
INTRODUCTION
Runx1 encodes a transcription factor that plays a vital role in development of the haematopoietic system [1]. It belongs to a three-membered family of mammalian gene products that bind a common DNA target sequence by virtue of the conserved Runt domain and share a common heterodimeric binding co-factor, CBFβ [2, 3]. Like their Drosophila homologue, Runt, the Runx proteins function as transcriptional regulators and are capable of activating or repressing target promoters through the recruitment of co-activators or co-repressors [4]. The RUNX1 (AML1) and CBFB genes are among the most commonly involved in human leukemias where they are affected by chromosomal translocations that frequently generate fusion oncoproteins [5].
Evidence that simple over-expression of any of the Runx gene family members can drive oncogenesis emerged first from mouse models, where it was shown that all three genes can act as targets for murine leukemia virus (MLV) insertional mutagenesis and transcriptional activation in lymphoma. Common targets in the Eµ-Myc lymphoma model include Runx1 and Runx3 [6], while all three members of the Runx family were identified as activation targets in CD2-MYC T-cell lymphomas [7-9]. The potent oncogenic effect of combining Myc and Runx over-expression is emphasised further in retroviral acceleration of lymphoma onset in Runx2 transgenic mice which frequently entails activation of c-Myc or N-Myc [10], while compound transgenic mice over-expressing Myc and Runx genes in the T or B-cell compartment display very rapid tumor onset [10-12]. However, the Runx family are not merely cofactors for Myc oncogenesis; CD2-Runx2 transgenic mice display dose-dependent predisposition to lymphoma [11, 13] and strongly synergistic lymphoma development in combination with other oncogenes such as Pim-1 and v-Myb, as well as with loss of p53 [10]. Notably, the combination of Runx2 and Myc oncogenes appears to overcome the need for mutational inactivation of p53 [14] despite the fact that both genes can trigger the p53 pathway and collaborate with p53 loss when over-expressed individually [15, 16].
In contrast to this catalogue of evidence of dominant oncogenic activity in lymphomagenesis, Runx1 deficient cells in chimeric mice develop T-cell lymphomas after treatment with ENU [17], suggesting that loss of Runx1 activity can also predispose to lymphoid malignancy. A similar dichotomy of observations exists for RUNX1 in human haematopoietic cancers. RUNX1 is among the most over-expressed genes in childhood ALL [18] and is highly amplified in a poor prognostic B-ALL subgroup [19] while presumptive loss-of-function RUNX1 mutations have been observed in a small proportion of T-ALLs where network analysis further implicated RUNX1 as a candidate tumor suppressor [20]. More extensive evidence of a tumor suppressor role for RUNX1 has come from myeloid malignancies where loss of function mutation is frequently observed in AML, and underlies familial platelet disorder with predisposition to AML [21, 22].
While the lymphomagenic effects of Runx over-expression have been amply demonstrated, the requirement for basal gene expression in tumor maintenance is an important and potentially far-reaching question that has been much less well investigated. In this study we tested the effects of ablating the endogenous Runx1 gene in the well-characterised Eµ-Myc lymphoma model system [23] where ectopic expression of Runx1 is known to drive lymphomagenesis [12]. We show that primary Eµ-Myc lymphomas have an increased requirement for Runx1, while this dependency is reduced, but not eliminated, in end-stage p53-deficient lymphoma cell lines. Our findings shed light on the paradoxical observation that Runx1 deficiency can also predispose to lymphoma but more importantly validate Runx1 function as a therapeutic target in p53 wild-type or mutant lymphomas.
RESULTS
Addiction to Runx1 in primary Eµ-Myc lymphoma cells is attenuated in established cell lines
Eµ-Myc mice develop lymphomas with highly variable onset (average 30 weeks) during which they acquire a range of secondary mutations in the Cdkn2a-p53 pathway [24]. To achieve more homogeneous tumor onset and facilitate tracking of p53 loss along with Runx1 deletion, we crossed these mice to a Trp53+/- background. Eµ-Myc/p53+/- mice succumb to B-cell lymphomas within a much narrower time window [25]. These mice were further crossed to generate Mx1Cre/Runx1fl/fl cohorts in which we could examine the ability of lymphoma cells to survive deletion of the endogenous Runx1 gene.
Surprisingly, we found no significant difference in the rate of onset of lymphoma in Eµ-Myc/p53+/-/Runx1fl/fl mice with active Cre recombinase (Figure 1A, 1B), initially suggesting that Runx1 loss had no effect on tumor onset. However, PCR analysis (Figure 1C) of tumor-bearing spleens showed that the intact Runx1 allele was strongly retained in the primary tumors, even in pIpC-treated mice. Spleens from mice with end-stage disease, which were markedly enlarged due to lymphoma expansion, showed levels of Runx1 deletion significantly lower than normal splenic lymphoid cells from age-matched Mx1Cre+/Runx1fl/fl mice without the Eµ-Myc oncogene, indicating an increased rather than a decreased requirement for Runx1 in primary lymphoma cells (Figure 1C, upper panel and lower right panel).
While the cell lines derived from these lymphomas almost uniformly retained the functional active Runx1 allele on initial establishment in culture (Figure 1D, upper left panel), they were able to survive excision of both wild-type alleles after treatment with IFNβ in vitro (Figure 1D, upper right panel). Moreover, single cell cloning of these lines after low dose IFNβ readily generated subclones with 0, 1 or 2 intact Runx1 alleles (Figure 1D, lower panel). Using these lines we validated the direct PCR assay as a consistent measure of the proportion of excised to non-excised allele. In this assay, the observed ratio of excised to non-excised allele is proportionate to input and independent of DNA concentration (Figure S1A, S1B).
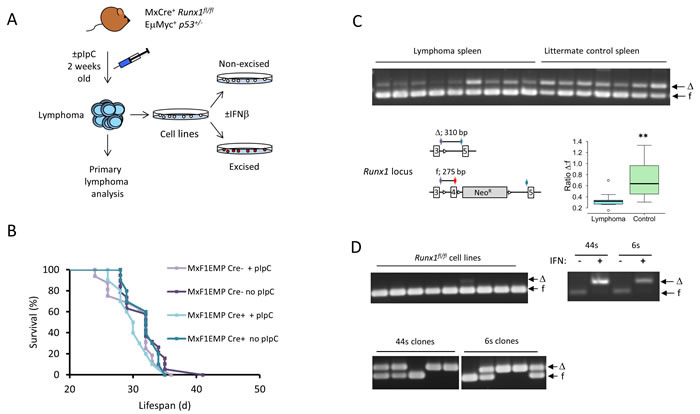
Figure 1 : Eµ-Myc lymphomas strongly resist deletion of Runx1 in vivo but not in vitro. A. Outline experimental design Lymphomas derived from Mx1Cre+/Runx1fl/fl/Eµ−Myc/p53+/- mice were analysed and also used to establish cell lines that could be treated in vitro with IFNβ to induce Runx1 excision. B. Survival curve for Runx1fl/fl/Eµ−Myc+/p53+/- mice with or without the Mx1Cre transgene and with or without pIpC treatment to excise Runx1. Statistical analysis by Wilcoxon Rank Sum test showed no significant difference in survival between the groups of mice. C. Upper panel : Runx1 excision PCR on genomic DNA derived from lymphoma spleen tissue of pIpC-treated Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- mice (lymphoma spleen) and age-matched Eµ-Myc- (lymphoma-free) littermate controls (littermate control spleen). Arrows indicate Runx1-floxed (f) and Runx1-deleted (∆) bands. The panel below left shows a diagram of the multiplex PCR for detection of deleted (∆) and floxed (f) Runx1. The cartoon shows loxP sites flanking exon 4 in the floxed allele before and after excision, location of primers (colored arrows) and size of PCR products. The panel below right shows the ratio of excised:non-excised band intensity determined by densitometry for the Runx1 excision PCR samples shown in 1C. Boxplot shows the distribution of all the ∆:f ratios with the box representing the 1st to 3rd quantiles (Q1 to Q3) and the midline representing the median. Whiskers represent the smaller of the most extreme data point, or 1.5x the Q1-Q3 interquantile range. Asterisks denote statistical significance, with p = 0.008 D. The upper left panel shows Runx1 excision PCR on genomic DNA from a series of independent cell lines derived from Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- lymphomas. The right panel shows Runx1 excision PCR on genomic DNA from samples of 44s and 6s Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- cell lines treated with IFNβ to excise Runx1 or with vehicle control; cell samples were taken 2 days after the start of IFNβ treatment. The lower panel shows Runx1 excision PCR on genomic DNA from single cell clones derived from the 44s and 6s Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- cell lines; cells were treated with a sub-optimal dose of IFNβ to induce partial excision of Runx1 and single cell cloned as detailed in Materials and Methods. Arrows indicate Runx1floxed (f) and Runx1-deleted (∆) bands.
