
INTRODUCTION
Colorectal cancer (CRC) is the most common gastrointestinal malignancy occurring fourth in males and third in females across the globe, accounting for over 1.2 million new cases and 0.6 million deaths per year [1]. A good deal of researches were carried out in the molecular pathogenesis of CRC, discovering that activation of multiple signaling pathways plays an important role in regulating cell proliferation, angiogenesis, cell motility, and apoptosis [2, 3]. KRAS, NRAS and BRAF are the downstream oncogenes and their mutation may lead to activation of mitogen-activated protein kinase (MARK) pathway independent of the function of upstream epidermal growth factor receptor (EGFR) [4–6]. Clinically, their mutations are important predictive and prognostic markers when determining candidacy of anti-EGFR treatment [7–9]. Besides MARK pathway, another important signal pathway is the phosphatidylinositol-3-OH (PI3K) pathway, often activated by mutation in PIK3CA gene [3, 10, 11]. PIK3CA is also considered as a predictive and prognostic marker toward anti-EGFR therapy [12, 13]. Lots of reports have documented KRAS, BRAF and PIK3CA mutation frequency in CRC [14–16]. Increasing evidence revealed the usefulness of a full molecular profile in making treatment strategy for CRC patients. The genome-scale analysis of 276 cases from the Cancer Genome Atlas (TCGA) in 2012 demonstrated a few frequently occurred genes [17]. At the same time, many more mutations that are much less frequent are also detected in many different genes [15, 18–23]. Those infrequent mutated gene might have a synergic or independent effect with mutations in KRAS, BRAF, NRAS and PIK3CA, though the clinical value of most of them still remains to be uncovered.
There have been researches regarding the population-based differences in the clinicopathological features and the genetic profile of the same cancer, as well as the response to anticancer treatment. For instance, for lung adenocarcinoma, the Northeast Asian population has a higher prevalence of activating mutation of the EGFR tyrosine kinase domain [24]. In Chinese CRC population, there are data regarding the prevalence and clinical significance of KRAS, BRAF, NRAS and PIK3CA mutations [25, 26]. But for those less frequently mutated genes whose significance is yet to be discovered, published data are quite limited among Chinese population.
The Sequenom platform has developed MassARRAY® gene profiling technique. It’s based on a matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) to detect multiple gene mutations with high sensitivity and accuracy [27]. The OncoCarta™ panel is a set of pre-designed and pre-validated assays by the parallel analysis of 238 possible mutations in 19 clinically relevant genes with as little as 500 ng DNA per sample, including frequent mutated genes such as KRAS, NRAS, BRAF and PIK3CA, which are most clinically relevant for CRC. In addition, it also contains assays for other infrequent mutations in genes, such as AKT1, EGFR, HRAS, NRAS, MET and others. Our center has been performing clinical molecular testing with OncoCarta™ Panel on metastatic colorectal cancer (mCRC) patients since 2014. This testing was performed on the group of mCRC patients for whom testing result would assist in identifying targeted therapies according to genotype pattern. We conducted this retrospective study to investigate the genetic profile in Chinese population, as well as to investigate the relationship between mutational status and the clinicopathological features. In addition, this study also explored the correlation between mutational profile and anti-EGFR treatment response.
RESULTS
Main patient characteristics
322 Chinese patients with metastatic colorectal cancer were considered eligible. Among the detected samples, 270 (83.9%) samples were from primary tumors, 38 (11.8%) from metastatic sites and the rest 14 (4.35%) were unknown. The main metastatic sites included liver in 188 (58.4%) patients, lung in 101 (31.4%), distant lymph node in 121 (37.6%), peritoneum in 95 (29.5%), and bone in 32 (9.9%). Other metastasis included uterus, ovary, adrenal gland, spleen, skeletal muscle and so on. Main patient characteristics are listed in Table 1.
