
INTRODUCTION
The development of pharmacologic strategies targeting anaplastic lymphoma kinase (ALK) reflects the increasing involvement of ALK in a subset of human malignancies where ALK is well accepted as an initiator and progression marker, representing a tractable oncogene for targeted therapy. The original discovery of ALK was in 1994, when Morris and colleagues first characterized the ALK gene as a fusion partner of nucleophosmin (NPM), in the NPM-ALK translocation found in a subset of anaplastic large cell lymphoma (ALCL) [1]. Further studies have revealed numerous different ALK fusion proteins in other tumors such as inflammatory myofibroblastic tumor (IMT), diffuse large B cell lymphoma (DLBCL) and non-small cell lung cancer (NSCLC) among others [2, 3].
First and second generation ALK inhibitors, such as crizotinib and ceritinib, which have been FDA approved [4], provide hope for a targeted therapy in patients with aberrant ALK activity. An additional ALK inhibitor, alectinib (CH5424802) has been approved in Japan for use in ALK-positive NSCLC [5]. While the above mentioned drugs are all ATP-competitive inhibitors of ALK, they differ in their binding properties and display differential activity in blocking the activity of the various ALK resistant mutant forms [3, 6, 7]. Thus, a complex picture of ALK inhibition is emerging, with an increasing number of reports suggesting distinct patterns of resistance mutations arising following primary treatment with particular ALK inhibitors.
The situation in pediatric neuroblastoma is further complicated by the fact that point mutations in ALK occur as primary, and potentially driver mutations in therapy naïve patients. Neuroblastoma, a tumor of the developing nervous system accounts for 15% of all pediatric oncology death [8, 9]. Neuroblastoma is a heterogeneous disease and while a subset may undergo spontaneous differentiation or regression with little or no therapy, the majority are difficult to cure with current regimes [8, 9]. The most common genetic features of neuroblastoma are amplification of the proto-oncogene MYCN, deletions of parts of chromosome arms 1p and 11q, gain of parts of 17q and triploidy [8–10], and mutations in the kinase domain of ALK which occur in both sporadic and familial forms of neuroblastoma [11–15]. A recent trial of crizotinib in ALK-positive pediatric cancers demonstrated excellent activity in pediatric ALCL, where the NPM-ALK fusion protein is the major driver, but suboptimal activity in neuroblastoma [16]. At the present time it is not clear whether this reflects suboptimal inhibitory properties or possibly reflects the heterogeneous nature of neuroblastoma in terms of chromosomal irregularities, since neuroblastoma exhibits both numerical and segmental chromosome alterations [8, 17–20]. ALK aberrations in neuroblastoma are predominantly point mutations in the context of full-length ALK however other variants, such as deletions have also been reported [21, 22]. ALK mutation occurs in about 5-7% of neuroblastoma cases but this percentage is increased significantly in the relapsed patient population where approximately 20-25% of patients have ALK mutations [23]. This has been verified in an additional study of relapsed tumors in which the percentage of ALK-positive relapsed patients was reported as greater than 40% [24, 25]. It has been reported that neuroblastoma patients bearing both MYCN amplification and ALK mutations are characterized by unfavorable aggressive neuroblastoma phenotype [26]. Activating ligands for ALK have recently been identified as FAM150A and FAM150B [27, 28]. These small secreted ligands are able to drive ‘super activation’ of activated ALK mutants from neuroblastoma suggesting dysregulation of the ALK ligands may play a role in neuroblastoma [27]. Further characterization of the FAM150 mediated ligand activation of ALK signaling should clarify the significance of the ligand-ALK interaction as a potential therapeutic target. Thus, in the context of neuroblastoma, a number of approaches are actively being explored for therapeutic intervention, with evaluation of new ALK inhibitors a high priority.
Brigatinib, also known as AP26113, is one of the most recently described second generation ALK inhibitors [6]. Clinical trial data reports that about 72% of crizotinib refractory ALK-positive NSCLC patients responded to treatment with brigatinib [29]. Based on these encouraging clinical responses in NSCLC, we decided to explore the therapeutic potential of brigatinib in the context of ALK-positive neuroblastoma.