Permissiveness for Runx1 deletion is lineage-dependent
In accord with previous reports [26], analysis of non-tumor bearing mice revealed lineage-dependence in permissiveness for Runx1 deletion. Healthy tissues showed a marked difference in the levels of spontaneous and enforced deletion of Runx1, with thymus consistently more resistant than kidney or spleen. However, even the permissive tissues showed at most 50% deletion (Figure 2A). Mono-allelic deletion in Runx1fl/fl cells has been reported previously in Runx1-dependent tissues from vav-CreRunx1fl/fl mice [26]. These results suggest that a strong homeostatic process selects for cells that retain at least one copy of Runx1. Consistent with this hypothesis, the cell type composition of spleen showed remarkably little change after enforced Runx1 deletion (Figure 2B). However, fractionation of spleen cells into lymphoid (B220+ or CD3+) or myeloid (Mac1+) confirmed the relative permissiveness of the myeloid compared to the lymphoid compartment for deletion of Runx1 [26] (Figure 2C).
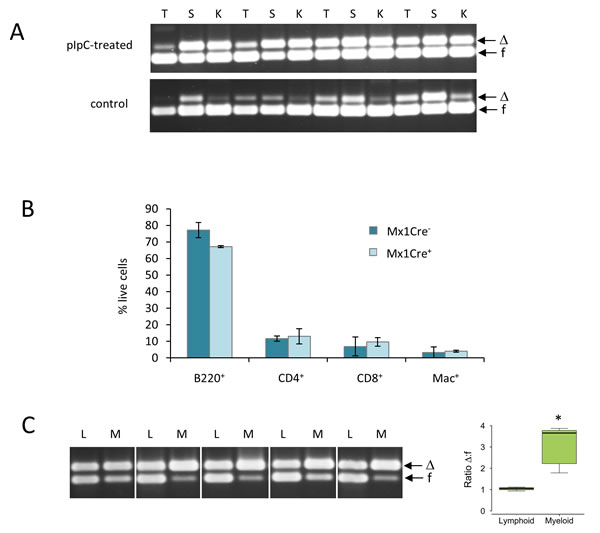
Figure 2: Healthy tissues display lineage-specific differences in permissiveness for Runx1 deletion. A. Runx1 excision PCR analysis of genomic DNA from whole thymus (T), spleen (S) and kidney (K) of non-lymphoma-bearing adult Runx1fl/fl mice treated (upper panel) or untreated (lower panel) with pIpC to induce Mx1Cre expression. Arrows indicate Runx1floxed (f) and Runx1-deleted (∆) bands. B. Flow cytometric analysis of spleen cell populations in pIpC-treated Mx1Cre- and Mx1Cre+ Eµ-Myc- healthy littermate control mice. Plot shows mean ± SD for measurements from 3 mice in each group; no statistically significant differences between the Mx1Cre+ and Mx1Cre- cell populations were found. C. Runx1 excision PCR analysis of genomic DNA from sorted lymphoid (B220+ or CD3+ cells; L) and myeloid (Mac1+; M) cells from non-lymphoma-bearing spleens of 5 pIpC-treated adult Runx1fl/fl mice. Boxplot shows the distribution of all the ∆:f ratios as described in Figure 1. Asterisk indicates that the difference is statistically significant (p = 0.01) .
Enforced deletion of Runx1 promotes the outgrowth of p53 null cells in Eµ-Myc/p53+/- lymphomas
Established lymphoma cell lines were found to have lost the wild-type Trp53 allele in most cases and displayed consistent de-repression of Cdkn2a/p19Arf [24] (Figure 3A, S2A). We therefore considered whether the contrasting behaviour of primary lymphomas and established cell lines with regard to retention of Runx1 was a consequence of loss of the wild-type Trp53 allele. To address this hypothesis further and examine the temporal order of events in vivo, we analysed the minor blastic fraction of cells from end-stage lymphoma-bearing spleens, a procedure that has been shown to enrich for the most rapidly proliferating cells within the tumor [27]. The fractionated blasts displayed strong enrichment for cells that had lost the wild-type Trp53 allele (Figure 3B). The fact that most lymphoma blast cells retained at least one Runx1 allele indicated that loss of p53 was an earlier event that preceded permissiveness for Runx1 loss, a conclusion reinforced by findings on newly established cell lines (Figure 1D, upper left panel). Enrichment for p53-deleted blasts was observed most strongly in pIpC-treated mice, suggesting that attempts to enforce deletion of Runx1 may have perturbed the tissue sufficiently to create a permissive niche for outgrowth of bystander p53-/- blasts. These results further emphasise the importance of Runx1 for survival of Eµ-Myc lymphoma cells in vivo, even though it becomes dispensable in established cell lines in vitro.
The hypothesis that Runx1 is required to counteract the growth suppressive effects of p53 was tested by transduction of a p53 null Eµ-Myc/Runx1fl/fl lymphoma cell line (3s) with a temperature-sensitive p53 allele (Val135). After transduction, interferon treatment was used to produce matched pairs of cell lines with or without endogenous Runx1 that could be tested for sensitivity to p53-induced death. However, the p53 “add-back” cells died by apoptosis at the permissive temperature (32°C) regardless of their Runx1 status or exposure to UV irradiation (Figure 3C, 3D, S2B). The functional integrity of the p53 pathway was maintained with respect to down-regulation of Cdkn2a/p19ARF in response to temperature shift but the major target for p53-mediated growth arrest, Cdkn1a/p21WAF1, was undetectable in these cells (Figure 3E), suggesting a partial functional deficit. As it has been reported that p53 can affect Runx1 expression in T-cells [28], we also examined Runx1 expression in 3s cells, but saw no evidence of modulation by p53 (Figure 3E).