Table 1: Main characteristics of 322 patients with metastatic colorectal cancer and the association of mutation profile with clinicopathological parameters
Clinicopathological features |
Total samples, N=322 |
n (%) |
KRAS |
NRAS |
BRAF |
PIK3CA |
Mutation numbers |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Mutation (%) |
P |
Mutations (%) |
P |
Mutations (%) |
P |
Mutations (%) |
P |
Single mutations (%) |
≥2 mutations (%) |
P |
|||
Sex |
|||||||||||||
Male |
195 (60.6) |
64 (32.8) |
.40 |
9 (4.6) |
>.99 |
4 (2.1) |
.12 |
17 (8.7) |
.56 |
176 (90.3) |
19 (9.7) |
>.99 |
|
Female |
127 (39.4) |
48 (37.8) |
5 (3.9) |
7 (5.5) |
14 (11.0) |
114 (89.8) |
13 (10.2) |
||||||
Age |
|||||||||||||
>60 |
95 (29.5) |
30 (31.6) |
.52 |
8 (9.4) |
.03 |
2 (2.1) |
.52 |
12 (12.6) |
.30 |
84 (88.4) |
11 (11.6) |
.54 |
|
≤60 |
227 (70.5) |
82 (36.1) |
6 (2.6) |
9 (4.0) |
19 (8.4) |
206 (90.7) |
21 (9.3) |
||||||
Median |
52 |
||||||||||||
Range |
15-82 |
||||||||||||
Tumor differentiation |
|||||||||||||
Well/Moderate |
213 (66.1) |
84 (39.4) |
.02 |
9 (4.2) |
>.99 |
8 (3.8) |
.76 |
26 (12.0) |
.03 |
186 (87.3) |
27 (12.7) |
.03 |
|
Poor |
109 (33.9) |
28 (25.7) |
5 (4.6) |
3 (2.8) |
5 (4.6) |
104 (95.4) |
5 (4.6) |
||||||
Tumor type |
|||||||||||||
Papillary/tubular adenocarcinoma |
288 (89.4) |
104 (36.1) |
.18 |
14 (4.9) |
.38 |
10 (3.5) |
>.99 |
31 (10.8) |
.06 |
256 (88.9) |
32 (11.1) |
.03 |
|
Mucinous/signet ring cell |
34 (10.6) |
8 (23.5) |
0 (0.0) |
1 (2.9) |
0 (0.0) |
34 (100) |
0 (0.0) |
||||||
Primary tumor site |
|||||||||||||
Right colon |
84 (26.1) |
36 (42.8) |
.20 |
4 (4.8) |
.91 |
4 (4.8) |
.79 |
14 (16.7) |
.04 |
73 (86.9) |
11 (13.1) |
.53 |
|
Left colon |
127 (39.4) |
40 (31.5) |
5 (3.9) |
5 (3.9) |
8 (6.3) |
115 (90.6) |
12 (9.4) |
||||||
Rectum |
84 (26.1) |
28 (33.3) |
4 (4.8) |
2 (2.4) |
6 (7.1) |
77 (91.7) |
7 (8.3) |
||||||
Multiple origin |
9 (2.8) |
5 (55.6) |
0 (0.0) |
0 (0.0) |
0 (0.0) |
9 (100) |
0 (0.0) |
||||||
Missing |
18 (5.6) |
||||||||||||
Family history |
|||||||||||||
With family history |
92 (28.6) |
35 (38.0) |
.60 |
5 (5.4) |
.76 |
6 (6.5) |
.11 |
10 (10.9) |
.53 |
83 (90.2) |
9 (9.8) |
>.99 |
|
No family history |
196 (60.9) |
68 (34.7) |
8 (4.08) |
5 (2.6) |
17 (8.7) |
175 (89.3) |
21 (10.7) |
||||||
Missing |
34 (10.6) |
||||||||||||
Metastasis |
Liver |
188 (58.4) |
72 (38.3) |
.25 |
8 (4.2) |
.77 |
5 (2.6) |
.20 |
19 (10.1) |
.67 |
169 (89.9) |
19 (10.1) |
.84 |
Lung |
101 (31.4) |
41 (40.6) |
.25 |
4 (4.0) |
>.99 |
3 (3.0) |
.75 |
9 (8.9) |
>.99 |
89 (88.1) |
12 (11.9) |
.55 |
|
Distant Lymph nodes |
121 (37.6) |
35 (28.9) |
.05 |
5 (4.1) |
>.99 |
7 (5.8) |
.21 |
9 (7.4) |
.42 |
109 (90.1) |
12 (9.9) |
>.99 |
|
Peritoneum |
95 (29.5) |
36 (37.9) |
.70 |
2 (2.1) |
.23 |
8 (8.4) |
.007 |
9 (9.5) |
>.99 |
85 (89.5) |
10 (10.5) |
>.99 |
|
Bone |
32 (9.9) |
11 (34.3) |
>.99 |
0 (0.0) |
.37 |
3 (9.4) |
.11 |
4 (12.5) |
.52 |
28 (87.5) |
4 (12.5) |
.76 |
|
Bold figures represent P<0.05.
Of these 322 patients, 80 (19.6%) patients received anti-EGFR treatment. Cetuximab was administrated as single agent or in combination with 5-FU/oxaliplatin/irinotecan regimen in palliative treatment. As first-line treatment 51 (63.8%) patients received cetuximab and second-line in 14 (17.5%). 15 (18.8%) patients were treated with cetuximab in third-line or beyond. No patient received panitumumab treatment.
Mutational profile
Out of 322 tumors, 166 (51.6%) were all wild-type, defined as no mutation in any of the 19 genes listed in Table 2. At least one mutation was identified in 156 (48.4%) cases. In total, there were 44 mutations in 11 genes detected in the OncoCarta™ Panel, in 156 cases (Table 3). KRAS was the most commonly gene (112; 34.8%), followed by PIK3CA (31, 9.6%) NRAS (14, 4.3%) and BRAF (11, 3.4%). No mutation was identified in ABL1, AKT2, CDK, FGFR3, FLT3, JAK, KIT or MET. One single mutation was present in 38.5% (124/322) of all tested cases, two concomitant mutations in 9.0% (29/322) and three mutations in 3 tumors (<1%). A schematic map of the 156 patients with at least one mutation in any of the 19 genes is shown in Figure 1.
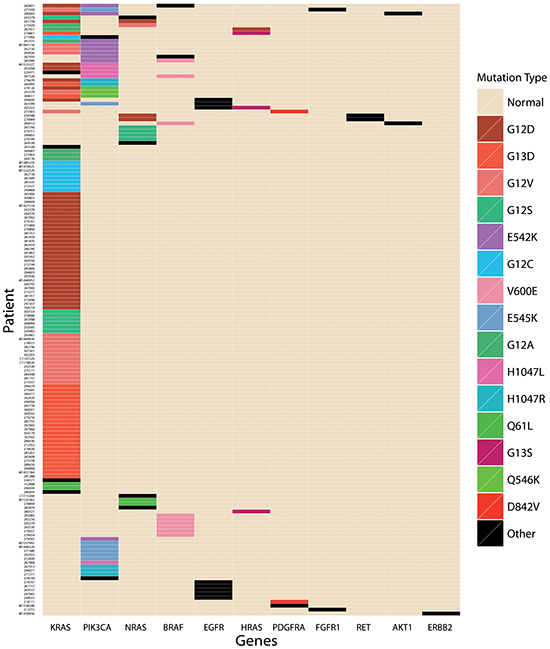
Figure 1: A schematic map of mutated genes in the 156 patients with at least one mutation in any of the 19 genes.
