
INTRODUCTION
Polycystic ovary syndrome (PCOS) is a complex and highly heterogeneous women endocrine disorder, the features include ovarian dysfunction, menstrual disorders, hyperandrogenemia, insulin resistance, abdominal obesity and infertility [1]. The familial clustering and inheritance indicates genetic factors contributed to PCOS development [2, 3], in addition to genetic predisposition, environment and life style factors; emerging evidence suggests that epigenetic modification/regulation contributed to the development of PCOS [4, 5]. PCOS is the most prevalent diseases an affects about 7% of the reproductive-age women [6]. However, the etiology remains unclear.
DNA methylation(5mC) as one of the best-studied DNA modification is a major epigenetic modification of the genome generally inhibit gene expression, whereas DNA hydroxymethylation(5hmC) associated with increasing gene expression [7, 8]. Different level in 5mC at single specific loci can be sufficient to regulate gene expression [9]. However, 5mC changes at specific loci cannot indicate the changes occurring at global levels. In adult mammals, the level of 5hmC exhibit rather variable and show tissues and cell types specific [10, 11]. It had been reported that peripheral blood DNA global methylation was not significantly altered in PCOS compared with healthy controls [12]. However, whether global DNA methylaiton and hydroxymethylation involved in PCOS development or not was needed to explore.
Epigenetic changes as a potential mechanism causing PCOS development has been investigated in many studies before. It had reported that genome-wide DNA methylation profile of PCOS peripheral blood revealed that 79 differentially methylated genes between PCOS-insulin resistance and PCOS-noninsulin resistance patients, and 40 differentially methylated genes between PCOS patients and healthy controls [13]. Further study had detected significant differences genome-wide DNA methylation and expression patterns exist between PCOS ovaries and normal ovaries [14, 15]. These studies indicated that DNA methylation may in part be responsible for the PCOS phenotype. Previous study has revealed that gene body DNA methylation of ovarian granulosa cells associated genes expression in PCOS patients [16]. The granulosa cells of PCOS ovaries were variable sensitivity to FSH, show lower apoptotic rate and higher proliferation rate coupling with 211 genes differentially expressed [17–19]. Ovary genome-wide DNA methylation may mask specific cell DNA methyaltion pattern, such as follicular granulosa cells, which play key roles during oocyte development through intercellular communication [20, 21]. Hence, it is essential to genome-wide profile the different DNA methylation between PCOS patients and healthy controls granulosa cells.
To confirm epigenetic abnormality and to recover a novel mechanism for granulosa cells in PCOS development and follicular development, we systemly study the granulosa cell global DNA methylation and hydroxymethylation of both PCOS patients’ and healthy controls’, and genome-wide profile the DNA methylaiton of obesity and non-obesity PCOS patients’. Analysis of the epigenetic phenotypes and identification of specific epigenetic changes of granulosa cells will help us illustrate the epigenetic etiology of PCOS.
RESULTS
Basic clinical characteristics of study PCOS patients and controls
All the clinical characteristics of study participants were shown in Table 1. The obesity PCOS patients and non-obesity patients showed significant higher luteinizing hormone (LH), testosterone (T) and Basic Follicular Number compared with healthy controls. Obviously, the obesity PCOS patients had highest body mass index (BMI). There were no differences between the BMI of the non-obesity PCOS patients and healthy controls. Blood glucose concentration showed no significance among the obesity PCOS patients, non-obesity patients and healthy controls. During Controlled Ovarian Hyperstimulation of clinical treatment, the gonadotropin (Gn) does and day’s treatment showed no significance. The free thyroxine 3 (FT3), free thyroxine 4 (FT4) and thyroid stimulating hormone (TSH) also showed no statistical significant among three groups. Ovarian granulosa cells from 24 patients were conducted to DNA methylation chip investigation and RT-qPCR.