RESULTS
Brigatinib inhibits ALK activity and abrogates proliferation of ALK addicted neuroblastoma cell lines
Brigatinib has been shown to inhibit ALK activity in NSCLC cell lines carrying the EML4-ALK fusion protein [6, 30]. In order to investigate the therapeutic efficacy of brigatinib in a neuroblastoma setting we employed several neuroblastoma cell lines, including CLB-BAR (MYCN amplification, ALK (Δ4-11) and amplified, ALK addicted), CLB-GE (MYCN amplification, ALK (F1174V) amplification, ALK addicted), IMR32 (MYCN amplification, WT ALK) and CLB-PE (MYCN amplified, WT ALK) [21, 31–33]. We have earlier shown that both CLB-BAR and CLB-GE cell lines are ALK addicted, while IMR32 and CLB-PE are not [34, 35]. ALK addicted cell lines were treated with either brigatinib or crizotinib, as a positive control for ALK inhibition, and their effect on ALK signaling analyzed by immunoblotting (Figure 1A, 1B). Brigatinib inhibited ALK phosphorylation in a dose-dependent manner, and at 50 nM brigatinib, phosphorylation of ALK was abolished (Figure 1A and 1B). Reduction of ALK phosphorylation by crizotinib was also observed, although at around 250 nM, in agreement with earlier published reports [7, 35]. In keeping with the inhibition of ALK phosphorylation, phosphorylation of downstream signaling targets such as ERK1/2, ERK5 and AKT were also affected and a decrease in MYCN levels was observed (Figure 1A, 1B).
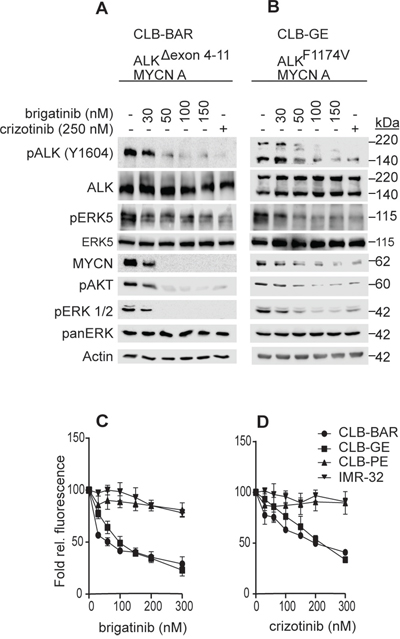
Figure 1: Brigatinib inhibits ALK activity and proliferation of ALK addicted neuroblastoma cell lines. A, B. CLB-BAR (ALK-Δ4-11) and CLB-GE (ALK-F1174V), both ALK addicted cell lines, were treated with increasing doses of brigatinib for 6 hours. Crizotinib (250 nM) was employed as a positive control. Cells lysates were resolved on SDS/PAGE followed by immunoblotting for pALK (Y1604) and additional downstream targets as indicated. In the CLB-BAR cell line there is a genomic deletion in ALK between exon 4-11, resulting in an ALK band of approximately 170 kDa [21]. The CLB-GE cell line expresses a mutant full length version of ALK (F1174V) which is cleaved resulting in the detection of two bands with the antibody employed here. C, D. CLB-PE (ALK-WT) and IMR32 (ALK-WT) are ALK non-addicted neuroblastoma cell lines. Neuroblastoma cells were treated with increasing concentration of either brigatinib (C) and crizotinib (D) for 72 hours and cell viability was assessed by resazurin assay (Sigma, Sweden). Plotted values are means +/- SE from growth curves from at least three independent experiments performed in triplicate.
While brigatinib and crizotinib both inhibited cell growth of ALK addicted neuroblastoma lines, they exhibited different IC50 values. The IC50 values observed for brigatinib and crizotinib in CLB-BAR were 75.27 ± 8.89 nM and 186.40 ± 17.28 nM, respectively, while in CLB-GE the IC50 values for brigatinib and crizotinib were 100.00 ± 17.53 nM and 225 ± 26, respectively (Figure 1C, 1D). Neither brigatinib nor crizotinib was able to inhibit growth of the non-ALK addicted neuroblastoma cell lines, IMR32 and CLB-PE, indicating that neither brigatinib nor crizotinib inhibitor was toxic to cells at the levels employed. Thus, brigatinib inhibits ALK activity in ALK addicted neuroblastoma cell lines more efficiently than crizotinib and displays a lack of toxicity at the cellular level.