Table 2: Genes included in the OncoCarta panel
Panel of genes analyzed |
||
|---|---|---|
ABL1 |
FGFR1 |
JAK |
AKT1 |
FGFR3 |
KIT |
AKT2 |
FLT3 |
MET |
BRAF |
KRAS |
PDGFRA |
CDK |
NRAS |
PIK3CA |
EGFR |
HRAS |
RET |
ERBB2 |
||
Table 3: Summary of mutations identifies in 322 patients with metastatic colorectal cancer
Mutation |
N% (322) |
||
|---|---|---|---|
Gene |
Mutation |
Cases |
|
KRAS |
112 |
34.8 |
|
NRAS |
14 |
4.3 |
|
HRAS |
4 |
1.2 |
|
BRAF |
V600E |
9 |
|
G464E |
1 |
||
G469A |
1 |
||
total |
11 |
3.4 |
|
PIK3CA |
C420R |
1 |
|
E542K |
9 |
||
E545K |
7 |
||
H1047L |
5 |
||
H1047R |
5 |
||
M1043I |
1 |
||
Q546K |
3 |
||
total |
31 |
9.6 |
|
EGFR |
L747-P753>S |
1 |
|
G719S |
1 |
||
P772-H773insV |
1 |
||
E709K |
1 |
||
H773-V774insNPH |
2 |
||
P753S |
1 |
||
D770-N771insG |
1 |
||
total |
8 |
2.5 |
|
PDGFRA |
D842V |
2 |
|
T674I |
1 |
||
total |
3 |
0.9 |
|
FGFR1 |
S125L |
1 |
|
I836del |
1 |
||
total |
2 |
0.6 |
|
RET |
C643Y |
2 |
0.6 |
AKT1 |
E17K |
2 |
0.6 |
ERBB2 |
G776S |
1 |
0.3 |
The family members of human RAS genes include HRAS, KRAS and NRAS genes. At least one gene mutation of the RAS family was identified in 125 (38.8%) tumors (details shown in Table 4). The most frequent mutation occurred in codon 12 for both KRAS and NRAS. One patient harbored a double KRAS mutation in both codon 12 and codon 59 (G12D, A59T). The distribution of mutation subtypes is summarized in Figure 2. Unlike the KRAS and NRAS genes, the status of HRAS mutation was detected in only 4 (1.2%) cases. Among them, G13S mutation in codon 13 was identified in 3 tumors, and G12D mutation in codon 12 in 1 case.
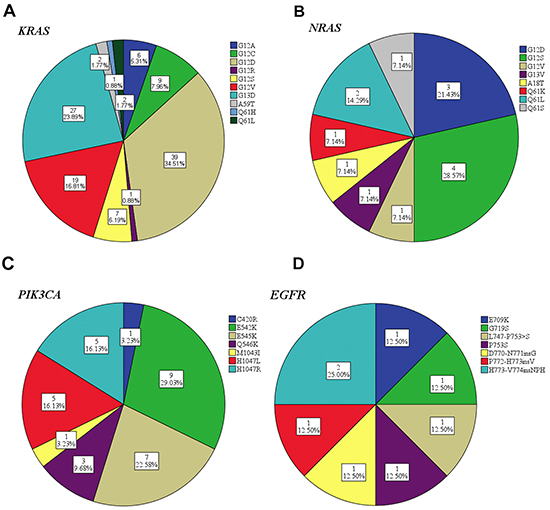
Figure 2: Mutation subtypes frequency distribution of KRAS A. NRAS B. PIK3CA C. and EGFR D.
Table 4: Frequency of mutation in RAS family in patients with metastatic colorectal cancer
Genes |
Cases with mutation (%) |
|
|---|---|---|
Total cases with RAS mutation |
125(38.8) |
|
Total cases with KRAS mutation |
112 (34.8) |
|
KRAS codon 12 |
G12A |
6 (1.9) |
G12C |
9 (2.8) |
|
G12D |
39 (12.1) |
|
G12R |
1 (0.3) |
|
G12S |
7 (2.2) |
|
G12V |
19 (5.9) |
|
KRAS codon 13 |
G13D |
27 (8.4) |
KRAS codon 59 |
A59T |
2 (0.6) |
KRAS codon 61 |
Q61H |
1 (0.3) |
Q61L |
2 (0.6) |
|
Total cases with NRAS mutation |
14 (4.4) |
|
NRAS codon 12 |
G12D |
3 (0.9) |
G12S |
4 (1.2) |
|
G12V |
1 (0.3) |
|
NRAS codon 13 |
G13V |
1 (0.3) |
NRAS codon 18 |
A18T |
1 (0.3) |
NRAS codon 61 |
Q61K |
1 (0.3) |
Q61L |
2 (0.6) |
|
Q61S |
1 (0.6) |
|
Total cases with HRAS mutation |
4 (1.2) |
|
HRAS |
G12D |
1 (0.3) |
G13S |
3 (0.9) |
Notably, in our study we identified 5 cases in which the different genes mutation in RAS family concomitantly existed. There were 3 patients detected with KRAS and NRAS concurrent mutation. The exact KRAS and NRAS mutation was G12A and G12V, G12D and G12D, G12S and A18T respectively for the 3 cases (Figure 1). In addition, 2 patients were detected with concurrent KRAS and HRAS mutation. One of them was identified KRAS G12A mutation and HRAS G12D mutation, and the other exhibited G13D for KRAS and G13S for HRAS. No concurrent NRAS and HRAS mutation was detected in our study. These findings reveal that the mutations in RAS family proto-oncogene are not mutually exclusive.
BRAF gene, another important incidence on EGFR pathway, was found to be mutant in exon 15 (9 cases) and exon 11 (2 cases), with V600E mutation as the most frequent subtype. In our study group, there was 1 patient detected with concurrent KRAS G12D mutation and BRAF G464E mutation, together with PIK3CA E452K mutation. However, we found no co-mutation of BRAF V600E spot with any KRAS.