Table 1: Clinical characteristic of PCOS patients and healthy controls
Basic Characteristic |
PCOS-obesity |
PCOS-nonobesity |
Control |
P value |
|---|---|---|---|---|
Age |
30.38±4.14 |
29.75±2.82 |
29.63±2.67 |
0.887 |
BMI(Kg/m2) |
28.95±0.74 |
21.95±2.18 |
21.21±2.83 |
3.43E-07 |
FSH(IU/L) |
5.64±1.78 |
5.92±1.25 |
6.55±1.69 |
0.51 |
E2(pg/ml) |
51.52±28.19 |
43.63±25.33 |
43.58±18.65 |
0.758 |
P(ng/ml) |
0.76±0.24 |
0.8±0.31 |
0.9±0.35 |
0.664 |
PRL(ng/ml) |
12.06±6.45 |
14.79±8.47 |
20.8±8.38 |
0.0964 |
LH(IU/L) |
7.5±3.34 |
9.54±5.16 |
4.28±1.47 |
0.0285 |
T(ng/ml) |
0.45±0.12 |
0.64±0.26 |
0.24±0.11 |
0.000976 |
Basic Follicular Number |
22.75±2.55 |
23.75±0.7 |
13.88±5.1 |
8.54E-06 |
Days of Gn |
12.13±2.42 |
10.75±1.98 |
10.63±1.69 |
0.289 |
Dose of Gn |
2248.44±934.47 |
1487.5±435.43 |
1651.56±484.14 |
0.0733 |
Blood Glucose(mmol/L) |
5.06±0.48 |
4.67±0.36 |
4.75±0.38 |
0.161 |
FT3(pmol/L) |
5.16±0.92 |
4.95±0.72 |
4.83±0.54 |
0.676 |
FT4(pmol/L) |
11.05±1.49 |
10.52±1.15 |
11.23625±1.64 |
0.597 |
TSH(miu/L) |
2±0.82 |
2.31±0.79 |
2.3275±0.93 |
0.687 |
Global DNA methylation and hydroxymethylation status of PCOS patients and healthy controls granulosa cells
Genome-wide global DNA methylation and hydroxymethylation may stimulate or activate gene expression activity. Although the global methylation of peripheral blood DNA was not significantly altered in PCOS compared with controls, it is still essential to investigation the global DNA methylation and hydroxymethylation in granulosa cells in PCOS [12]. In our study, the granulosa cell global DNA methylation of PCOS patients showed significant higher level than the controls’ (58.82 ± 16.1% VS. 45.76 ± 13.1%, P<0.05, Figure 1A). However, the global DNA hydroxymethylation was very low and showed no statistical difference between PCOS and control (2.40 ± 0.42% VS. 2.37±0.61%, P>0.05, Figure 1B). We further summarized the hypermethylation and hypomethylation in obesity PCOS patients, non-obesity patients and healthy controls. Our results showed that PCOS-obesity and PCOS-nonobesity patients harbored much more hypermethylated CpG sites (DNA methylation lever above 80%) than control (20.5% and 19.0% VS. 12.9%, Figure 1C), this further supported that granulosa cell DNA global methylation level of PCOS was higher than controls’.
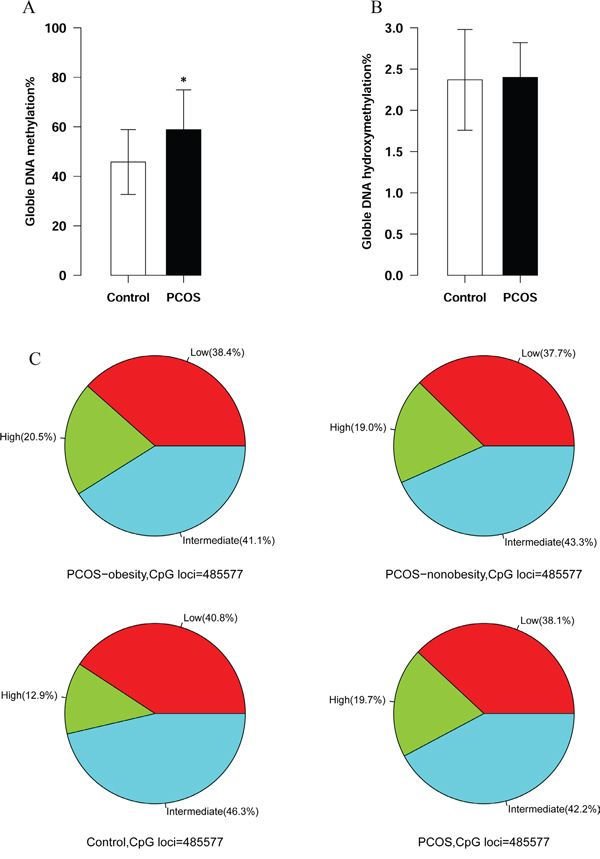
Figure 1: Global DNA methylation and hydroxymethylation between PCOS patients’ and Controls’ ovary granulosa cell. A. Global DNA methylation of granulosa cell between PCOS patients and controls. The number of PCOS patients was 20 and controls were 20. P value <0.05 B. Global DNA hydroxymethylation of granulosa between PCOS patients and controls. The number of PCOS patients was 20 and controls were 20. P value >0.05 C. Hypermethylation CpGs distribution among PCOS-nonobesity, PCOS-obesity and Controls. (The hypermethylaiton of DNA methylation was defined lever above 0.8, the number of PCOS-obesity patients was 8, controls was 8 and PCOS-nonobesity was 8.)