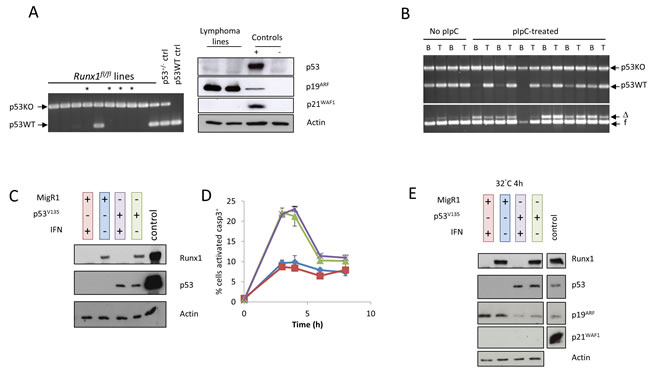
Figure 3: Loss of p53 precedes permissiveness for Runx1 deletion during establishment of lymphoma cell lines. A. Left panel: p53 allele PCR on genomic DNA from a series of independent cell lines derived from lymphomas in Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- mice; arrows indicate p53null (p53KO) and wild type (p53WT) alleles in control samples for p53wt/wt and p53+/-; asterisks indicate cell lines established from pIpC-treated mice. Right panel: western blot analysis of total protein extracted from two established and independent lymphoma cell lines derived from lymphomas in Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- mice that did not receive pIpC treatment. Extracts were probed with antibodies to p53, p19ARF and p21WAF1. Actin was used as a loading control. Positive controls are listed in Materials & Methods and used in all subsequent analyses. B. Upper panel shows p53 allele PCR on genomic DNA from sorted blast cells (B) or whole tissue (T) from lymphoma-bearing spleens of untreated and pIpC-treated Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- mice, while the lower panel shows Runx1 excision PCR on the same DNA samples. Blast cells were sorted using CD45/SSC on B220+ cells as detailed in Materials and Methods. Arrows indicate p53null (p53KO) and wild type (p53WT) alleles, and Runx1floxed (f) and Runx1-deleted (∆) bands. C. Cells transduced with vector expressing temperature-sensitive p53V135 or the MigR1 control vector were treated with IFNβ to excise endogenous Runx1, and expression of p53 and Runx1 examined by western blot analysis. D. Paired Runx1+ and Runx1null p53 addback cell lines treated with 5J/m2 UV were grown at 32°C to activate temperature sensitive p53 and stained for intracellular activated caspase 3 after 3, 4, 6 and 8h incubation. The percentage of cells expressing activated caspase 3 was determined by flow cytometry. Line colors are indicated by the color-coding of the western blots samples in 3C. E. Western blot analysis of total protein extracted from the experiment as shown in (D) 4h after UV treatment. Extracts were probed as before and loading confirmed by actin expression.
Runx1 deficient cells display down-regulation of genes involved in growth and proliferation along with de-repression of Rag genes
Changes in gene expression resulting from deletion of Runx1 were examined using Affymetrix gene expression microarrays (Affymetrix GeneChip MTA 1.0). Three biological replicates of the 3s cell line were compared. Possible confounding effects of IFNβ treatment and Cre recombinase induction were controlled by comparison with a phenotypically matched Eµ-Myc lymphoma cell line (30s) of the same genotype apart from the non-deletable Runx1wt/wt allele. Treatment with IFNβ had no effect on Runx1 expression in these cells (Figure S3A, S3B).
Many genes showed changes in expression specific to the Runx1fl/fl genotype. To focus on the most prominent changes we applied a statistical significance threshold of p = < 0.05 and a fold change cut-off of 1.25. After subtraction of genes significantly changed in the 30s control, this left 123 genes of which 70 were up-regulated and 53 down-regulated on excision of Runx1 (Figure 4A). As can be seen from the heat map in Figure 4B, the control 30s cells showed relatively modest changes after IFNβ treatment compared to Runx1fl/fl 3s cells. The gene set specific for Runx1 loss was subjected to Ingenuity Pathway Analysis which revealed significant associations with cancer and in particular with proliferation, apoptosis and differentiation of lymphocytes (Figure 5A). The direction of changes on deletion indicated that Runx1 is acting to sustain proliferation and survival while impeding differentiation in these cells (Figure 5B). Rag1 and Rag2 were common components of all four pathway clusters (Figure 5B) and were among the most strongly de-repressed genes in Runx1-deleted cells. Validation of the changes in Rag genes and other signature genes by quantitative real-time PCR confirmed the array findings and revealed larger fold changes in most cases, consistent with the precision but systematic underestimation of differences by this methodology [29] (Figure 5C).
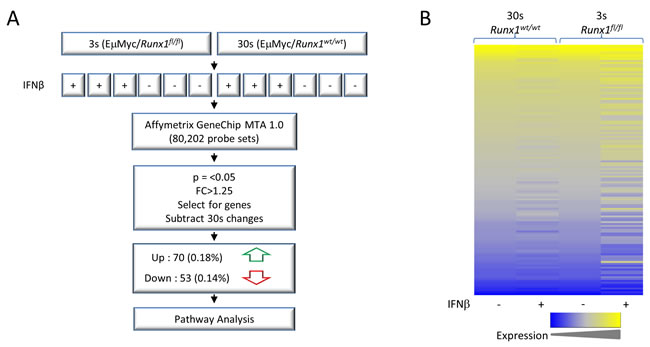
Figure 4: Gene expression microarray analysis of Eµ-Myc lymphoma cells before and after Runx1 excision A. Process overview. 3s+ and 30s+ cell lines derived from Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- mice were treated with IFNβ, excising Runx1 from 3s cells (Runx1fl/fl) but not 30s controls where interferon responses induce Cre expression but deletion does not occur on the Runx1wt/wt background (see also Figure S3). RNA was extracted and gene expression examined by Affymetrix microarray. Significantly changed genes were selected by p-value and fold-change and a subset that were also significantly changed in 30s cells were subtracted to exclude non-Runx1 related changes. This gene list was then used in downstream analyses such as pathway analysis. B. Heat map of all genes significantly changed in 3s cells, defined as p < 0.05 and fold change > |1.25|. Shown is raw intensity for 3s and 30s cells. Low expression is shown in blue and high expression in yellow. 30s gene expression was normalised to 3s control (-IFNβ) expression.
As recent studies have defined Runx1/RUNX1 gene expression signatures in other contexts, we examined our array data for similar changes in the key genes. An RNA-Seq study of normal haematopoietic stem and progenitor cells using the same Runx1fl/fl allele revealed a ribosome biogenesis gene expression signature associated with loss of Runx1 [30]. However, we saw no significant difference in these genes. A subset of the most changed genes from the signature gene set is shown in Figure S3C. Nor did we note any obvious change in cell size or morphology in Runx1-excised Eµ-Myc lymphoma cell lines (Figure S3D). We also examined a mitotic checkpoint gene signature that was reported in Kasumi/AML cells after knockdown of RUNX1 [31]. The genes shown in Figure S3E were all significantly downregulated in Kasumi knockdown cells but were mostly unchanged in Runx1null Eµ-Myc lymphoma cells. Only Nek6 was significantly down-regulated while two of the genes showed a significant increase (Nek2, Bub1b).