The frequency of PIK3CA mutation is the second highest (31/322, 9.6%), following KRAS mutation. Mutations in exon 9 coding for the helical domain (C420R, E542K, E545K and Q546K) were found in 20 patients. Exon 20 mutations coding for the kinase domain (H1047L, H1047R and M1043I) were found in 11 patients (shown in Figure 2). Mutations in PIK3CA tended to be accompanied with a second or even a third mutation. Notably, PIK3CA exon 9 mutation were significantly associated with KRAS mutation (11/112 [9.8%] vs 9/210 [4.3%] P=.04), whereas PIK3CA exon 20 didn’t show such an association (5/112 [4.5%] vs 6/210 [2.9%], P=.52). As for PIK3CA exon 9 and BRAF co-mutation, this significant association also existed (3/11 [27.3%] in BRAF mutant vs 17/311 [5.5%] in BRAF wild-type, P=0.03), while no significant correlation existed between PIK3CA exon 20 and BRAF (1/11 [9.1%] in BRAF mutant vs 10/311 [0.3%] in BRAF wild-type, P=.32), thereby suggesting that PIK3CA exon 9 may occur as a second mutation in the later stage in the carcinogenesis.
In our study, less frequent mutations in CRC were also found in EGFR (8), PDGFRA (3), RET (2), AKT1 (2), FGFR1 (2), and ERBB2 (1). Some of these genes were presented as single mutation, while some co-occurred with KRAS, NRAS, BRAF and PIK3CA. 3 of the 8 EGFR mutation (Figure 2) were accompanies with KRAS, HRAS and PIK3CA mutations, all of which exist in the downstream signal pathways of EGFR. Interestingly, there were 2 cases identified with RET C643Y mutation, and there seemed to be a strong correlation with NRAS mutation (2/14 [14.3%] in NRAS mutant vs 0/308 [0%] in NRAS wild-type, P=.002). Both cases identified with AKT1 mutation had concomitant RAS/RAF mutation, one with BRAF V600E mutation, the other with KRAS codon 12 and PIK3CA exon 9 mutations. Only one ERBB2 mutation was detected, presenting a frequency of 0.3%. The concomitant and exclusive relationship among all 11 genes is visualized in Figure 3.
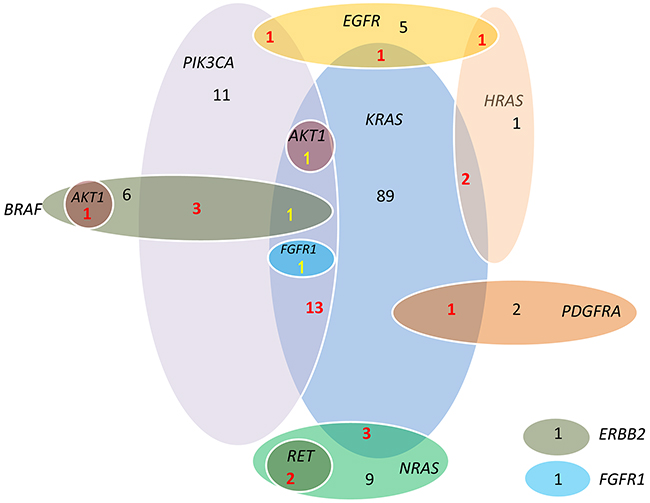
Figure 3: Associations among KRAS, NRAS, HRAS, BRAF, PIK3CA, EGFR, FGFR1, PDGFRA, RET, AKT1 and ERBB2 mutations. Mutations of three genes from RAS family are not mutually exclusive, neither were the KRAS and BRAF. RET mutations co-occurred with NRAS mutation. AKT1 mutation co-occurred with RAS/RAF.
Exploratory analysis of mutation profile and clinicopathological characteristics
An exploratory analysis of the clinicopathological characteristics by genetic profile was performed. KRAS mutation rate was higher in patients with histologically well/moderate grade tumor (39.4% vs 25.7%, P=.02) or in patients without distant lymph node metastasis (40.8% vs 28.9%, P=.05). NRAS showed a higher mutation frequency in patients older than 60 years (9.4% vs 2.6%, P=.03). BRAF mutation trended toward an association with peritoneum implantation (8.4% vs 1.5%, P=.007). PIK3CA mutation frequency was higher in well/moderate differentiation tumors (12.0% vs 4.6%, P=.03), and right colon as the primary tumor site (P=.04). They trended toward a positive association with papillary or tubular adenocarcinoma (10.8% vs 0.0%, P=.06). Patients with more than one mutation in any of the genes detected in OncoCarta™ Panel were associated with better differentiation (12.7% vs 4.6%, P=.03) and papillary or tubular adenocarcinoma (11.1% vs 0.0%, P= .03). Mutation profile and clinical correlations are summarized in Table 1.
Anti-EGFR treatment response by mutation profile
Among the 80 patients treated with cetuximab, the objective response was a CR in 1 (1.3%) patient, PR in 30 (37.5%), stable disease (SD) in 18 (22.5%) and progression disease (PD) in 14 (17.5%). In 17 patients, the objective response couldn’t be evaluated, either because the medical documentation was incomplete or the patients were initiating cetuximab treatment at the time when the information was collected. All those 80 patients were RAS wild-type. In our analysis, better objective response rate (ORR) was strongly correlated with patients who presented wild-type in all the 19 genes (31/56 [55.3%] in all wild-type vs. 0/7 [0%] in any mutation, P=.006). During a relatively short follow-up time (median 8.2 months, range 2.2-42.0 months), survival information was collected successfully collected in only 15 patients. However in the survival analysis, the small group sample size did not present any significant result between mutation profile and survival benefit (overall survival and progression-free survival), including mutation number analysis (data not shown).