Brigatinib effectively inhibits constitutively active ALK mutants and neurite outgrowth
We next examined the ability of brigatinib to abrogate the activity of mutated ALK variants found in neuroblastoma cases. Previously validated gain-of-function ALK mutants, such as ALK-G1128A, ALK-I1171N, ALK-F1174L, ALK-R1192P, ALK-F1245C, ALK-R1275Q and ALK-Y1278S were ectopically expressed in PC12 cells, in parallel with wildtype ALK, and inhibition of ALK phosphorylation by brigatinib or crizotinib was assessed by immunoblotting for pALK-Y1604 (Figure 2A, 2B). All loading ALK control blots for this experiment are shown as Supplementary Figure 1. We also included analysis of the ALK-G1269A mutant, which has so far not been observed as a neuroblastoma mutation but has been described as a more resistant mutation arising in EML4-ALK from NSCLC patients treated with crizotinib [36]. We observed that ALK-WT and ALK-F1174L were most sensitive to crizotinib whereas ALK-I1171N and the EML4-ALK secondary mutation mimic ALK-G1269A required relatively high doses of crizotinib for effective inhibition of ALK-Y1604 phosphorylation (Figure 2A, 2B, Table 1). Interestingly, brigatinib inhibited the activation of ALK gain-of-function alleles with a general IC50 of 5-35 fold less than that observed with crizotinib (Figure 2A, 2B, Table 1). These results suggest a superior inhibition profile for brigatinib as compared with crizotinib in a neuroblastoma setting. It is notable that ALK-I1171N and ALK-G1269A are more resistant to crizotinib with IC50s of 111 and 119 nM, respectively. In contrast, brigatinib inhibits both the ALK-I1171N and the ALK-G1269A mutant receptors at low nM levels, 10 and 4, respectively.
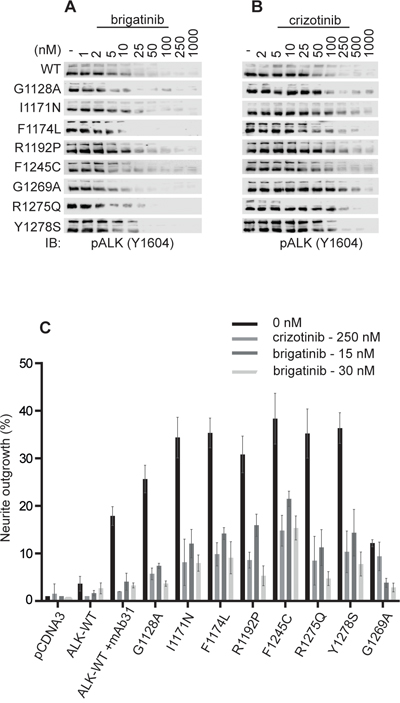
Figure 2: Brigatinib effectively blocks ALK activation and ALK-mediated neurite outgrowth in PC12 cells. A, B. PC12 cells were transfected with ALK (0.75 μg). 48 hours post-transfection cells were treated with increasing amounts of either brigatinib or crizotinib. Wild type (WT) ALK was transfected with 1.5 μg pcDNA3-ALK-WT. Whole cell lysates were blotted for pALK (Y1604) to measure ALK activation. C. PC12 cells were co-transfected with pEGFPN1 together with the indicated ALK mutant. Wild type (WT) ALK was stimulated with 1 μg/ml of mAb31. GFP-positive PC12 cells were scored for neurite outgrowth after 48 hours treatment with either brigatinib or crizotinib (control) at the indicated concentrations.
Table 1: IC50 values for inhibition of ALK Y1604 phosphorylation by brigatinib or crizotinib in PC12 cells
ALK mutations |
IC50 (nM) |
Fold difference |
|
|---|---|---|---|
brigatinib |
crizotinib |
||
WT |
2.6± 0.2 |
19.68 ± 9 |
8 |
G1128A |
2.0 ± 0.6 |
41.82 ± 5 |
21 |
I1171N |
10.2 ± 5.7 |
110.90 ± 18 |
11 |
F1174L |
1.5 ± 0.4 |
25.35 ± 10 |
17 |
R1192P |
2.5 ± 0.4 |
64.0 ± 12 |
26 |
F1245C |
6.6 ± 0.3 |
34.07 ± 0 |
5 |
G1269A |
3.4 ± 2.0 |
118.9 ± 23 |
35 |
R1275Q |
4.2 ± 0.7 |
32.58 ± 0 |
8 |
Y1278S |
4.6 ± 0.2 |
72.44 ± 2 |
16 |
IC50 values were determined by quantification of Y1604 phosphorylation from the immunoblots in Figure 2A and 2B. Values represent mean ± SD from at least two independent experiments.