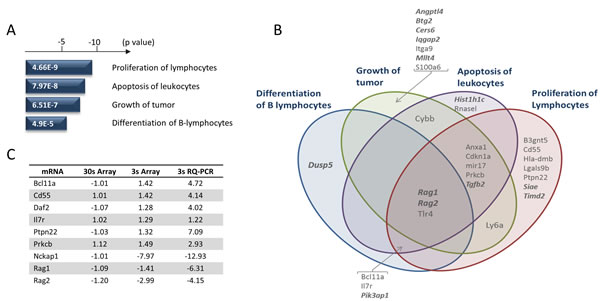
Figure 5: Gene expression changes specific for Runx1 deletion in Eµ-Myc lymphoma cells. A. Ingenuity Pathway Analysis of the most changed genes (Figure 4) revealed significant enrichment for genes involved in the processes depicted. P-values are shown for top sub-processes in relevant categories. B. Venn diagram showing the overlap of the gene signatures for the significant processes in A. Genes in bold italics show increased expression on loss of Runx1, while the remainder are down-regulated. C. Validation of key target genes contributing to the Runx1 deletion signatures. Fold changes estimated by microarray and RQ-PCR are shown alongside comparable array values for the 30s control cells.
Negative correlation of RUNX1 and RAG mRNA expression in a human leukemia panel
Aberrant RAG activity has recently been reported as a major source of cancer driver mutations in TEL-RUNX1 t(12;21) B-cell leukemias [32], leading us to consider whether there may be a wider role for RUNX1 in RAG mis-regulation in human leukemia/lymphomas. Using Partek Genomics Suite 6.6 we examined the relationship between expression of RUNX1 and RAG1/RAG2 expression in a panel of acute and chronic leukemias from the MILE study (Microarray Innovations in LEukemia), a global microarray study comprising gene expression analysis of > 4000 patients [33]. As shown in Figure S4, RUNX1 mRNA expression was relatively uniform compared to the RAG genes that displayed markedly higher expression in ALLs compared to normal tissues and myeloid leukemias. Also, expression of RAG1 was significantly higher in t(12;21) ALLs than in other ALL types (Figure S4). Comparison of the levels of RUNX1 and the RAG genes within the dataset (n = 750) identified significant, negative correlations between RUNX1/RAG1 and RUNX1/RAG2 (Figure 6). This pattern was evident in the total ALL dataset with or without the t(12;21) subset, where interpretation is more complex due to the detection of TEL-RUNX1 as well as RUNX1 mRNAs by the RUNX1 probe sets. While the lack of significant correlation between RUNX1 and RAG1 and the weaker correlation between RUNX1 and RAG2 in t(12;21) cells is therefore difficult to interpret, the strongly negative correlation in other ALL sets is a robust observation.
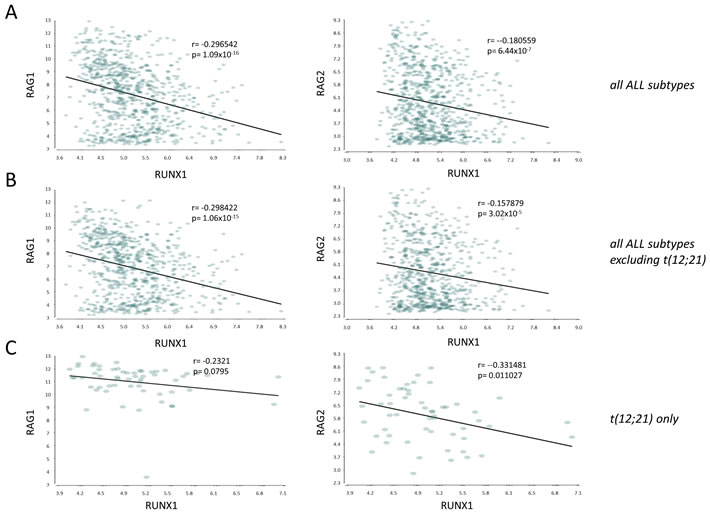
Figure 6: RUNX1 mRNA expression is negatively correlated with RAG1 and RAG2 in a large panel of human ALLs. Pearson correlation plots of gene expression (probeset intensities) of RUNX1 vs RAG1 or RAG2 in A all ALL subtypes within the MILE database (ref [33]) (n = 750), B. all ALL subtypes with the exception of the t(12;21)/ETV6-RUNX1 subset (n = 692), C. the t(12;21) translocation only (n = 58). Plots show p-value and r-value.
Eµ-Myc lymphoma cells lacking Runx1 display growth impairment and increased sensitivity to genotoxic stress and dexamethasone-induced apoptosis
Careful observation suggested that the Runx1null cell lines grew more slowly, and this suspicion was confirmed by serial passage of clonal Eµ-Myc/Runx1fl/fl cell lines in which partial excision had been induced. Cells retaining Runx1 consistently outgrew their null clonal siblings (Figure 7A). This disadvantage could be accounted for by the observed lengthening of doubling time in Runx1-deleted cells (Figure 7B) operating over the prolonged culture period. Marked differences were also noted when cells were exposed to genotoxic stresses, where Runx1null cells displayed more rapid death in the presence of doxorubicin or ethanol (Figure 7C, 7D). The effects of ethanol were particularly potent on Runx1null cells (Figure 7D), possibility reflecting the fact that ethanol elicits wider oxidative stress-induced effects in addition to DNA damage [34, 35]. In light of our previous findings that ectopic Runx1 expression suppresses glucocorticoid growth inhibition in murine fibroblasts [36] we also tested the effects of dexamethasone. Again, Runx1-deficient Eµ-Myc lymphoma cells displayed significantly greater induction of cell death (Figure 7E)
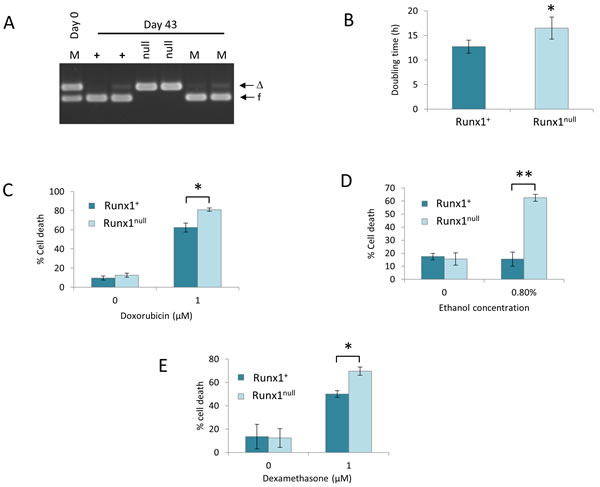
Figure 7: Excision of Runx1 impairs proliferation and survival of Eµ-Myc lymphoma cells in vitro. A. Runx1 excision PCR analysis of genomic DNA from duplicate samples of Runx1+ (+) and Runx1null (null) cells or of a 1:1 mixture of these cells (M) on day 0 and after culture for 43 days. Arrows indicate Runx1 floxed (f) and Runx1-deleted (∆) bands. Results shown are from a single experiment representative of 3 independent experiments. B. Increased doubling time of Runx1null cells. Runx1+ and Runx1null cells were plated at a density of 2x105 cells/ml and cultured for 24h before counting. Doubling time was calculated as described in Materials and Methods. C.-E.Increased sensitivity of Runx1null cells to chemotherapeutic agents and oxidative stress. Runx1+ and Runx1null were plated at 2x105 cells/ml and treated with 1.0µM doxorubicin (C), 0.8% ethanol (D) or 1.0µM dexamethasone (E) for 24-30 hours before viability counting. Plots show mean ± SD for a single experiment carried out in triplicate and are representative of at least 3 independent experiments; * = p < 0.05, ** = p < 0.01.