Genome-wide differentially DNA methylation genes profiling in PCOS
Genome-wide DNA methylation analysis showed 6936 differentially methylated CpG sites (p<0.005) respectively between control and PCOS-obesity group and 2664 CpG sites exhibited highly significant (P<0.001, Supplementary Dataset 1A). There were 12245 differential methylated CpG sites (P<0.005) between control and PCOS-nonobesity group and 1582 CpG sites exhibited highly significant (P<0.001, Supplementary Dataset 1B). 5202 methylated CpG sites (P<0.005) were detected significantly differential between PCOS-obesity and PCOS-nonobesity group and 1186 CpG sites showed highly significant (P<0.001, Supplementary Dataset 1C). We further explored the different methylated CpG sites in methylated CpG content and neighborhood context (Shelf, Shore, Island and Open Sea); in particular, the different methylated sites between groups were residing in CpG islands (Control VS. PCOS-obesity: 31.17%, Control VS. PCOS-nonobesity: 46.14% and PCOS-obesity VS. PCOS-nonobesity: 43.76%, Figure 2A.) and Shore (Control VS. PCOS-obesity: 33.94%, Control VS. PCOS-nonobesity: 27.16% and PCOS-obesity VS. PCOS-nonobesity: 24.78%, Figure 2A), indicating that the gene expression may be activated or inactivated by DNA methylaiton located in CpG islands. However, no significant difference in methylated CpG sites residing in chromosomal locations was detected (Chr1-22 and XY) (Figure 2B). The different methylated CpG sites across the gene structure (TSS1500, TSS200, 5’UTR, First exon, Gene body, 3’UTR, and intergenic genomic region(IGR)) (Figure 2C), indicating the different methylated sites were located in gene boby and IGR, which would contributed to the gene expression alternative splicing. Overall, our data suggested the existence of a marked difference in methylation and regulation patterns, both of which are implicated in the development of PCOS. To further explore the different methylated sites whole genome distribution, we genome-wide display the different methylated sites across whole-genome (shown in Supplementary Figure 1, 2 and 3).
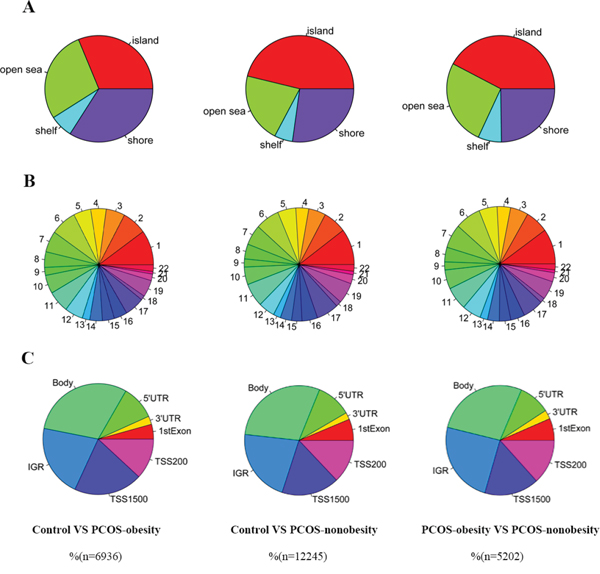
Figure 2: Different DNA methylation CpGs of granulose cells distribution in PCOS-obesity, PCOS-nonobesity and Control. A. CpG content and neighbourhood context of CpG Island, shore, shelf and open sea distribution of different methylation CpG sites B. Different DNA methylation CpGs sites distribution across autosome (chromosome 1 to 22) C. Different DNA methylation CpGs sites gene structure: 5’UTR, 3’UTR, 1stExon, TSS200, TSS1500, IGR and gene body distribution. The number of PCOS-obesity patients was 8, controls were 8 and PCOS-nonobesity was 8.
Functional analysis of genome-wide different DNA methylated sites
In order to explore the biological function of genes relevant in regulatory network of PCOS-obesity and PCOS-nonobesity, we further performed GO and KEGG analysis of the differentially methylated genes associated with PCOS-obesity and PCOS-nonobesity with DAVID. The all GO analysis results of three groups was shown in supplementary dataset 2 (Dataset 2A, Dataset 2B and Dataset 2C). The top significant GO terms associated with PCOS were shown in Figure 3. GO enrichment analysis for different DNA methylation sites of genes in GCs between Control and PCOS-nonobesity showed that development protein (False Discovery Rate (FDR) =1.64E-12), transcription factor activity (FDR=1.70E-11), embryonic morphogenesis (FDR=2.25E-10), sequence-specific DNA binding (FDR=3.52E-10) and embryonic development ending in birth or egg hatching (FDR=3.49E-07) were highly significant enriched. The GO categories splice variant (FDR=7.70E-09), alternative splicing (FDR= 7.99E-09), phosphoprotein (FDR= 4.53E-07) were the most significant enrichment between Control and PCOS-obesity. The GO categories developmental protein (FDR= 1.15E-05), sequence-specific DNA binding (FDR= 2.72E-05) and regulation of transcription DNA dependent (FDR=2.87E-04) were enrichment between PCOS-nonobesity and PCOS-obesity.