DISCUSSION
During the past decades, molecular testing has been attached greater and greater significance with the progress in targeted treatment. In colorectal cancer, data have been accumulated in genetic profiling [15, 18-20, 28, 29]. In this cohort of 322 mCRC patients, our study described a genotype distribution picture among Chinese population, with an accurate and sensitive multiple gene detection technique on MassARRAY® platform.
Among the 19 genes tested in the OncoCarta™ Panel, KRAS and NRAS belong to the most clinically relevant genes in CRC. In our current study, the frequency of KRAS mutations was 34.8%, consistent with most reports from other populations [29–32], suggesting that such an alternation exists in 30-40% of colorectal cancer. Compared to the Catalogue of Somatic Mutations in Cancer (COSMIC) database and other publication [33], the frequency and specific amino acid mutations detected here were similar to western countries [34]. However, the correlation between KRAS and clinicopathological parameters remains controversial according to different reports. Our finding suggested that KRAS mutation is more frequent in well and moderately differentiated tumors than poorly differentiated ones. This finding conflicted with some publications in which the KRAS mutation is associated with older age, deeper invasion, and poorer differentiation [35, 36]. The difference may be explained by the distinction of selected patients and sample sources. In our study, only metastatic patients were included and the tested samples came from both surgery and biopsy, both primary tumor and metastasis, both pre-treatment and post-treatment stage. More reliable data are needed to confirm the KRAS correlation with clinicopathological features. The prevalence of NRAS mutation in our study is 4.4%, in accordance with reported range in literature between 2.6% and 7% [8, 37–39]. We observed that NRAS mutations were more common in patients older than 60 years, similar to previous publications [20]. In melanomas and leukemia, this association was also discovered [40]. Unlike KRAS and NRAS from the same family, the clinical significance of HRAS mutation has not been convincingly elucidated so far. We detected a HRAS mutation frequency of 1.2%, slightly higher than the estimated frequency of lower than 1 % in a previous review [41]. No significant correlation with clinicopathological features was identified probably due to the limited number of HRAS mutation. Some observations deserve further discussion. First of all, one patient with both KRAS codon 12 and codon 59 mutations caught our attention, indicating that concurrent mutations may exist in different KRAS exons. This suggests heterogeneity may occur in one single tumor lesion. Secondly, to our knowledge, this is the first report showing that KRAS, NRAS and HRAS mutations are not mutually exclusive. This phenomenon conflicted with several previous reported suggesting mutually exclusive mutations in RAS family [8, 35]. One possible explanation may be the low detection rate of KRAS exon3 and 4, NRAS and HRAS mutation limited the discovery of potential co-occurrence. Hopefully with accumulated data in multiple gene profile, relationship among RAS family members may be better elucidated.
We identified 11 BRAF mutations among 322 samples. As a poor prognostic mutation recognized widely, lots of researches reported the associations between BRAF mutation with primary tumor site, age, gender and differentiation grade [35, 42, 43]. But in our study we didn’t find the association, possibly because of the bias resulted from the limited number of detected mutations in our sample. Yet the higher prevalence in patients with peritoneum implantation somehow indicated the poor predictive value. As for the relationship between KRAS and BRAF mutations, we identified a patient with concomitant KRAS G12D mutation and BRAF G464E mutation. This test result shared a similar phenomenon in a previous report: though KRAS and BRAF V600E mutations are mutually exclusive, it is possible that BRAF non-V600E mutation can co-exist with KRAS mutation in the same case; this condition is rarely observed mainly because of the low frequency of BRAF non-V600E mutation [44].
Some previously published studies have suggested that PIK3CA mutations were associated with KRAS mutation [8, 25, 45]. In agreement with the conclusion from a large retrospective European study that analyzed 1022 CRC samples [8], we detected a strong association between exon 9 PIK3CA and KRAS/BRAF mutations. As expected, this association didn’t apply to PIK3CA exon 20. This can be putatively explained by the research finding that PIK3CA exon 9 mutations (coding for helical domain) adjusted function though a Ras-GTP dependent pattern, whereas the gain of function induced by PIK3CA exon 20 mutations (coding for kinase domain) didn’t require the involvement of Ras [46].
In our study, we also identified 6 more genes in CRC involved different signaling pathways, including RAS/MAPK and PI3K/AKT pathway. Previous reports about these gene mutations are quite limited in CRC. EGFR, an upstream element in signaling pathway, is a significant predictive and prognostic indicator in lung adenocarcinoma to guide the decision of targeted treatment. In our study of CRC, 8 EGFR mutations were detected, with frequency of 2.5%. This prevalence is much lower than that in lung adenocarcinoma in Asian populations, about 30% [24]. EGFR mutations are not exclusive with downstream mutations according to our study. PDGFRA mutations are often studied in gastrointestinal stromal tumor (GIST) [47]. To our knowledge, this is the first report of PDGFRA mutations in CRC, with a frequency of 0.9%. Preclinical researches have elucidated the role of FGFR1 in regulating CRC cell behavior and FGFR1 is considered a putative therapeutic target in early phase trials [48, 49]. Previous reports showed FGFR1 gene amplification rate was 3.8% in 291 CRC cases [50], whereas our study suggested a 0.9% FGFR1 mutation rate. Oncogenic RET point mutations and RET fusions occur mainly in papillary thyroid cancer and lung adenocarcinoma [51]. The RET gene mutation frequency was 0.6% in our study. To be noted, all the 2 RET mutations had concurrent NRAS mutation. This phenomenon has not been reported before, deserving more exploration in the correlation of NRAS and RET mutation. AKT1 is an important component in PI3K/AKT/mTOR pathway. Our findings were quite similar to the conclusion of a previous study that AKT1 frequency was 0.7% in CRC and tended to co-occur with RAS/RAF-activating mutation [21]. The detection of ERBB2 mutation was also reported in TCGA [17] and these patients may benefit from target treatment with ERBB2 antibody. Although clinical significance of these infrequent genes is not yet uncovered, the occurrence of such mutation gives us more insight into the complexity of cancer cell genotype and offers more clues as treatment target.