Eµ-Myc lymphoma cells are tumorigenic but circulating tumor cells display increased sensitivity to dexamethasone
The unexpected survival of Eµ-Myc lymphoma cell lines after Runx1 excision in vitro led us to consider whether Runx1 is required for lymphoma re-establishment in vivo. This was tested by inoculation of NOD-SCID/ γCnull (NSG) mice with cell lines with and without excision of Runx1 (Figure 8). Retrospective analysis showed that the Runx1-excised input cells had a small residual fraction of non-excised cells, while the non-excised input cells were virtually pure (Figure 8A). Notably, the non-excised cells appeared to have a significant advantage in cells obtained from ascites but not in tumors that arose within the skin at the injection site, where mainly excised cells were observed. As these mice were not treated with pIpC, the non-excised cells had clearly undergone spontaneously induced deletion in the skin tumor deposits. While this phenomenon may also reflect local production of endogenous IFNβ in skin e.g. by plasmacytoid dendritic cells [37] the key observation for this study is that tumors can develop in vivo with no detectable Runx1. This analysis also shows that there is a strong selective advantage for retention of Runx1 in cells growing in suspension in the peritoneal cavity.
We also tested the effects of administration of dexamethasone on tumor formation. The most striking observation here was in circulating cells in blood, where dexamethasone treatment had a much greater effect in eliminating excised cells compared to their non-excised counterparts (Figure 8B). This observation illustrates the fact that the growth requirements of cells in vivo cannot be fully elucidated in vitro and suggests that free cells in circulation are most dependent on Runx1 for their survival. Notably, some splenic lymphomas appeared to consist of exclusively Runx1null cells (Figure 8B, lower panel), again indicating attenuation of Runx1-dependency compared to primary lymphomas.
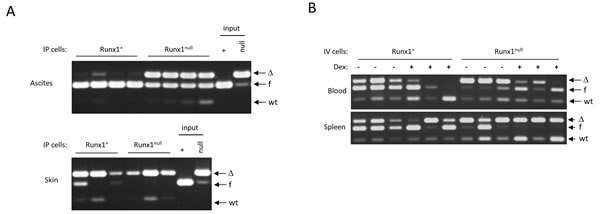
Figure 8: Runx1null lymphoma cells are tumorigenic but are out-competed by Runx1fl/fl cells in ascitic fluid and in the blood of in dexamethasone-treated mice. A. Runx1 excision PCR on genomic DNA from vehicle control (Runx1+) or IFNβ-treated (Runx1null) 3s cell line cells (“input”), and from cells from ascites fluid (upper panel) and tumors in skin (lower panel) derived from 4 mice transplanted IP with each of the input cell types. B. Runx1 excision PCR on genomic DNA of cells derived from blood and spleen of 12 mice comprising 6 mice transplanted intravenously with Runx1+(3s+) and 6 with Runx1null (3s-) cells, and 3 mice from each of these groups treated with dexamethasone or vehicle control as detailed in Materials and Methods.
DISCUSSION
Previous studies have shown that ectopic expression of the Runx genes as a result of retroviral insertional mutagenesis or transgenic over-expression is potently synergistic with over-expressed Myc or loss of p53 in lymphomagenesis [9-11, 38, 39]. The present study shows that basal expression of the normal Runx1 gene is vital for maintenance of primary Myc-driven lymphoma in vivo and that this dependence is stronger than in normal lymphoid cells, providing evidence of oncogene addiction in vivo. Surprisingly, lymphoma-derived cell lines were able to proliferate indefinitely and remained tumorigenic after complete excision of Runx1. This is a telling observation, as it demonstrates that Runx1 is not merely a structural component of the transcriptional apparatus that confers B-cell identity and sustains viability. However, Runx1null lymphoma cells displayed growth impairment compared to Runx1 non-excised controls and were hypersensitive to DNA damaging agents and glucocorticoids. On transplantation, cells retaining Runx1 also had a selective advantage when growing as non-adherent cells in the peritoneal cavity and as circulating cells in the blood of mice treated with glucocorticoid. These observations suggest that Runx1null cells may be partially protected by supportive sites of high cell density in vivo.
The ability of p53null Eµ-Myc lymphoma cell lines to survive Runx1 deletion suggested a possible functional link whereby Runx1 protects against Myc-driven induction of p53 and/or downstream responses, and becomes superfluous after loss of the intact Tp53 allele during in vitro establishment. However, reintroduction of a temperature-sensitive p53 expression construct (Val135) into cell lines did not discriminate between Runx1 positive and negative cells, which succumbed to apoptosis with similar kinetics after temperature shift with or without irradiation. It is conceivable that another change, secondary to Trp53 allele loss, allows the established cells to survive without Runx1. This hypothesis is consistent with the observation that the Trp53null blast cell fraction in vivo showed substantial retention of the intact Runx1 allele. However, we cannot exclude the possibility that ectopic p53 in vitro fails to recapitulate fully the behaviour of endogenous p53 in vivo. For example, we were surprised to observe a lack of induction of detectable CDKN1A/p21Waf1 protein expression in response to irradiation and p53 activation. This observation implies that established Eµ-Myc lymphoma cells may have lost the capacity to undergo cell cycle arrest in response to p53 induction and choose cell death as the default pathway. This phenomenon could account for the increased selection against wild-type p53 in cultured lymphoma cells compared to their in vivo counterparts [14].
The cell-type specific regulatory processes controlled by the Runx transcription factors are underlined by transcriptome analysis of Eµ-Myc lymphoma cells before and after Runx1 excision, which revealed many changes that were not evident in control Runx1wt/wt lymphoma cells with inducible Cre expression. The Runx1 gene expression signature we observed in Eµ-Myc lymphoma cells was significantly enriched for regulators of lymphocyte proliferation, survival and differentiation, with changes consistent with the observed growth advantage and chemo-resistance associated with intact Runx1 expression.
The marked up-regulation of Rag1 and Rag2 after Runx1 deletion is of particular interest. These genes are regulated in a complex, lineage-specific manner in T- and B-cells and undergo waves of expression during B-lymphocyte development [40]. The promoters of both genes harbour multiple Runx binding sites, while further Runx motifs are essential for the function of an intergenic Rag silencer in T-cells which is over-ridden in double positive thymocytes by an anti-silencer upstream of Rag2 [41]. We also noted strong inverse correlations between RUNX1 and RAG1/RAG2 mRNA expression in a panel of human leukemias and lymphomas. This is interesting in light of evidence that driver mutations are frequently induced by aberrant RAG activity in t(12;21) TEL-RUNX1 leukemias [42] which express unusually high levels of RAG1, but also in other genotypes that have no known lesion in RUNX1 [43, 44]. The weaker correlation between RUNX1 and RAG expression in t(12;21) leukemias may be an artefact of the detection of mRNA for both TEL-RUNX1 and the untranslocated RUNX1 allele but may also be a function of direct interference with RAG repression by the fusion oncoprotein. Moreover, the increased incidence of T-cell lymphomas in ENU-treated chimeric mice [17] and the occurrence of apparent loss-of-function RUNX1 mutations in a subset of ALLs [20] might be explained at least in part by dysregulation of recombinase gene expression.