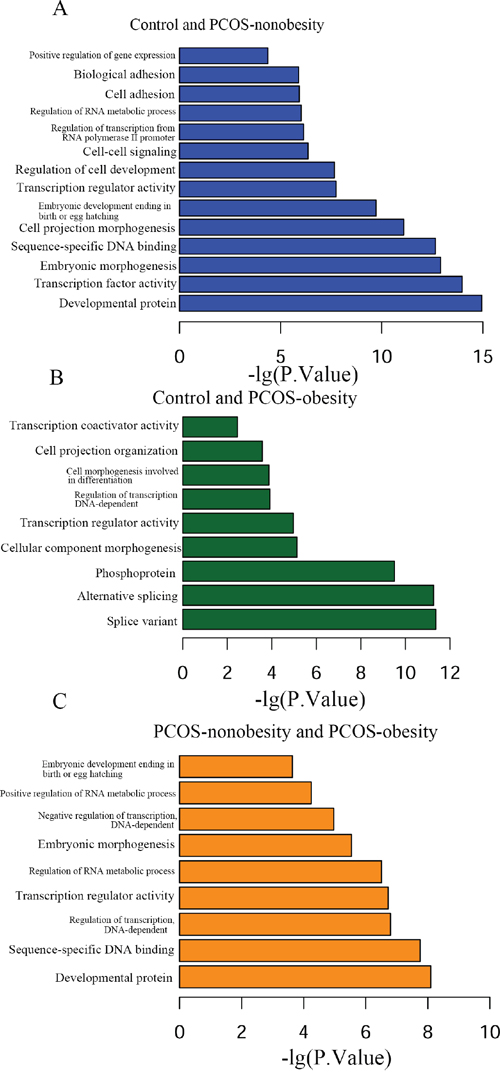
Figure 3: Functional analysis of different methylation sites of PCOS-obesity, PCOS-nonobesity and Control. A. Different methylation sites between control and PCOS-nonobesity functional analysis using DAVID B. Functional analysis of different methylation sites between control and PCOS-obesity using DAVID C. Different methylation sites between PCOS-obesity and PCOS-nonobesity functional analysis using DAVID.
Different DNA methylated site validation and target gene expression
We used pyrosequencing to validate the methylated sites detected in our study. The cg27227742 of MATN4 (Chr20:43935292-43935291), cg21498547 of DLGAP2 (Chr8:1651128-1651129), and cg08292959 of MGAT5B (chr17:74878420-74878421) were selected in our study, and our pyrosequencing results was in consonance with our Infinium HumanMethylation 450 BeadChip data (Figure 4). In addition, we detected a site (MATN4 Chr20:43935281-43935280) showed higher level of methylation in PCOS-nonobesity than the other two groups (Figure 4B). DNA hypermethylation was known to down regulate gene expression, then Quantitative Real Time PCR was performed to evaluate gene expressions patterns of the nine genes previously reported different expressed in PCOS patients [19]. And our data showed that a different methylated site located in potential regulated region of those genes (Supplementary Table 2).We detected five genes were different expressed (Figure 5), YWHAQ, NCF2, DHRS9 and SCNA only up-regulation in PCOS-obesity patients with no significance different between control and PCOS-nonobesity patients. However, PYHIN showed down-regulation in PCOS-obesity patients.
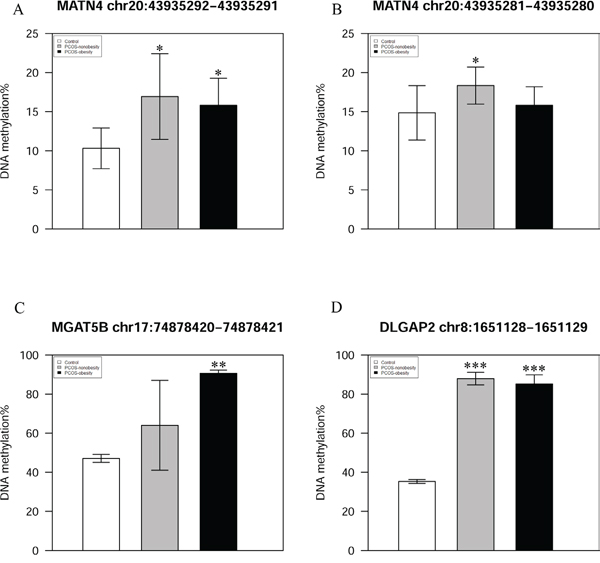
Figure 4: Validation of MATN4、 MGAT5B and DLGAP2 methylation site using pyrosequencing. A. Validation of MATN4 Chr20:43935292-43935291 DNA methylation level B. DNA methylaiton level of MATN4 Chr20:43935281-43935280 C. Validation of MGAT5B chr17:74878420-74878421 DNA methylation level D. Validation of DLGAP2 chr8:1651128-1651129 DNA methylation level.