Since anti-EGFR monoclonal antibody became an effective treatment, mutation detection of KRAS and NRAS has been recommended by the National Comprehensive Cancer Network (NCCN) and the European Society for Medical Oncology (ESMO) guidelines to avoid the inefficacy in RAS mutant groups. However, up to 65% of patients with KRAS wild-type tumors are resistant to anti-EGFR monoclonal antibodies. Our finding suggested that the treatment response of cetuximab was more obvious in patients who presented wild-type in all the 19 genes than those who harbored at least one mutation in any genes. According to an European Consortium report, the all-wide-type patients (KRAS, BRAF, NRAS and PIK3CA-exon 20 wide-type) reached the highest ORR from anti-EGFR therapy in chemo-refractory setting. Our finding supplemented this conclusion, with expansion to more infrequent genes in addition to KRAS, BRAF, NRAS and PIK3CA, and expansion to all cetuximab settings other than chemo-refractory condition. This conclusion suggests that in order to improve the ORR, multiple gene detection can be recommended to decide the all wide-type patients. Larger samples are needed to further confirm the predictive value of multiple mutation profiling.
There are several limitations in our study that merits further discussion. This study is retrospective and exploratory in nature and the drawing of more convincing conclusion is thus limited. Due to the restrictions of medical record documentation and short follow-up time, we failed to collect adequate treatment and survival information in anti-EGFR treated patients. In addition, the OncoCarta™ panel based on MassARRAY® platform identified 19 genes that were predetermined with a bias toward detecting hot-spot mutations which may be considered for targeted treatment. This may miss other potential mutations throughout the coding sequence.
In conclusion, we conducted a retrospective study to describe a Chinese mCRC mutational profile and performed exploratory analysis to make clinical correlations. These findings supplemented the limited data of mCRC in Chinese population, and offered us a clearer image of multiple gene mutational profile in not only clinically prognostic KRAS, NRAS, BRAF and PIK3CA gene, but also less frequent mutated genes such as HRAS, EGFR, PDGFRA, RET, AKT1, FGFR1, and ERBB2. Knowledge of these gene mutation patterns may give clues in exploring interesting accompanying co-occurrence relationship or mutually exclusive relationship between mutated genes. It may also help to predict benefit of all-wild-type patients from anti-EGFR treatment. Hopefully the genotype landscape will advocate the development of precision medicine.
MATERIALS AND METHODS
Study population and clinical data
After approval from the local Institutional Review Board, we retrospective investigated 322 mCRC patients who received clinical molecular testing as part of their standard care at Sun Yat-sen University Cancer Center (Guangzhou, China) between August 2014 and July 2015. Patients were chosen to undergo testing at the discretion of their treating physician. Principal inclusion criteria were as follows: histologically confirmed papillary/tubular adenocarcinoma, signet ring carcinoma and mucinous carcinoma of the colon or rectum; and presence of unresectable metastatic disease. Patient’s nationality of China was also required to be included into this population-specific study. CRC diagnosis was confirmed by hematoxylin and eosin (HE) staining and histological analysis. Informed consent was obtained from all individual participants included in the study, giving their authorization to access their clinical information and tumor samples for research purpose.
Clinical data were retrieved by medical record archive, including age, sex, previous resection and site of primary tumor, number and type of received treatments, exposure to EGFR inhibitors or antiangiogenics, treatment response, type and date of metastasectomy. Pathologic data consisted of tumor size, tumor location and grade, histological type, lymphovascular and perineural invasion. Tumors were classified for histological type and grade using the current World Health Organization criteria by two independent pathologists. Since all patients included in this study were with unresectable metastasis considered as Stage IV, the TNM stage of each patient was omitted. Objective tumor response was evaluated every 6 weeks by computing tomography scan according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1). Responders were defined as the patients who had a complete response (CR) or partial response (PR).
Tissues and mutation detection
Each patient included in the analysis has provided available and adequate FFPF tumor specimens for molecular analysis. These specimens were resected at various cancer hospitals and sent to our center. Surgery primary CRC samples, resected metastasis or small biopsies samples were accepted. Routinely processed HE staining slides were reviewed by a pathologist to determine tumor adequacy and to select the area of highest tumor percentage. Sections (4–6μm) were cut and transferred to 1.5 mL Eppendorf tubes for DNA extraction. DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. The quantity and quality of the isolated DNA were tested using a Nanodrop ND-2000 Spectrophotometer (Thermo Scientific, Niederelbert, Germany). The final DNA samples were diluted to 10ng/μL for analysis.
For mutation detection, the OncoCarta Panel v. 1.0. (Sequenom Inc., San Diego, CA, USA) were used and the protocol provided by the manufacture was followed. As was described by Zhang [27], 20 ng of DNA was amplified using 24 sets of OncoCarta™ polymerase chain reaction (PCR) primers. An extension reaction based on the OncoCarta™ extension primers was then conducted. After salts were removed with a cation exchange resin, the products were spotted onto a 384-well SpectroChipII using the MassARRAY® Nanodispenser RS1000 (Sequenom Inc.) and analyzed on a MALDI-TOF mass spectrometer (Sequenom Inc.). In each experiment, HPLC-purified water was selected as the blank control and normal human somatic cells as the negative control. The OncoCarta™ Panel has the capacity to detect 238 mutations in 19 genes, listed in Table 2. A successful experiment should satisfy the standard of that the sample figure was typical and the blank control had no peak. Preliminary analysis of mutation data was performed by the software MassARRAY TYPER 4.0 (Sequenom Inc., San Diego, USA). The results of mutation frequency with more than 5% integrated with medium or high credibility were re-analyzed, while those with 5-10% and medium credibility were validated using fluorescent qRT-PCT. The false-positive results generated by ion disturbance were excluded from the analysis.