In agreement with the inhibition profiles observed for ALK-Y1604 phosphorylation, inhibition of ALK driven PC12 cell neurite outgrowth was also blocked by brigatinib. PC12s are a clonal rat adrenal pheochromocytoma cell line with enteric cell origin, which has the ability to differentiate and extend neurites upon extended ERK1/2 stimulation [37]. We and others have previously shown that activation of both human and mouse ALK triggers differentiation of PC12 cells into sympathetic-like neurons, a process that is characterized by extension of neurites [38]. This assay offers a convenient read out for ALK activity in vitro. Stimulated wild type ALK mediates neurite outgrowth which was abrogated by both crizotinib and brigatinib (Figure 2C). Further, brigatinib could abrogate neurite outgrowth of all tested ALK mutants. It should be noted that brigatinib, tested at both 15 and 30 nM blocks ALK-G1269A, ALK-R1275 and ALK-R1192P more efficiently than crizotinib (250 nM) (Figure 2C). Thus, brigatinib shows inhibition of multiple gain-of-function ALK neuroblastoma mutations including the “hot-spot” mutations as well as the crizotinib resistant ALK-G1269A mutation.
Brigatinib blocks activity of the ALK gain-of-function alleles F1174L and R1275Q in vivo
To test if brigatinib can inhibit ALK gain-of-function mutations in vivo, we employed Drosophila melanogaster as a model organism by using the Gal4-UAS-system for tissue specific expression of the alleles F1174L and R1275Q in the eye imaginal disc [39]. pGMR-Gal4 and pGMR-Gal4>UAS-ALK controls displayed a hexagonal arrangement of ommatidia similar to the wild type w1118 and show that the ectopic expression of pGMR-Gal4 or human ALK wild type does not lead to an eye phenotype (Supplementary Figure 2B, 2C). Transgenic flies expressing two gain-of-function variants of the human ALK receptors were generated to confirm their ligand-independent characteristics and also for testing of ALK inhibition as we have previously reported for NVP-TAE684 [7]. ALK gain-of-function “hot spot” mutations were ectopically expressed in the Drosophila eye disc, employing the pGMR-Gal4 driver line, which directs protein expression in developing photoreceptors during larval stages (Figure 3). The expression of human ALK proteins was confirmed by immunostaining of eye discs using anti-human ALK antibody (Supplementary Figure 2A, 2B). Ectopic expression of the ALK gain-of-function alleles F1174L and R1275Q led to disrupted eye morphology in all offspring (100%), a so called ‘rough eye’ phenotype, which is characterized by disorganized ommatidia and missing interommatidial bristles, reflecting their robust ligand-independent activity (Figure 3) [38, 39, 41]. All larvae that were grown on brigatinib inhibitor-containing food developed into flies which showed a concentration-dependent improvement of the rough eye phenotype (Figure 3). Thus, brigatinib has the ability to abrogate rough eye phenotype in Drosophila ectopically expressing two different neuroblastoma “hot-spot” mutations.
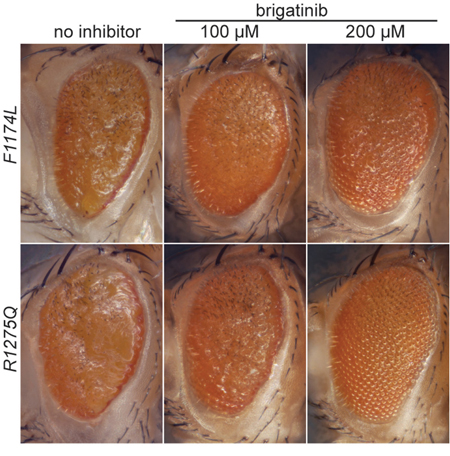
Figure 3: Effect of brigatinib on ALK gain-of-function rough eye phenotypes in a Drosophila ALK model. Ectopic expression of gain-of-function “hot spot” ALK mutations ALK-F1174L and ALK-R1275Q in Drosophila eye imaginal discs with the Gal4-UAS-system results in a rough eye phenotype that is 100% penetrant [7, 38, 40]. Offspring that were grown on food containing brigatinib displayed a concentration dependent improvement of the rough eye phenotype.