We noted very little overlap with changes associated with Runx1 deficiency in murine haematopoietic precursor cells where ribosomal biogenesis was implicated recently as a Runx1-directed function [45]. Apart from their separation in the differentiation hierarchy, Eµ-Myc cells differ from HPSCs in over-expression of Myc, a known driver of ribosome biogenesis [46]. It is therefore conceivable that the effect of Runx1 deletion on ribosomal gene expression in HSPCs is mediated indirectly through loss of signalling to Myc. We also noted diametrically opposite effects of Runx1 deficiency on responses to DNA damage, where HSPCs were reported to display increased resistance [47], while we noted increased sensitivity of Runx1null Eµ-Myc cells. While this difference might be related to cell transformation, the relative permissiveness to Runx1 deletion of the myeloid compartment in normal spleen suggests that lineage-specific factors are likely to be involved. Moreover, the increased fragility of Myc-driven lymphoma cells lacking Runx1 indicates that targeting of Runx pathways is likely to be of therapeutic benefit in the context of Myc-driven lymphoma. We also noted little overlap with the mitotic checkpoint signature observed after knockdown of RUNX1 in Kasumi t(8;21) AML cells [31]. While lineage-specific differences may again be invoked to explain this discrepancy, the AML cells also express the RUNX1-ETO fusion protein which is likely to modulate at least some of the key promoters and enhancers vacated by RUNX1 knockdown.
The finding that basal Runx1 activity is critical for Myc-driven lymphoma maintenance in vivo and that dependence is only partially attenuated in established cell lines lacking p53 is encouraging for ongoing efforts to target the Runx genes and their downstream effectors in cancer therapy [48, 49]. Moreover, the increased sensitivity of Runx1-deleted cells to components of standard chemotherapeutic regimens in current use for lymphoma therapy suggests that these may be combined with Runx inhibition for greater efficacy.
MATERIALS AND METHODS
Generation of transgenic crosses and animal experiments
Mx1Cre+/Runx1fl/fl mice [26] were crossed with Eµ-Myc mice and p53-/- mice to produce highly tumour-prone Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/- mice and littermate controls lacking Mx1Cre and/or Eµ-Myc. Litters were treated at two weeks of age with 8.5mg/kg pIpC injected intraperitoneally (IP; two injections two days apart) or left untreated; animals were humanely culled when they showed signs of tumour development. Runx1wt controls (Mx1Cre+/ Runx1wt/wt/Eµ-Myc+/p53+/-) were treated in the same way. Adult Eµ-Myc-/Runx1fl/fl mice were treated with 6 injections of 600µg pIpC 2-3 days apart at 5 weeks of age. Transplantation assays were performed in NOD/SCID/γCnnull (NSG) mice, which were transplanted IP or intravenously (IV) with 5 x 106 Runx1+ or Runx1null cells; IV-transplanted mice were treated with 20mg/kg Dexadresone® IP or vehicle control daily, Monday-Friday for 3 weeks and 5-10µl blood for flow cytometric analysis of B220+ cell counts were removed weekly by tail-tipping. Animal protocols used in this work were evaluated and approved by the University of Glasgow Ethics and Welfare Committee and were carried out under Home Office License (approval granted September 2012, license number PPL 60/4408) as governed by the Animal Scientific Procedures Act, 1986.
Statistical analysis
Survival curves were compared using the Wilcoxon rank sum test. All other statistical comparisons were performed using the Student’s t-test.
Flow cytometry and cell sorting
Phenotyping of spleen cells was performed using the following antibodies: anti-B220-phycoerythrin, anti-CD4-phycoerythrin, anti-Mac1-PerCPCy5.5 (all BD Biosciences) and anti-CD8-FITC (Serotec). Spleen cell suspensions in PBS + 0.1% BSA (wash buffer) were stained with combinations of antibodies or isotype controls for 30 minutes at 4°C, red cells were lysed using Pharmlyse (BD Biosciences) and cells were analysed using an Accuri C6 flow cytometer and Cflow Sampler software (BD Biosciences). Analysis of activated caspase 3 expression was performed by flow cytometry using the PE Active Caspase 3 Apoptosis Kit (BD Biosciences) according to manufacturer’s instructions. Sorting of spleen lymphoid and myeloid cells was performed using a combination of anti-B220-phycoerythrin and anti-CD3-phycoerythrin to identify lymphoid cells and anti-Mac1-PerCPCy5.5 to identify myeloid cells. Sorting of blast cells from primary lymphomas was performed by staining cells with anti-B220-phycoerythin and anti-CD45-FITC; in the B220+ population, blast cells were identified using CD45 and SSC as reported in [27]. All sorting was performed using a FACSAria (BD Biosciences).
Genomic DNA extraction and PCR
Genomic DNA was extracted from cell lines using the DNeasy kit (Qiagen) and from tissues using the illustra Nucleon BACC2 DNA extraction kit (GE Life Sciences). Determination of DNA concentration was carried out using a Nanodrop 2000 (Thermo Scientific, Walatham, MA. USA). Analysis of Runx1 excision in primary cells/tissues and cell lines was carried out using previously described primers and cycling conditions [50]; master mix was prepared with 2x ReddyMix, 1.6µM each primer and 10ng template DNA. Validation of this assay for quantitative determination of Runx1 excision was performed by analysing excision in standards containing mixtures of 0-100% excised cells. PCR for p53 wild-type and null alleles was performed using 20ng template DNA, 2x ReddyMix (ThermoFisher Scientific) and primers directed against p53 intron 4 (WP53: GTGTTTCATTAGTTCCCCAC), exon 5 (UP3: ATGGTGGGGGCAGCGTCTCA), and the null allele (NP5: CGGTCTTGTCGATCAGGATG); cycling conditions were 5 minutes at 94°C, 35 cycles of 1 minute at 94°C, 1 minute at 55°C and 1 minute at 72°C, then 7 minutes at 72°C, generating PCR products of 242bp (p53 wt allele) or 470bp (p53 null allele). All PCR products were separated on a 1.5 or 2% agarose gel and visualised with ethidium bromide and UV transillumination. Densitometry was carried out using ImageJ software (http://imagej.nih.gov/ij/); images shown were adusted only for contrast.
Cell culture, constructs and retroviral transductions
The 3s, 6s and 44s cell lines were established from spleen tissue from primary lymphomas in Mx1Cre+/Runx1fl/fl/Eµ-Myc/p53+/- transgenic mice and grown in RPMI 1640 supplemented with 10% FCS, 100U/ml penicillin and streptomycin, 2mM L-glutamine and 50µM 2-mercaptoethanol (complete RPMI; all reagents from Life Technologies). Paired Runx1null and Runx1+ cells were created from Runx1fl/fl parental cell lines by treatment with 5-50U/ml IFNβ (R&D Systems) or vehicle control (PBS+0.1% BSA); Runx1 excision in IFNβ-treated lines was confirmed by Runx1 excision PCR. The p53V135-GFP was constructed by ligating the 1.4kb EcoR1 fragment containing murine p53V135 [51] into the Mig-R1 vector [52]. Viral supernatants were prepared following transient transfection of GP86+E or 293T cells respectively and used to infect 3s cells as described previously [53]. Infected cells were sorted for GFP+ cells. Doubling time assays were performed in 12-well cell culture plates with 2x105cells/ml and counted 24 hours after initiation of culture; doubling times were calculated using the formula Td = ti x (log(2)/log(q2-q1)) where Td = doubling time, ti = incubation time, q1 = number of cells at start of assay and q2 = number of cells at end of assay. Competition assays were performed by mixing equal numbers of excised and non-excised cells; cells were cultured in duplicate in complete RPMI, passaging three times per week, returning 105 cells to 10ml culture at each passage; Runx1 excision in the culture was monitored weekly by Runx1 excision PCR. Ethanol, doxorubicin (Stratech Scientific #S1208 10mM in DMSO) or dexamethasone (Sigma D2915) treatment of pairs of excised and non-excised cell lines was carried out in 12-well plates using 2x105 cells/ml and with the addition of ethanol (0.8%), doxorubicin (1.0µM) or dexamethasone (1.0µM). Single cell cloning of cell lines was performed by partially excising Runx1 by sub-optimal IFNβ treatment and sorting single cells into 96-well plates in complete RPMI; growing clones were transferred to larger culture dishes until sufficient cells to analyse were obtained.