Statistical analysis
Since this analysis was exploratory, no sample size was calculated. Descriptive analysis for clinical and molecular data was performed. Statistical analysis was carried out by the IBM SPSS® Statistics 21.0.0 package software (SPSS Inc). Frequency distributions for categorical variables and mean with standard deviation, 25th and 75th percentiles for continuous variables were calculated. Pearson’s Chi-square (χ2) test was used to compare the proportion of gene mutations among groups with different clinicopathologic variables. All the P values were two-tailed, and the statistical significance was set at P<0.05.
ACKNOWLEDGMENTS
The authors gratefully thank staff members in the Department of Medical Oncology and Department of Molecular Diagnostics at Sun Yat-sen University Cancer Center for their suggestions and assistance.
FUNDING INFORMATION
This work was supported by The National Natural Science Foundation of China (No.81372570), The Science and Technology Department of Guangdong Province, China (No.2012B031800088) and The Science and Technology Department of Guangdong Province, China (No.C2011019).
CONFLICTS OF INTEREST
All authors declared that there is no conflict of interest.
Abbreviations
CRC = colorectal cancer, mCRC = metastatic colorectal cancer, MARK = mitogen-activated protein kinase, EGFR = epidermal growth factor receptor, PI3K = phosphatidylinositol-3-OH kinase, TCGA = the Cancer Genome Atlas, MALDI-TOF MS = matrix-assisted laser desorption ionization–time of flight mass spectrometry, FFPE tissue = formalin-fixed paraffin-embedded tissue, HE staining = hematoxylin and eosin staining, RECIST = Response Evaluation Criteria in Solid Tumors, CR = complete response, PR = partial response, SD = stable disease, PD = progression disease, PCR = polymerase chain reaction, ORR = objective response rate, COSMIC = Catalogue of Somatic Mutations in Cancer, GIST = gastrointestinal stromal tumor, NCCN = the National Comprehensive Cancer Network, ESMO = the European Society for Medical Oncology.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
2. De Roock W, De Vriendt V, Normanno N, Ciardiello F and Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011; 12:594-603.
3. Berg M and Soreide K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer: implications for targeted therapy. Discov Med. 2012; 14:207-214.
4. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F and Wittinghofer A. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997; 277:333-338.
5. Trahey M and McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987; 238:542-545.
6. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R and Cancer Genome P. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004; 116:855-867.
7. De Roock W, Lambrechts D and Tejpar S. K-ras mutations and cetuximab in colorectal cancer. N Engl J Med. 2009; 360:834; author reply 835-836.
8. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11:753-762.
9. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S and Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008; 26:5705-5712.
10. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9:550-562.
11. Samuels Y, Diaz LA, Jr., Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B and Velculescu VE. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005; 7:561-573.
12. Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S and Bardelli A. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009; 69:1851-1857.
13. Barault L, Veyrie N, Jooste V, Lecorre D, Chapusot C, Ferraz JM, Lievre A, Cortet M, Bouvier AM, Rat P, Roignot P, Faivre J, Laurent-Puig P and Piard F. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer. 2008; 122:2255-2259.
14. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949-954.
15. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318:1108-1113.
16. Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S, Jr., Duval A, Carneiro F, Machado JC, Hamelin R and Seruca R. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005; 41:1649-1654.
17. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
18. Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Lee JC, Nicoletti R, Hatton C, Goyette M, Girard L, Majmudar K, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007; 39:347-351.
19. Stachler MD, Rinehart E, Lindeman N, Odze R and Srivastava A. Novel molecular insights from routine genotyping of colorectal carcinomas. Hum Pathol. 2015; 46:507-513.
20. Russo AL, Borger DR, Szymonifka J, Ryan DP, Wo JY, Blaszkowsky LS, Kwak EL, Allen JN, Wadlow RC, Zhu AX, Murphy JE, Faris JE, Dias-Santagata D, Haigis KM, Ellisen LW, Iafrate AJ, et al. Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer. 2014; 120:1482-1490.
21. Hechtman JF, Sadowska J, Huse JT, Borsu L, Yaeger R, Shia J, Vakiani E, Ladanyi M and Arcila ME. AKT1 E17K in Colorectal Carcinoma Is Associated with BRAF V600E but Not MSI-H Status: A Clinicopathologic Comparison to PIK3CA Helical and Kinase Domain Mutants. Mol Cancer Res. 2015; 13:1003-1008.
22. Gawlick U, Lu KC, Douthit MA, Diggs BS, Schuff KG, Herzig DO and Tsikitis VL. Stage III & IV colon and rectal cancers share a similar genetic profile: a review of the Oregon Colorectal Cancer Registry. Am J Surg. 2013; 205:608-612; discussion 612.
23. Atreya CE, Sangale Z, Xu N, Matli MR, Tikishvili E, Welbourn W, Stone S, Shokat KM and Warren RS. PTEN expression is consistent in colorectal cancer primaries and metastases and associates with patient survival. Cancer Med. 2013; 2:496-506.
24. Ma BB, Hui EP and Mok TS. Population-based differences in treatment outcome following anticancer drug therapies. Lancet Oncol. 2010; 11:75-84.
25. Mao C, Zhou J, Yang Z, Huang Y, Wu X, Shen H, Tang J and Chen Q. KRAS, BRAF and PIK3CA mutations and the loss of PTEN expression in Chinese patients with colorectal cancer. PLoS One. 2012; 7:e36653.