Brigatinib displays potent anti-tumor growth in a xenograft model of neuroblastoma
We have earlier shown that crizotinib exhibits limited efficacy as a single agent treatment in orthotopic xenograft tumor growth and in the TH-ALKF1174L/TH-MYCN mouse model [42]. Therefore we examined the therapeutic effect of brigatinib as a single agent in BalbC/NUDE mice injected with human neuroblastoma CLB-BAR cells, (ALK Δ4-11 and MYCN amplified, ALK addicted) subcutaneously. Animals were treated for 14 days with brigatinib (50 mg/kg body weight, once daily via oral gavage) or vehicle control (Figure 4A). Examination of the tumor volume showed that brigatinib considerably inhibits proliferation of tumor growth in vivo (p=0.0004) (Figure 4A and 4B), and tumor weight was significantly reduced in the brigatinib treated group as compared with controls (p=0.01) (Figure 4C). Further, we observed an increase in tumor volume/growth when brigatinib treatment was discontinued after 14 days and for 8 days (n=2), indicating that tumor growth inhibition was due to brigatinib treatment (Figure 4A). The body weight of mice did not show any variation during treatment (Figure 4D) (p=0.9), and no obvious side effect of brigatinib treatment in vivo was observed. Analysis of excised tumor material with the Ki-67 proliferation marker indicated a significantly reduced proliferation in tumors from mice treated with brigatinib compared with controls (Figure 4E). Immunoblotting analysis of tumor material revealed decreased phospho-ALK levels in the brigatinib treated group, in line with our earlier findings (Figure 2). No obvious change in total ALK protein levels compared to control group were observed, however, MYCN, which is a downstream target of ALK activity showed decreased protein levels upon treatment with brigatinib (Figure 4F). Phosphorylated ERK5 and AKT, other downstream targets of ALK, exhibited reduced phosphorylation in the brigatinib treated group compared with the control group (Figure 4F). Therefore, brigatinib shows a robust and potent anti-tumor activity in this xenograft neuroblastoma mouse model.
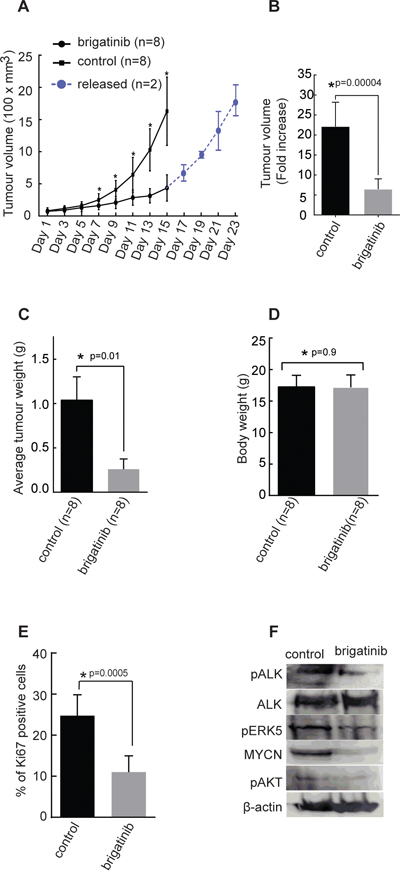
Figure 4: Effect of brigatinib in a xenograft neuroblastoma model. 2.5×106 CLB-BAR cells were injected into mice subcutaneously. In the first investigation mice (n=3) were employed and in the second investigation mice (n=5) were employed. A. Tumor growth curves represented as the average volume of either control group or brigatinib group (n=8 for each group, p=0.0002). In the second investigation where 5 mice were employed a subset of mice (n=2) were released from treatment after 14 days, for a further 8 days (blue curve). B. Average tumor volume in control and brigatinib treated groups after 14 days, p=0.00004. C. Average tumor weights in control and brigatinib treated groups after 14 days (p=0.01). D. Average body weight on the day at which mice were sacrificed after 14 days treatment with either brigatinib or vehicle control (p=0.9). E. Immunohistochemical staining of tumors with Ki-67 as a measure of proliferation rate as indicated, Ki-67-positive cells were counted manually per field of vision and quantitative results presented as mean ± SD (p=0.0005). Data are means +/- SE from 8 tumours, p values indicated; Student’s t test. F. Immunonblotting analysis of indicated proteins from tumors collected after 8 days of treatment with vehicle or brigatinib.