RNA extraction and microarray analysis
Total RNA was isolated by RNeasy kit (Qiagen, Manchester, UK) from three cultures each of established lymphoma lines 3s (Mx1Cre+/Runx1fl/fl/Eµ-Myc+/p53+/-)
and 30s (Mx1Cre+/Runx1wt/wt/Eµ-Myc+/p53+/-), treated with and without IFNβ to excise endogenous Runx1 (Figure 4A). RNA was tested for quality on the Agilent 2100 Bioanalyser (Agilent Technologies, Stockport, UK) and NanoDrop 2000 (Thermo Scientific, Walatham, MA. USA) before screening against Affymetrix GeneChip Mouse Transcriptome Array 1.0 (High Wycombe, UK, 2014) by ATLAS Biolabs (Berlin, Germany) according to standard protocols. RMA normalisation followed by probe annotation and statistical analysis to generate p-values and fold changes was performed using Partek Genomics Suite 6.6 (Partek Inc., St.Louis, MO, USA). Microarray data are available at the Gene Expression Omnibus (GEO) repository, accession number GSE78001.
Quantitative real-time PCR
cDNA was prepared from 1µg aliquots of RNA using a Quantitect Reverse Transcription kit (Qiagen) and diluted 1 in 20 in DEPC-treated water to give a working stock. For quantitative real-time PCR, 12.5ng aliquots of cDNA were amplified in triplicate on an ABI 7500 real-time PCR system using Power SYBR Green PCR master Mix (Thermo Fisher Scientific, UK), and primers for murine Bcl11a, Cd55, Daf2, Il7r, Nckap1, Ptpn22, Prkcb, Rag1, Rag2 or endogenous control 18S rRNA (Qiagen QuantiTect Primer Assays). Relative quantification was carried out and calibrated to vector control samples where appropriate. Data were analysed using the standard software for the ABI 7500 real-time PCR system.
Western blotting and antibodies
Preparation of whole cell protein extracts was performed as described previously [54]. Samples equivalent to 50μg of protein (Bio-Rad protein assay) were resolved on 8-12% SDS polyacrylamide gels and transferred to enhanced chemiluminescence (ECL; Fischer Scientific), nitrocellulose membranes. The antibodies used were α Runx1 (#8229 New England Biolabs), α p53(FL393), α p21WAF1, α Actin (sc-6243, sc-471 and sc-1616, Santa Cruz Biotechnology) and α CDKN2A/p19ARF (ab80, Abcam). Positive controls were as follows: Runx1 (NIH3T3 transduced with pBabeRunx1 P1 [15], p53 and p21WAF1 (UVC-treated wild type MEF extract), p19ARF (SV3T3 cell extract).
CONCLUSIONs
Primary lymphoma cells from Eµ-Myc mice show evidence of addiction to Runx1 in vivo, but become permissive for deletion in vitro. Loss of p53 function appears to be necessary but not sufficient for this process. In this context Runx1 controls a network of genes involved in lymphocyte proliferation, survival and differentiation, shedding light on its dualistic behaviour in lymphomagenesis. While the ability of Myc-driven lymphoma cells to grow in the absence of Runx1 is surprising, their impaired proliferation and increased chemo-sensitivity validates Runx1 function as a candidate target in future combination therapies.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by a joint programme grant from Bloodwise (grant number 13046) and Cancer Research UK (grant number A11951). We thank Nancy Speck (University of Pennsylvania) for kind provision of Runx1fl/fl mice. We thank Jennifer Cassels (University of Glasgow) for helpful assistance with cell sorting.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
REFERENCES
1. North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marin-Padilla M, Speck NA: Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development 1999;126:2563-2575.
2. Levanon D, Groner Y: Structure and regulated expression of mammalian RUNX genes. Oncogene 2004;23:4211-4219.
3. Ito Y: RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res 2008;99:33-76.
4. Ito Y, Bae SC, Chuang LS: The RUNX family: developmental regulators in cancer. Nat Rev Cancer 2015;15:81-95.
5. Blyth K, Cameron ER, Neil JC: The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 2005;5:376-387.
6. Mikkers H, Allen J, Knipscheer P, Romeijn L, Hart A, Vink E, Berns A: High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet 2002;32:153-159.
7. Stewart M, Terry A, Hu M, O’Hara M, Blyth K, Baxter E, Cameron E, Onions DE, Neil JC: Proviral insertions induce the expression of bone-specific isoforms of PEBP2alphaA (CBFA1): evidence for a new myc collaborating oncogene. Proc Natl Acad Sci U S A 1997;94:8646-8651.
8. Wotton S, Stewart M, Blyth K, Vaillant F, Kilbey A, Neil JC, Cameron ER: Proviral insertion indicates a dominant oncogenic role for Runx1/AML-1 in T-cell lymphoma. Cancer Res 2002;62:7181-7185.
9. Stewart M, Mackay N, Cameron ER, Neil JC: The common retroviral insertion locus Dsi1 maps 30 kilobases upstream of the P1 promoter of the murine Runx3/Cbfa3/Aml2 gene. J Virol 2002;76:4364-4369.
10. Blyth K, Terry A, Mackay N, Vaillant F, Bell M, Cameron ER, Neil JC, Stewart M: Runx2: a novel oncogenic effector revealed by in vivo complementation and retroviral tagging. Oncogene 2001;20:295-302.
11. Vaillant F, Blyth K, Terry A, Bell M, Cameron ER, Neil J, Stewart M: A full-length Cbfa1 gene product perturbs T-cell development and promotes lymphomagenesis in synergy with myc. Oncogene 1999;18:7124-7134.
12. Blyth K, Slater N, Hanlon L, Bell M, Mackay N, Stewart M, Neil JC, Cameron ER: Runx1 promotes B-cell survival and lymphoma development. Blood Cells Mol Dis 2009;43:12-19.
13. Vaillant F, Blyth K, Andrew L, Neil JC, Cameron ER: Enforced expression of Runx2 perturbs T cell development at a stage coincident with beta-selection. J Immunol 2002;169:2866-2874.
14. Blyth K, Vaillant F, Hanlon L, Mackay N, Bell M, Jenkins A, Neil JC, Cameron ER: Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res 2006;66:2195-2201.
15. Wotton SF, Blyth K, Kilbey A, Jenkins A, Terry A, Bernardin-Fried F, Friedman AD, Baxter EW, Neil JC, Cameron ER: RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene 2004;23:5476-5486.
16. Blyth K, Terry A, O’Hara M, Baxter EW, Campbell M, Stewart M, Donehower L, Onions DE, Neil JC, Cameron ER: Synergy between a human c-myc transgene and p53 null genotype in murine thymic lymphomas: contrasting effects of homozygous and heterozygous p53 loss. Oncogene 1995;10:1717-1723.
17. Kundu M, Compton S, Garrett-Beal L, Stacy T, Starost MF, Eckhaus M, Speck NA, Liu PP: Runx1 deficiency predisposes mice to T-lymphoblastic lymphoma. Blood 2005;106:3621-3624.