26. Ma BB, Mo F, Tong JH, Wong A, Wong SC, Ho WM, Wu C, Lam PW, Chan KF, Chan TS, Tsui WM, Tsang AK, Fung MN, Chan AT and To KF. Elucidating the prognostic significance of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese patients with metastatic colorectal cancer. Asia Pac J Clin Oncol. 2015; 11:160-169.
27. Zhang ZC, Fu S, Wang F, Wang HY, Zeng YX and Shao JY. Oncogene mutational profile in nasopharyngeal carcinoma. Onco Targets Ther. 2014; 7:457-467.
28. Fumagalli D, Gavin PG, Taniyama Y, Kim SI, Choi HJ, Paik S and Pogue-Geile KL. A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer. 2010; 10:101.
29. Yanus GA, Belyaeva AV, Ivantsov AO, Kuligina E, Suspitsin EN, Mitiushkina NV, Aleksakhina SN, Iyevleva AG, Zaitseva OA, Yatsuk OS, Gorodnova TV, Strelkova TN, Efremova SA, Lepenchuk AY, Ochir-Garyaev AN, Paneyah MB, et al. Pattern of clinically relevant mutations in consecutive series of Russian colorectal cancer patients. Med Oncol. 2013; 30:686.
30. Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, Qian ZR, Nishihara R, Meyerhardt JA, Haigis KM, Fuchs CS and Ogino S. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res. 2012; 18:4753-4763.
31. Nakanishi R, Harada J, Tuul M, Zhao Y, Ando K, Saeki H, Oki E, Ohga T, Kitao H, Kakeji Y and Maehara Y. Prognostic relevance of KRAS and BRAF mutations in Japanese patients with colorectal cancer. Int J Clin Oncol. 2013; 18:1042-1048.
32. Palomba G, Colombino M, Contu A, Massidda B, Baldino G, Pazzola A, Ionta M, Capelli F, Trova V, Sedda T, Sanna G, Tanda F, Budroni M, Sardinian Translational Oncology G, Palmieri G, Cossu A, et al. Prevalence of KRAS, BRAF, and PIK3CA somatic mutations in patients with colorectal carcinoma may vary in the same population: clues from Sardinia. J Transl Med. 2012; 10:178.
33. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA and Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008; Chapter 10:Unit 10 11.
34. Neumann J, Zeindl-Eberhart E, Kirchner T and Jung A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol Res Pract. 2009; 205:858-862.
35. Kawazoe A, Shitara K, Fukuoka S, Kuboki Y, Bando H, Okamoto W, Kojima T, Fuse N, Yamanaka T, Doi T, Ohtsu A and Yoshino T. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer. 2015; 15:258.
36. Shen Y, Wang J, Han X, Yang H, Wang S, Lin D and Shi Y. Effectors of epidermal growth factor receptor pathway: the genetic profiling ofKRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS One. 2013; 8:e81628.
37. Irahara N, Baba Y, Nosho K, Shima K, Yan L, Dias-Santagata D, Iafrate AJ, Fuchs CS, Haigis KM and Ogino S. NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol. 2010; 19:157-163.
38. Bokemeyer C, Kohne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, Beier F, Duecker K, van Krieken JH and Tejpar S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer. 2015; 51:1243-1252.
39. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, Blasinska-Morawiec M, Smakal M, Canon JL, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013; 369:1023-1034.
40. Hacker E, Nagore E, Cerroni L, Woods SL, Hayward NK, Chapman B, Montgomery GW, Soyer HP and Whiteman DC. NRAS and BRAF mutations in cutaneous melanoma and the association with MC1R genotype: findings from Spanish and Austrian populations. J Invest Dermatol. 2013; 133:1027-1033.
41. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003; 3:11-22.
42. Zlobec I, Bihl MP, Schwarb H, Terracciano L and Lugli A. Clinicopathological and protein characterization of BRAF- and K-RAS-mutated colorectal cancer and implications for prognosis. Int J Cancer. 2010; 127:367-380.
43. Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K and Yatabe Y. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011; 104:856-862.
44. Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, Futreal PA, Stratton MR, Wooster R and Leung SY. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002; 62:6451-6455.
45. Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, Fu S, Luthra R, Garrido-Laguna I and Kurzrock R. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011; 6:e22769.
46. Zhao L and Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008; 105:2652-2657.
47. Daniels M, Lurkin I, Pauli R, Erbstosser E, Hildebrandt U, Hellwig K, Zschille U, Luders P, Kruger G, Knolle J, Stengel B, Prall F, Hertel K, Lobeck H, Popp B, Theissig F, et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011; 312:43-54.
48. Goke F, Goke A, von Massenhausen A, Franzen A, Sharma R, Kirsten R, Bohm D, Kristiansen G, Stenzinger A, Wynes M, Hirsch FR, Weichert W, Heasley L, Buettner R and Perner S. Fibroblast growth factor receptor 1 as a putative therapy target in colorectal cancer. Digestion. 2013; 88:172-181.
49. Lee CK, Lee ME, Lee WS, Kim JM, Park KH, Kim TS, Lee KY, Ahn JB, Chung HC and Rha SY. Dovitinib (TKI258), a multi-target angiokinase inhibitor, is effective regardless of KRAS or BRAF mutation status in colorectal cancer. Am J Cancer Res. 2015; 5:72-86.
50. Kwak Y, Nam SK, Seo AN, Kim DW, Kang SB, Kim WH and Lee HS. Fibroblast Growth Factor Receptor 1 Gene Copy Number and mRNA Expression in Primary Colorectal Cancer and Its Clinicopathologic Correlation. Pathobiology. 2015; 82:76-83.
51. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. 2014; 14:173-186.