DISCUSSION
There has been a significant increase in targeted therapies available for the management of ALK-positive cancers including NSCLC, inflammatory myofibroblastic tumors (IMT), and anaplastic large cell lymphoma (ALCL). This improvement is due to the development of ALK inhibitors such as crizotinib, alectinib and ceritinib in the treatment of these ALK-related diseases [3, 42, 43]. Generally, treatment with ALK inhibitors shows an initial strong response in tumors harboring ALK fusion oncogenes, with a less encouraging response observed in ALK-positive neuroblastoma patients [16]. This low response in neuroblastoma patients may reflect the heterogeneity of the disease, and may also reflect the need for more potent ALK inhibitors in this tumor type. Neuroblastoma exhibits both numerical and segmental chromosome alterations [8, 17–20]. Further, MYCN and ALK are both located on the same chromosome, 2p, in close proximity to each other [3]. Therefore, amplification of MYCN can also involve ALK amplification, and neuroblastoma patients bearing both MYCN amplification and ALK mutations are characterized by an unfavorable aggressive neuroblastoma phenotype [26]. Finally, the recently reported ligands, FAM150A and FAM150B activate not only the wild type ALK receptor, but also super-activate constitutive active ALK neuroblastoma variants, offering additional options for the inhibition of ALK activity in NB [27].
Aberrant ALK activity occurs in about 5-7% of neuroblastoma cases but this percentage increases significantly in the relapsed patient population [8, 23, 24], thereby adding weight to the clinical relevance of ALK in neuroblastoma pathogenesis. The selection of intrinsic rare resistant clones by, and/or the emergence of acquired resistance to, crizotinib give credence to the need for second-generation ALK inhibitors such as brigatinib. Brigatinib, an ALK inhibitor, has been tested in crizotinib-refractory EML4-ALK-positive NSCLC patients, where about 72% of these patients showed encouraging responses [29, 44].
Treatment with brigatinib resulted in abrogation of cell growth and proliferation, as observed in this study. However, brigatinib inhibits ALK addicted neuroblastoma cell line proliferation with roughly 2 to 3-fold lower IC50 values compared with crizotinib. Previously described downstream targets of ALK such as ERK5 [35], AKT and ERK1/2 were clearly less phosphorylated upon treatment with brigatinib [7, 35]. MYCN amplification is associated with poor prognosis in NB patients, and there is evidence to the effect that MYCN cooperates with ALK in a synergistic manner to promote tumor growth [34]. Here we show that abrogation of ALK activity upon treatment with brigatinib also results in reduction of MYCN levels.
Multiple ALK point mutations have been identified in neuroblastoma, however, not all of them are classified as gain-of-function mutations, as some of the mutations are kinase dead and some seem to behave (at least in terms of activation) as wild type ALK [3]. It is clear that the constitutive active ALK mutations show differential sensitivity to ALK inhibitors such as crizotinib in both preclinical and also in clinical studies [45, 46]. Notably, ALK-G1269A, which is a secondary mutation found in a EML4-ALK-positive crizotinib-refractory NSCLC patients [36], together with the ALK-I1171N mutation, are more resistant to crizotinib. Brigatinib abrogated the activities of these mutant alleles with very low IC50 values. These observations indicate that the more resistant ALK-G1269A phenotype observed with crizotinib can be overcome by brigatinib. Brigatinib exhibits strong activity towards the most difficult to inhibit ALK neuroblastoma mutation among those investigated here (ALK-I1171N), inhibiting its phosphorylation with an observed IC50 value of 10 nM which is still low. The increased activity of brigatinib compared to crizotinib, is in the range of 5-35 fold based on our examination of the various constitutively active ALK mutations.
ALK mutations at residues F1174, F1245 and R1275 are often referred to as ‘hot spot’ mutations, together accounting for more than 80% of all sporadic ALK mutations found in neuroblastoma [14, 15, 26]. The robust rescue of the rough-eye phenotype caused by the ectopic expression of the hot-spot ALK gain-of-function mutations, ALK-F1174L and ALK-R1275Q, in Drosophila model system, confirms the ability of brigatinib to block aberrant ALK activity in an in vivo model system. Crizotinib is unable to rescue the rough-eye phenotype as observed with brigatinib [7]. In agreement, in an ALK/MYCN-driven mouse xenograft neuroblastoma model, brigatinib potently inhibited tumor growth, highlighting tumor inhibition efficacy in vivo. The abrogation of ALK phosphorylation in the excised tumor indicates that brigatinib does indeed block ALK activity within the tumor. Furthermore, the reduced levels of MYCN protein observed in tumors from mice receiving brigatinib are in keeping with our previous observations in cultured cells.
