis. The fluorescence intensity of the cell populations was quantified using the staining index (SI) formula: [mean fluorescence intensity (MFI) obtained for a given mAb minus MFI obtained with a control mAb]/[2 times the standard deviation of the MFI obtained with the same control mAb] [19].
CFSE labeling
Cell division was assessed by CFSE labeling as previously described [31]. Briefly, purified MBCs were washed and re-suspended at a concentration of 106 cells/ml in PBS/0.1% BSA with 10 μM CFSE (Molecular Probes, Eugene, OR, USA), incubated at 37°C for 10 minutes and extensively washed before culture. At day 4 of culture, cells were washed and labeled with anti-PB-CD20 and anti-PerCP-Cy5.5-CD38 for flow cytometry analysis. Cell divisions were quantified using the ModFit LT software (Verity Software House, Topsham, ME, USA).
Western blot analysis
Cells were lysed in RIPA buffer (Cell Signaling Technology, Beverly, MA, USA) supplemented with 1 mM phenylmethylsulfonyl fluoride immediately before use. Lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% gels) and transferred to nitrocellulose membranes using an iBlot® Gel Transfer Device (InVitrogen). Non-specific membrane sites were blocked by incubation at room temperature in 140 mM NaCl, 3 mM KCl, 25 mM Tris-HCl (pH 7.4), 0.1% Tween 20 (tris-buffered saline Tween-20), 5% non-fat milk for 2 h, and then immunoblotted with rabbit polyclonal antibodies against Ikaros (Santa Cruz Biotechnology, Dallas, TX, USA), Aiolos (Cell Signaling Technology) or Cereblon (Sigma-Aldrich). As a control for protein loading, a mouse monoclonal anti-β-actin antibody (Sigma-Aldrich) was used. The primary antibodies were visualized with peroxidase-conjugated goat anti-rabbit (Sigma-Aldrich) or goat anti-mouse (Jackson ImmunoResearch, West grove, PA, USA) antibodies and an enhanced chemiluminescence detection system. Western blots were quantified by densitometry using the NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA) and protein levels were normalized according to those of β-actin.
Analysis of Ig secretion
ELISA. Flow cytometry-sorted PCs (106 cells/ml) were cultured for the indicated time with increasing lenalidomide concentrations or the largest DMSO concentration used to dilute lenalidomide (DMSO) and culture supernatants harvested. IgA and IgG concentrations were assessed by ELISA using human IgA and IgG ELISA kits from Bethyl Laboratories (Montgomery, TX, USA), according to the manufacturer's recommendations.
Detection of malignant and normal PCs in BM of patients with MM
Malignant and normal PCs were counted using the multi-parameter flow cytometry technique reported previously [29]. Briefly, erythrocyte-lysed BM samples were labeled with anti-CD19, CD20, CD38 and CD45 monoclonal antibodies (mAbs) in association with anti-CD138, CD27, CD56, CD117, CD200 mAbs or isotype controls. Cells were then fixed and permeabilized with the Cytofix/Cytoperm kit (BD Biosciences, Le Pont De Claix, France), and labeled with anti-Ig Kappa and anti-Ig Lambda mAbs. Data were acquired with a Cyan flow cytometer, driven by the Summit 4.3 software (Beckman Coulter, Fullerton, CA, USA). B-lymphocytes were defined as [CD19+CD20+CD45+CD38-/+ and (Kappa+ or Lambda+)] cells and PCs as [CD38high and (Kappa+ or Lambda+) and (not B lymphocytes)] cells. MMCs were identified based on the monoclonal expression of Kappa or Lambda light chains together with the aberrant expression of one or several myeloma markers: CD20 and/or CD56 and/or CD117 and/or CD200, lack of CD19, and lack/weak CD27 and/or CD45 expression. [32–35] Healthy PCs were identified based on the polyclonal expression of Kappa or Lambda light chains together with the expression of CD19, CD27. The acquisition of at least 100 events was required to define MMCs or healthy PCs as detectable [36]. Data were analyzed with the FlowJo 9.1 software (Tree star, Ashland, OR).