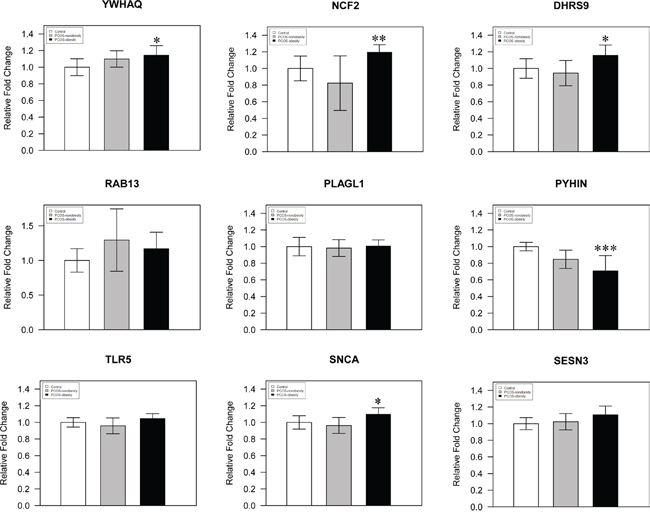
Figure 5: A histogram showing qPCR results of three groups for 9 selected genes. The YWHAQ, NCF2, DHRS9, RAB13, PLAGL1, PYHIN, TLR5, SNCA and SESN3 were analyzed by Q-RT-PCR, using total 24 samples from PCOS-obesity patients, controls and PCOS-nonobesity, each group 8 samples. YWHAQ, DHRS9 and SCNA showed significantly higher in PCOS-obesity patients (P<0.05), NCF2 showed highly significant up-regulation in PCOS-obesity groups(P<0.01), while PYHIN exhibited most significantly down-regulation in PCOS-obesity groups(P<0.001).
DISCUSSION
PCOS affects approximately 7% of women of childbearing age, previous studies suggested that obesity plays a critical role in PCOS development and may reduce the pregnancy rate and results in cancellation rate during in vitro fertilization treatment [22]. Despite many factors have been proved play important role in PCOS development over the past decades, DNA modification in ovary granulosa cell remain unclear. In this study, we firstly system investigated the PCOS-obesity and PCOS-nonobesity ovary granulosa cell DNA modifications including global genome DNA methylation and DNA hydroxymehytlation, genome-wide DNA methylation and their potential regulation function on gene expression.
Obesity really showed obvious effects on both the development and the clinical manifestation of PCOS, mainly by increasing androgen availability, alternating function of granulosa cell and disturbing follicle development [23]. In our study, the PCOS-obesity patients showed higher BMI, LH and T compared with PCOS-nonobesity and control, which in accordance with previous studies [24]. Different gene expression patterns were detected in human PCOS granulose cell, indicating that PCOS profound affected the function of granulosa cell and may resulted in follicular development abnormal [19, 25, 26], the genetic and environmental roles in the development of the PCOS and dysregulation of gene expression remain unclear. Global DNA methylation and hydroxymethylation are key for maintaining tissue-specific gene expression regulation, indicated global DNA methylation and hydroxymethylation may be involved in gene activity [27]. Our results suggested higher global methylated DNA level in PCOS-obesity and PCOS-nonobesity patients, and harbored much more hypermethylated CpG sites, suggesting that genes expression were dysregulated in PCOS patients’ granulosa cell, DNA methylation may partlylead to gene expression alteration in PCOS patients granulose cell. However, global DNA hydroxymethylation level was very low in PCOS and control, and showed no significance, our data indicated that DNA methylation not hydroxymethylation may contribute to gene activity in PCOS ovary granulose cell. DNA hydroxymethylaiton was key epigenetic modification, may be involved in follicular development and oocyte maturation, but DNA hydroxymethylaiton showed low lever in human granulosa cell and may not contribute to PCOS progression. Hence, we concluded that DNA methylation alteration in human granulosa cell could contribute to obesity and non-obesity PCOS phenotype.
Methylation of the promoter region is well identified as a silencer; also the gene body has been proved as a regulator of gene expression in many tissues [28]. Our data showed that many different methylation sites were located in CpG islands and gene body, which indicated that these sites associated with gene expression. Functional enrichment analysis showed that the different methylated sites between three groups were related to development protein, splice variant, embryonic morphogenesis and transcription factor activity. Our global DNA methylation level and different methylated genes functional enrichment analysis indicated that DNA methylation was a key factor in development of PCOS and may affect follicular development and embryonic morphogeneis during IVF. Furthermore, our data showed that different methylated sites enriched in splice variant functional term, indicating that DNA methylation contributed to different genome-wide gene expression pattern between control and PCOS. We selected 9 genes which were shown different expression between PCOS insulin resistant and non-resistant previously published [19] and harbored different methylated sites, and 5 of them were observed different expressed potentially regulated by DNA methylation located in regulation region, which further support that DNA methylation contributed to different gene expression in granulosa cell between control and PCOS. YWHAQ, NCF2 and DHRS9 may be regulated by the different methylated sites, whereas there would other mechanism regulated PYHIN and SCNA expression. In our study, the selected previously 9 genes didn’t show significant obvious difference between groups, ethnicity and markedly insulin resistance may affect the gene expression.