18. Niini T, Vettenranta K, Hollmen J, Larramendy ML, Aalto Y, Wikman H, Nagy B, Seppanen JK, Ferrer SA, Mannila H, Saarinen-Pihkala UM, Knuutila S: Expression of myeloid-specific genes in childhood acute lymphoblastic leukemia - a cDNA array study. Leukemia 2002;16:2213-2221.
19. Harewood L, Robinson H, Harris R, Al-Obaidi MJ, Jalali GR, Martineau M, Moorman AV, Sumption N, Richards S, Mitchell C, Harrison CJ: Amplification of AML1 on a duplicated chromosome 21 in acute lymphoblastic leukemia: a study of 20 cases. Leukemia 2003;17:547-553.
20. Della GG, Palomero T, Perez-Garcia A, Ambesi-Impiombato A, Bansal M, Carpenter ZW, De KK, Sole X, Xu L, Paietta E, Racevskis J, Wiernik PH, Rowe JM, Meijerink JP, Califano A, Ferrando AA: Reverse engineering of TLX oncogenic transcriptional networks identifies RUNX1 as tumor suppressor in T-ALL. Nat Med 2012;18:436-440.
21. Osato M, Yanagida M, Shigesada K, Ito Y: Point mutations of the RUNx1/AML1 gene in sporadic and familial myeloid leukemias. Int J Hematol 2001;74:245-251.
22. Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, Loh M, Felix C, Roy DC, Busque L, Kurnit D, Willman C, Gewirtz AM, Speck NA, Bushweller JH, Li FP, Gardiner K, Poncz M, Maris JM, Gilliland DG: Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet 1999;23:166-175.
23. Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA: Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009;457:887-891.
24. Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL: Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 1999;13:2658-2669.
25. Hsu B, Marin MC, El-Naggar AK, Stephens LC, Brisbay S, McDonnell TJ: Evidence that c-myc mediated apoptosis does not require wild-type p53 during lymphomagenesis. Oncogene 1995;11:175-179.
26. Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG: Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005;106:494-504.
27. Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, Yeh JE, Chen JY, Iruela-Arispe ML, Varella-Garcia M, Wu H: Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008;453:529-533.
28. Shimizu K, Yamagata K, Kurokawa M, Mizutani S, Tsunematsu Y, Kitabayashi I: Roles of AML1/RUNX1 in T-cell malignancy induced by loss of p53. Cancer Sci. 2013; 104:1033-8.
29. Abdueva D, Skvortsov D, Tavare S: Non-linear analysis of GeneChip arrays. Nucleic Acids Res 2006;34:e105.
30. Cai X, Gao L, Teng L, Ge J, Oo ZM, Kumar AR, Gilliland DG, Mason PJ, Tan K, Speck NA: Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015;17:165-177.
31. Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, Lotem J, Tanay A, Groner Y: Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep 2013;4:1131-1143.
32. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van LP, Cooke SL, Marshall J, Martincorena I, Hinton J, Gundem G, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 2014;46:116-125.
33. Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De VJ, Hernandez JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, Kipps TJ, Rassenti LZ, Yeoh AE, Papenhausen PR, Liu WM, Williams PM, Foa R: Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 2010;28:2529-2537.
34. Wang Z, Wei D, Xiao H: Methods of cellular senescence induction using oxidative stress. Methods Mol Biol 2013;1048:135-144.
35. Abraham J, Balbo S, Crabb D, Brooks PJ: Alcohol metabolism in human cells causes DNA damage and activates the Fanconi anemia-breast cancer susceptibility (FA-BRCA) DNA damage response network. Alcohol Clin Exp Res 2011;35:2113-2120.
36. Wotton S, Terry A, Kilbey A, Jenkins A, Herzyk P, Cameron E, Neil JC: Gene array analysis reveals a common Runx transcriptional programme controlling cell adhesion and survival. Oncogene 2008;27:5856-5866.
37. Gregorio J, Meller S, Conrad C, Di NA, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, Gilliet M: Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med 2010;207:2921-2930.
38. Stewart M, Terry A, O’Hara M, Blyth K, Baxter E, Cameron E, Onions D, Neil JC: Proviral insertions induce the expression of bone-specific isoforms of PEBP2alphaA (CBFA1): evidence for a new myc collaborating gene. Proc Natl Acad Sci USA 1997;94:8646-8651.
39. Wotton S, Stewart M, Blyth K, Vaillant F, Kilbey A, Neil JC, Cameron ER: Proviral insertion indicates a dominant oncogenic role for Runx1/AML-1 in T-cell lymphoma. Cancer Res 2002;62:7181-7185.
40. Yu W, Misulovin Z, Suh H, Hardy RR, Jankovic M, Yannoutsos N, Nussenzweig MC: Coordinate regulation of RAG1 and RAG2 by cell type-specific DNA elements 5’ of RAG2. Science 1999;285:1080-1084.
41. Yannoutsos N, Barreto V, Misulovin Z, Gazumyan A, Yu W, Rajewsky N, Peixoto BR, Eisenreich T, Nussenzweig MC: A cis element in the recombination activating gene locus regulates gene expression by counteracting a distant silencer. Nat Immunol 2004;5:443-450.
42. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-421.
43. Mendes RD, Sarmento LM, Cante-Barrett K, Zuurbier L, Buijs-Gladdines JG, Povoa V, Smits WK, Abecasis M, Yunes JA, Sonneveld E, Horstmann MA, Pieters R, Barata JT, Meijerink JP: PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 2014;124:567-578.
44. Navarro JM, Touzart A, Pradel LC, Loosveld M, Koubi M, Fenouil R, Le NS, Maqbool MA, Morgado E, Gregoire C, Jaeger S, Mamessier E, Pignon C, et al.: Site- and allele-specific polycomb dysregulation in T-cell leukaemia. Nat Commun 2015;6:6094.
45. Cai X, Gao L, Teng L, Ge J, Oo ZM, Kumar AR, Gilliland DG, Mason PJ, Tan K, Speck NA: Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015;17:165-177.
46. van Riggelen J., Yetil A, Felsher DW: MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 2010;10:301-309.
47. Venkata JK, An N, Stuart R, Costa LJ, Cai H, Coker W, Song JH, Gibbs K, Matson T, Garrett-Mayer E, Wan Z, Ogretmen B, Smith C, Kang Y: Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014;124:1915-1925.
48. Marugan JJ, Xiao J, Zheng W, Southall N, Dehdashti S, Cunningham L, Liu PP: ML223: A Small Molecule Probe With In Vivo Activity Against Acute Myeloid Leukemia Subtype M4Eo. Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-2011.
49. Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S, Zhou Y, Boulton A, Kuntimaddi A, Gao Y, Rajewski RA, Guzman ML, Castilla LH, Bushweller JH: Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science 2015;347:779-784.
50. Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA: Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009;457:887-891.
51. Michalovitz D, Halevy O, Oren M: Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell 1990;62:671-680.
52. Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D: Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 1998;92:3780-3792.
53. Blyth K, Slater N, Hanlon L, Bell M, Mackay N, Stewart M, Neil JC, Cameron ER: Runx1 promotes B-cell survival and lymphoma development. Blood Cells Mol Dis 2009;43:12-19.
54. Wotton S, Terry A, Kilbey A, Jenkins A, Herzyk P, Cameron E, Neil JC: Gene array analysis reveals a common Runx transcriptional programme controlling cell adhesion and survival. Oncogene 2008;27:5856-5866.