Statistical analysis
Statistical comparisons were made with the non-parametric Mann-Whitney test, unpaired or paired Student’s t-test using the SPSS software. P-values ≤ .05 were considered as significant.
ACKNOWLEDGMENTS
This work was supported by grants from Celgene Corporation (R11083FF) and the European Community (FP7-OVERMYR).
CONFLICTS OF INTEREST
This study was partly supported by research funding from Celgene Corporation. RC and PS are employees of Celgene and hold stock options in Celgene. GC has received honoraria from Celgene. The remaining authors declare no conflict of interest.
REFERENCES
1. Barlogie B, Desikan R, Eddlemon P, Spencer T, Zeldis J, Munshi N, Badros A, Zangari M, Anaissie E, Epstein J, Shaughnessy J, Ayers D, Spoon D, Tricot G. Extended survival in advanced and refractory multiple myeloma after single-agent thalidomide: identification of prognostic factors in a phase 2 study of 169 patients. Blood. 2001; 98: 492–4.
2. Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T, Stoppa AM, Hulin C, Benboubker L, Garderet L, Decaux O, Leyvraz S, Vekemans MC, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012; 366: 1782–91.
3. Lacy MQ, McCurdy AR. Pomalidomide. Blood. 2013; 122: 2305–9.
4. Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: immunological, molecular and epigenetic targets in cancer therapy. Oncogene. 2013; 32: 4191–202.
5. Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010; 327: 1345–50.
6. Zhu YX, Braggio E, Shi C-X, Bruins LA, Schmidt JE, Van Wier S, Chang XB, Bjorklund CC, Fonseca R, Bergsagel PL, Orlowski RZ, Stewart AK. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011; 118: 4771–9.
7. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014; 343: 301–5.
8. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science. 2014; 343: 305–9.
9. Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, Ito T, Ando H, Waldman MF, Thakurta A, Klippel A, Handa H, Daniel TO, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4 CRBN. Br J Haematol. 2014; 164: 811–21.
10. Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, Georgopoulos K, Kuchroo VK. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nature Immunology. 2012; 13: 770–7.
11. Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu S, Zhao H, Zeng Y, Chen B, Epstein J, Staudt LM. IRF4 addiction in multiple myeloma. Nature. 2008; 454: 226–31.
12. Lopez-Girona A, Heintel D, Zhang L-H, Mendy D, Gaidarova S, Brady H, Bartlett JB, Schafer PH, Schreder M, Bolomsky A, Hilgarth B, Zojer N, Gisslinger H, et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br J Haematol. 2011; 154: 325–36.
13. Zhu YX, Braggio E, Shi C-X, Kortuem KM, Bruins LA, Schmidt JE, Chang XB, Langlais P, Luo M, Jedlowski P, Laplant B, Laumann K, Fonseca R, et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood. 2014; 124: 536–45.
14. Cortes M. Aiolos Is Required for the Generation of High Affinity Bone Marrow Plasma Cells Responsible for Long-Term Immunity. J Exp Med. 2004; 199: 209–19.
15. Tangye SG. Staying alive: regulation of plasma cell survival. Trends in Immunology. 2011; 32: 595–602.
16. Tarte K, De Vos J, Thykjaer T, Zhan F, Fiol G, Costes V, Reme T, Legouffe E, Rossi JF, Shaughnessy J, Orntoft TF, Klein B Generation of polyclonal plasmablasts from peripheral blood B cells: a normal counterpart of malignant plasmablasts. Blood. 2002; 100: 1113–22.
17. Tangye SG, Avery DT, Hodgkin PD. A division-linked mechanism for the rapid generation of Ig-secreting cells from human memory B cells. J Immunol. 2003; 170: 261–9.
18. Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, Radbruch A, Dorner T. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood. 2009; 113: 2461–9.
19. Jourdan M, Caraux A, De Vos J, Fiol G, Larroque M, Cognot C, Bret C, Duperray C, Hose D, and Klein B. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. 2009; 114: 5173–81.