In summary, our current study discloses global DNA methylation, hydroxymethylation and specific DNA methylation patterns related to CpG Island, Open Sea, Shelf and Shore in the granulosa cells of PCOS obesity and nonobesity women. Considering these different DNA methylaiton genes are also linked to development protein, transcription factor activity, alternative splicing, sequence-specific DNA binding and embryonic morphogenesis, we concluded that these genes may be related to embryo development and metabolic disorders associated with the different clinical phenotypes of PCOS.
MATERIALS AND METHODS
Study patients recruitment
Totally 40 PCOS patients and 40 healthy controls were recruited from the Reproductive Center of the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China). PCOS was defined by the revised Rotterdam diagnostic criteria for PCOS (Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group, 2004). Other causes of oligomenorrhea or hyperandrogenism (for example, nonclassical 21-hydroxylase deficiency, Cushing’s syndrome, hypothyroidism, significant elevations in serum prolactin) were excluded on clinical grounds in our study. The healthy control patients were those only with tubal blockage or male infertility coming for in vitro fertilization. Those patients were recruited based on the following criteria: (i) ≥25 and ≤35 years and (ii) normal karyotypes (46, XX) were shown in chromosome examinations for all of them. All our study was conducted in accordance with the ethical standards and according to the Helsinki Declaration of 1975. All patients were informed consents.
Ovarian stimulation protocols, oocyte retrieval and granulosa cells (GCs) collection
All patients were using the GnRH agonist standard long protocol, ovarian stimulations were treated with recombinant FSH/HMG after pituitary down-regulation with GnRH agonist. The pituitary suppression was achieved with daily 0.1 mg triptorelin (Decapeptyl; Ferring Pharmaceutical, Kiel, Germany) from the mid-luteal phase of the cycle, after ovarian suppression was done; the dose of Decapeptyl was reduced to 0.05mg till the day of HCG administration. When the absence of dominant follicular development was confirmed sonographically and serum E2 < 30 mIU/mL, LH < 3 mIU/mL, recombinant FSH (Gonal-F, Serono, Switzerland) administration was initiated at daily dosages ranging from 150 to 300 IU, or in combination with hMG (hMG, Lizhu, China). Final follicular maturation was triggered by the administration of 4,000-10,000 IU human chorionic gonadotropin (hCG) when the largest follicles were more than 20 mm in diameter, as well as more than two thirds of the dominant follicles were greater than 16 mm in diameter. Transvaginal ultrasound-guided oocyte retrieval was scheduled 36-37h after HCG administration [29, 30].
The follicular fluid (FF) samples were carefully collected from the first aspiration follicle of each ovary, and only FF samples which did not contain any visible blood contamination were used in our study. The FF samples were then centrifuged for 15 min at 450g, and the supernatants were discarded. The granulose cells (GCs) were isolated from the blood cells and cellular debris using Percoll gradient centrifugation (Sigma) according to the user guide. The GCs were then for DNA and RNA extraction.
Granulosa cell DNA/RNA extraction and global methylation/hydroxymethylation quantification
The GCs DNA and RNA were extracted using the AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany). All the manipulations were under the user guide. Global DNA methylation and hydroxymethylation status were detected using MethylFlashTM Methylated and Hydroxymethylated DNA Quantification Kit (Epigentek, Epigentek Group Inc., USA), both of which are ELISA-like reactions. For methylated DNA status, according to the protocol, input DNA amount is 100 ng (18 samples, including 9 PCOS and 9 controls); while 200 ng for hydroxymethylation status detecting (17samples, including 9 PCOS and 8 controls). The absorbance was read on a microplate reader (Thermo MK3,) at 450 nm within 10 min after the color reaction. Global Methylation/Hydroxymethylation(%) = (OD (sample-blank) /2)/OD (positive control-blank) ×100%.
Granulosa cell DNA bisulfite conversion and genome-wide DNA methylation profile using Illumina Infinium HumanMethylation 450 BeadChip
Bisulphite treatment of granulose cell DNA was performed using the EZ DNA CT Conversion Reagent following the manufacturer’s protocols. A total of 1 μg of genomic DNA was used to bisulfite conversion using the EZ DNA Methylation Gold Kit (Zymo Research Corporation, Irvine, CA).
The genome-wide DNA methylation microarray screening was performed in 8 obesity-PCOS patients, 8 non-obesity-PCOS cases with normal BMI and 8 cases of control women. About 600 ng of the bisulfite-converted DNA was analyzed on Infinium HumanMethylation 450 BeadChip (Illumina, San Diego, CA) following the manufacturer’s guidelines. These Chips feature more than 450,000 methylation sites within and outside CpG islands. Methylation values for individual CpG sites were obtained as β-values, calculated as the ratio of the methylated signal intensity to the sum of both methylated and unmethylated signals after background subtraction. Data were normalized to background intensity levels and displayed using Genome Studio software version 2010(Ilumina Inc.).