Brain metastases, particularly in non-small cell lung cancer patients, are among the common causes of disease progression with brain metastasis frequency in ALK inhibitor naive patients ranging from about 25 to 40%, which is high in patients with history of chemotherapy [47, 48]. However, in retrospective investigations of ALK-rearranged NSCLC patients treated with ALK inhibitors, mainly crizotinib, show a high incidence of CNS metastases, from about 45 to 70% [49–51]. Crizotinib has been reported to have poor penetration through the blood-brain barrier hence, the high incidence of brain metastasis in crizotinib refractory patients [52]. In a recent clinical trial of brigatinib in ALK-positive NSCLC patients, a radiological review of patients with baseline CNS metastases showed response in 50% (6/12) of patients, while in another subgroup of patients, 31% (8/26) with non-measureable CNS lesions had disappearance of all lesions [29], indicating that brigatinib is able to penetrate the blood-brain barrier. The overall incidence of brain metastasis in neuroblastoma patients ranges from 1.7 to 11.7% [53], making brigatinib a putative better treatment option than crizotinib.
In summary, we show that brigatinib abrogates ALK activation in neuroblastoma cell lines, in vitro biochemical assays as well as exogenously expressed ALK activity in flies and mice xenografts. Taken together, our data indicate that brigatinib is a potent inhibitor against ALK, supporting further exploration of brigatinib in an ALK-positive neuroblastoma setting.
MATERIALS AND METHODS
Generation of human ALK mutants
ALK mutants employed in this study were created in pcDNA3 by Eurofins MWG/operon (Ebersberg, Germany) as described by Chand et al. [38].
Cell lines, antibodies and inhibitors
Neuroblastoma cell lines used were CLB-BAR (amplified MYCN/ALK, Δ4-11), CLB-GE (amplified MYCN/ALK, ALK-F1174V), CLB-PE (amplified MYCN, WT ALK) and IMR-32 (amplified MYCN, WT ALK) and PC12 cells were cultured and grown as previously reported [7, 35]. Primary antibodies employed for immunoblotting were: phospho-ALK (Y1604), phospho-AKT (S473), MYCN, phospho-ERK1/2 (1:2000), phospho-ERK5 (1:1000), ERK5, panERK, Actin (1:5000) from Cell Signaling Technology (Danvers, MA). Monoclonal antibody 135 (anti-ALK) was produced in the Hallberg laboratory against the extracellular domain of ALK [38]. Horseradish peroxidase conjugated secondary antibodies; goat anti-rabbit IgG and goat anti-mouse, IgG (1:5000) were obtained from Thermo Scientific. Brigatinib, was obtained from Ariad Pharmaceuticals and crizotinib was from Haoyuan Chemexpress Co., Limited, Shanghai.
Cell culture, transfection, lysis, and immunoblotting
Neuroblastoma cell lines were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C, 5% CO2, 95% humidity. ALK addicted neuroblastoma cell lines, CLB-BAR and CLB-GE, were seeded into 6-well plates (0.5 × 106 cells/well). Cells were treated with varying concentrations of brigatinib (from 0 to 150 nM) and with 250 nM crizotinib, in starvation medium (RPMI-1640 without FBS), for 6 hours. Cells were washed with cold 1X PBS and then harvested in lysis buffer (25 mM of Tris-Cl, pH 7.5, 150 mM of NaCl, 1% (v/v) Triton X-100, 1 mM DTT, protease inhibitor cocktail tablet (Roche)). Cell lysates were cleared by centrifugation at 14,000 rpm for 15 minutes at 4°C, after which the samples were boiled in SDS sample buffer, and subsequently analyzed by immunoblotting.
PC12 cells (3 × 106) were transfected by electroporation in an Amaxa electroporator using 0.75 μg of ALK constructs (with the exception of 1.5 μg for WT ALK) and 100 μL of Ingenio electroporation solution (Mirrus Bio LCC). Cells from four electroporations were pooled together, mixed and equally seeded into 20 wells of 24-well plate with serial dilutions of the indicated inhibitors, for 4 hours. Cells were washed with cold 1X PBS and lysed with 1X SDS sample buffer (prepared from 4X SDS sample buffer - 3% SDS, 100mM Tris (pH6.8) 100 mM DTT, 50 mM EDTA, 40% glycerol). Samples were boiled at 95°C for 5 minutes and analyzed by immunoblotting. Phospho-ALK (Y1604) antibody was used to detect ALK phosphorylation. Intensity of pALK (Y 1604) and total (ALK) bands were quantified with Image Studio Lite 3.1. Data were normalized to the 0 nM inhibitor samples. GraphPad Prism 6.0 was used to calculate IC50 values by fitting data to a log (inhibitor concentration) versus normalized response (variable slope) equation.