20. Jourdan M, Caraux A, Caron G, Robert N, Fiol G, Reme T, Bollore K, Vendrell JP, Le Gallou S, Mourcin F, De Vos J, Kassambara A, Duperray C, et al. Characterization of a transitional preplasmablast population in the process of human B cell to plasma cell differentiation. J Immunol. 2011; 187: 3931–41.
21. Cocco M, Stephenson S, Care MA, Newton D, Barnes NA, Davison A, Rawstron A, Westhead DR, Doody GM, Tooze RM. In Vitro Generation of Long-lived Human Plasma Cells. J Immunol. 2012; 189: 5773–85.
22. Jourdan M, Cren M, Robert N, Bollore K, Fest T, Duperray C, Guilloton F, Hose D, Tarte K, Klein B. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia. 2014; 28: 1647–56.
23. Caraux A, Klein B, Paiva B, Bret C, Schmitz A, Fuhler GM, Bos NA, Johnsen HE, Orfao A, Perez-Andres M for the Myeloma Stem Cell Network (MSCNET). Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica. 2010; 95: 1016–20.
24. Chen N, Lau H, Kong L, Kumar G, Zeldis JB, Knight R, Laskin OL. Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J Clin Pharmacol. 2007; 47: 1466–75.
25. Escoubet-Lozach L, Lin I-L, Jensen-Pergakes K, Brady HA, Gandhi AK, Schafer PH, Muller GW, Worland PJ, Chan KWH, Verhelle D. Pomalidomide and lenalidomide induce p21 WAF-1 expression in both lymphoma and multiple myeloma through a LSD1-mediated epigenetic mechanism. Cancer Res. 2009; 69: 7347–56.
26. Huff CA, Matsui W. Multiple myeloma cancer stem cells. J Clin Oncol. 2008; 26: 2895–900.
27. Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45 low/- CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia. 2012; 26: 2530–7.
28. Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, Chung KC, Tiedemann RE. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013; 24: 289–304.
29. Caraux A, Vincent L, Bouhya S, Quittet P, Moreaux J, Requirand G, Veyrune JL, Olivier G, Cartron G, Rossi JF, Klein B. Residual malignant and normal plasma cells shortly after high dose melphalan and stem cell transplantation. Highlight of a putative therapeutic window in Multiple Myeloma? Oncotarget. 2012; 3: 1335–47. doi: 10.18632/oncotarget.650.
30. Amé-Thomas P, Maby-El Hajjami H, Monvoisin C, Jean R, Monnier D, Caulet-Maugendre S, Guillaudeux T, Lamy T, Fest T, Tarte K. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: role of stromal cells in follicular lymphoma pathogenesis. Blood. 2007; 109: 693–702.
31. Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc. 2006; 2: 2049–56.
32. Paiva B, Almeida J, Pérez-Andrés M, Mateo G, López A, Rasillo A, Vídriales MB, López-Berges MC, San Miguel JF, Orfao A. Utility of flow cytometry immunophenotyping in multiple myeloma and other clonal plasma cell-related disorders. Cytometry B Clin Cytom. 2010; 78: 239–52.
33. Klein B, Seckinger A, Moehler T, Hose D. Molecular pathogenesis of multiple myeloma: chromosomal aberrations, changes in gene expression, cytokine networks, and the bone marrow microenvironment. Recent Results Cancer Res. 2011; 183: 39–86.
34. Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009; 23: 10–24.
35. Ria R, Todoerti K, Berardi S, Coluccia AML, De Luisi A, Mattioli M, Ronchetti D, Morabito F, Guarini A, Petrucci MT, Dammacco F, Ribatti D, Neri A, Vacca A. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin Cancer Res. 2009; 15: 5369–78.
36. Domingo E, Moreno C, Sánchez-Ibarrola A, Panizo C, Páramo JA, Merino J. Enhanced sensitivity of flow cytometry for routine assessment of minimal residual disease. Haematologica. 2010; 95: 691–2.