Differential methylation genes screen and functional enrichment analysis
The overall different methylation loci were calculated using ChAMP [31] and Genome Studio software version 2010(Ilumina Inc.). DNA methylation profiling data were processed by the ChAMP package. Probes with a detection P>0.05 were removed, and β-values were compared between each two groups using two-sided Student’s t-test. The p-value for differential methylation between two groups was calculated by limma package [32, 33]. Only P<0.05 were considered as statistical differences. GO analysis of differenti ally methylated genes between each two groups were performed using DAVID Bioinformatics Resources Version 6.7 according to previously published protocol [34, 35].
Reverse transcription and real-time quantitative PCR
Total RNA of granulose cell was converted to cDNA with the QuantiTect Reverse Transcriptase Kit (Qiagen), using 1μg of total RNA and 1μl of random primer mix provided in the kit. Real-time PCR was performed using the 1μl cDNA in a total reaction volume of 20 μl containing 10 nM of forward and reverse primers each and 10 μl QuantiNova SYBR Green PCR Master Mix (Qiagen) and 0.1μl QN ROX Reference Dye (Qiagen). For each biological sample, Real-time PCR was performed in triplicate and the real-time quantitative PCR reaction were done on an ABI 7500 Sequence Detection System (Applied Biosystems; 40 cycles of 5 seconds of melting at 95°C followed by 30 seconds of annealing and extension at 60°C). 9 genes were quantitative analyzed in our study, the primers sequence could be obtained in Supplementary Table 1. The results were normalized using the β-actin as housekeeping gene. The data were analyzed using the ΔΔCt method.
DNA methylation validation using pyrosequencing
To validation the different methylation sites of Infinium HumanMethylation 450 BeadChip, we used pyrosequencing reactions according to the manufacturer’s instructions, using the PSQ 96 SNP Reagent Kit (Biotage AB). 3 CpG site located in MATN4、 DLGAP2 and MGAT5B were selected in our study, the primer of MATN4 F was GTGGTAGAGAGTGGATTAAAATTTATT, the primer R was CCTACTAAACAAATATAAACTTCCCAC and the primer S was GTAGAGAGTGGATTAAAATTTATTG. The primer of DLGAP2 F was TTGTAGAGGGGTTGGGGATAT, the primer R was ACCCCAATACCTAATCTTCCTTCC and the primer S was GATATTGTAAAGTGTAAATTAAGG. The primer of MGAT5B F was TGGTTAGGTTGGAGAATAGTAGTGA, the primer R was ATACCACAATATAATCCAAATCTTCTC, and the primer S was GTGGGGAGGAGGTATA. The methylation level of each CpG site was analyzed by AQ Software. 8 samples were selected from the PCOS-obesity, PCOS-nonobesity and control groups.
Statistical analysis
All the characters of PCOS patients and healthy controls were given as number of mean ± SD. The clinical characters were statistic by one-way-ANOVA. The global DNA methylation and hydroxymethlation was compared by Mann-Whitney U test. All probability values were two-sided, and P<0.05 was considered significant. * means P<0.05 and ** means P<0.01. *** means P<0.001. All the analyses were performed with R software (Version 3.2.2).
ACKNOWLEDGMENTS
Contributors JWX and YPS designed the study; XB, JWX and ZFP performed the experiments; ZFP, MXZ, LLW, QLD and JZ analysed the data; JWX and XB wrote the manuscript; YPS revised the manuscript.
CONFLICTS OF INTEREST
The authors have nothing to disclose.
GRANT SUPPORT
This work was supported by the National Natural Science Foundation of China (31271605 and 31471404 to Yingpu Sun.), the National Natural Science Foundation of China (3150090202 to Jiawei Xu) and the Youth Innovation Fund of the First Affiliated Hospital of Zhengzhou University (to Jiawei Xu), and their supports are gratefully acknowledged.
REFERENCES
1. Norman RJ, Dewailly D, Legro RS, Hickey TE. Polycystic ovary syndrome. Lancet. 2007; 370:685-697.
2. Evian Annual Reproduction Workshop G, Fauser BC, Diedrich K, Bouchard P, Dominguez F, Matzuk M, Franks S, Hamamah S, Simon C, Devroey P, Ezcurra D, Howles CM. Contemporary genetic technologies and female reproduction. Human reproduction update. 2011; 17:829-847.
3. Diamanti-Kandarakis E, Kandarakis H, Legro RS. The role of genes and environment in the etiology of PCOS. Endocrine. 2006; 30:19-26.
4. Qu F, Wang FF, Yin R, Ding GL, El-Prince M, Gao Q, Shi BW, Pan HH, Huang YT, Jin M, Leung PC, Sheng JZ, Huang HF. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: hyperandrogenism induces epigenetic alterations in the granulosa cells. Journal of molecular medicine. 2012; 90:911-923.
5. Wang P, Zhao H, Li T, Zhang W, Wu K, Li M, Bian Y, Liu H, Ning Y, Li G, Chen ZJ. Hypomethylation of the LH/choriogonadotropin receptor promoter region is a potential mechanism underlying susceptibility to polycystic ovary syndrome. Endocrinology. 2014; 155:1445-1452.
6. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nature reviews Endocrinology. 2011; 7:219-231.
7. Guibert S, Weber M. Functions of DNA methylation and hydroxymethylation in mammalian development. Current topics in developmental biology. 2013; 104:47-83.
8. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013; 38:23-38.
9. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nature reviews Genetics. 2013; 14:204-220.
10. Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PloS one. 2010; 5:e15367.
11. Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic acids research. 2010; 38:e181.
12. Xu N, Azziz R, Goodarzi MO. Epigenetics in polycystic ovary syndrome: a pilot study of global DNA methylation. Fertility and sterility. 2010; 94:781-783 e781.
13. Shen HR, Qiu LH, Zhang ZQ, Qin YY, Cao C, Di W. Genome-wide methylated DNA immunoprecipitation analysis of patients with polycystic ovary syndrome. PloS one. 2013; 8:e64801.
14. Wang XX, Wei JZ, Jiao J, Jiang SY, Yu DH, Li D. Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget. 2014; 5:6603-6610. doi:10.18632/oncotarget.2224.
15. Yu YY, Sun CX, Liu YK, Li Y, Wang L, Zhang W. Genome-wide screen of ovary-specific DNA methylation in polycystic ovary syndrome. Fertility and sterility. 2015; 104:145-153 e146.
16. Yu B, Russanova VR, Gravina S, Hartley S, Mullikin JC, Ignezweski A, Graham J, Segars JH, DeCherney AH, Howard BH. DNA methylome and transcriptome sequencing in human ovarian granulosa cells links age-related changes in gene expression to gene body methylation and 3'-end GC density. Oncotarget. 2015; 6:3627-3643. doi:10.18632/oncotarget.2875.
17. Erickson GF, Magoffin DA, Garzo VG, Cheung AP, Chang RJ. Granulosa cells of polycystic ovaries: are they normal or abnormal? Human reproduction. 1992; 7:293-299.
18. Das M, Djahanbakhch O, Hacihanefioglu B, Saridogan E, Ikram M, Ghali L, Raveendran M, Storey A. Granulosa cell survival and proliferation are altered in polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2008; 93:881-887.
19. Kaur S, Archer KJ, Devi MG, Kriplani A, Strauss JF, 3rd, Singh R. Differential gene expression in granulosa cells from polycystic ovary syndrome patients with and without insulin resistance: identification of susceptibility gene sets through network analysis. The Journal of clinical endocrinology and metabolism. 2012; 97:E2016-2021.
20. Canipari R. Oocyte--granulosa cell interactions. Human reproduction update. 2000; 6:279-289.
21. Sugiura K, Pendola FL, Eppig JJ. Oocyte control of metabolic cooperativity between oocytes and companion granulosa cells: energy metabolism. Developmental biology. 2005; 279:20-30.
22. Heijnen EM, Eijkemans MJ, Hughes EG, Laven JS, Macklon NS, Fauser BC. A meta-analysis of outcomes of conventional IVF in women with polycystic ovary syndrome. Human reproduction update. 2006; 12:13-21.
23. Pasquali R, Gambineri A, Pagotto U. The impact of obesity on reproduction in women with polycystic ovary syndrome. BJOG. 2006; 113:1148-1159.
24. Dale PO, Tanbo T, Vaaler S, Abyholm T. Body weight, hyperinsulinemia, and gonadotropin levels in the polycystic ovarian syndrome: evidence of two distinct populations. Fertility and sterility. 1992; 58:487-491.
25. Lan CW, Chen MJ, Tai KY, Yu DC, Yang YC, Jan PS, Yang YS, Chen HF, Ho HN. Functional microarray analysis of differentially expressed genes in granulosa cells from women with polycystic ovary syndrome related to MAPK/ERK signaling. Scientific reports. 2015; 5:14994.
26. Coskun S, Otu HH, Awartani KA, Al-Alwan LA, Al-Hassan S, Al-Mayman H, Kaya N, Inan MS. Gene expression profiling of granulosa cells from PCOS patients following varying doses of human chorionic gonadotropin. Journal of assisted reproduction and genetics. 2013; 30:341-352.
27. Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature reviews Genetics. 2002; 3:662-673.
28. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature reviews Genetics. 2012; 13:484-492.
29. Cheung LP, Lam PM, Lok IH, Chiu TT, Yeung SY, Tjer CC, Haines CJ. GnRH antagonist versus long GnRH agonist protocol in poor responders undergoing IVF: a randomized controlled trial. Human reproduction. 2005; 20:616-621.
30. Depalo R, Jayakrishan K, Garruti G, Totaro I, Panzarino M, Giorgino F, Selvaggi LE. GnRH agonist versus GnRH antagonist in in vitro fertilization and embryo transfer (IVF/ET). Reproductive biology and endocrinology. 2012; 10:26.
31. Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics. 2014; 30:428-430.
32. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015; 43:e47.
33. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004; 3:Article3.
34. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009; 4:44-57.
35. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009; 37:1-13.