Proliferation assay
Cells were seeded at 15,000 per well with serial dilutions of the indicated inhibitors. After 72 hours cell viability was assessed by resazurin (Sigma, Sweden). IC50 values were calculated with GraphPad Prism 6.0 by fitting data to a log (inhibitor concentration) vs. normalized response (variable slope) equation. Each experiment was performed in duplicate and repeated at least three times.
Neurite outgrowth assay
PC12 cells (2 x106) co-transfected, in 100 μl of Ingenio electroporation solution (Minus Bio LCC), with pEGFPN1 (Clonetech) (0.5 ng) and ALK-mutant (0.75 ng) or ALK-wt (0.75 ng) as indicated were seeded sparingly into 24-well plates [38]. ALK-wild type was stimulated with 1 μg/ml mAb31. After 48 hours post transfection, the fraction of GFP-positive and neurite carrying cells versus GFP-positive cells were analyzed under a Zeiss Axiovert 40 CFL microscope. A cell was considered as neurite-carrying if it had neurite(s) that reached at least twice the length of the diameter of a normal cell body. The experiments were performed at least three times in triplicates.
Drosophila transgenic lines and brigatinib treatment
The following Drosophila stocks from Bloomington Drosophila Stock Center (Indiana University) were employed: w1118 and pGMR-Gal4 (stock numbers 5905 and 9146, respectively). Generation of the Drosophila transgenic ALK lines was as described previously [7]. Transgenic Drosophila lines carrying the UAS-ALKF1174L or UAS-ALKR1275Q gain-of-function mutations were crossed with the pGMR-Gal4 transgenic driver line to drive expression of the ALK mutations ectopically in the eye imaginal discs [38, 40]. Progeny of this cross were transferred at first instar larvae stage to food containing either 100 μM or 200 μM brigatinib (stock solution 10 mM in molecular grade ethanol dissolved at 25°C) in 2% molecular grade ethanol. Controls were grown in food containing 2% molecular grade ethanol. Over 100 larvae were included in this experiment, which was conducted at 25°C. Adult flies were collected and frozen at -25°C until microscopic analysis.
Immunostaining of Drosophila eye imaginal discs
Imaginal discs from third instar larvae were fixed for 20 min in phosphate buffered saline (PBS) containing 4% formaldehyde. After 3 washes in PBS, discs were permeabilized for 10 min in PBS containing 1% Triton-X-100 and subsequently blocked for 1 hour in PBT (PBS containing 0.5% Triton-X-100 and 4% fetal bovine serum) at room temperature. Samples were incubated with anti-ALK antibody (monoclonal rabbit anti-ALK (D5F3; Cell Signaling #3633BC, dilution 1:200) in PBT overnight. After three washes in PBS secondary antibody (Alexa Flour A488, Jackson Immuno Research #111-545-144, dilution 1:1000) was applied in PBT for 2 h at room temperature. After three washes in PBS, discs were mounted in Flouromount G.
Xenograft neuroblastoma model in vivo
Female Balbc/nude mice (Charles River, Germany) at 5-6 weeks age were maintained in a pathogen-free environment. For xenografts, 2.5 million CLB-BAR cells (MYCN amplified, ALK (Δ4-11) amplified) were injected subcutaneously. Once tumor volume achieved about 50mm3, drug was given at 50 mg/kg body weight, by oral gavage, once daily, continuously for 14 days. Mice were grouped by tumor volume randomly. Tumor volume was calculated with the following equation: V=0.5236×L×W2 (V=volume, L=longest, W=width). Brigatinib was formulated in 90% polyethylene glycol and 10% 1-methyl-2-pyrrolidinone solution.
Immunohistochemistry
Mice were sacrificed and tumors were collected. Tumors were fixed in 4% PFA, followed by 15% and 30% sucrose in PBS. Tumors were embedded in OCT compound (VWR chemicals) and frozen at –80°C. For immunohistochemistry, samples were sectioned at 10 μm, blocked with 10% non-fat milk before incubating with primary antibody to detect Ki-67 (Cell Signaling Tech., Danver, MA) overnight at 4°C. Secondary antibodies (Cell Signaling Tech., Danver, MA) were applied for 2 hours at room temperature. Impact DAB (Vector Laboratories, SK 4105) was employed for staining followed by hematoxylin cross-staining (Sigma-Aldrich). After mounting sections, images were taken using an Axio zoom V16 stereo microscope (Zeiss).
Ethical permission
All in vivo experiments were approved by Swedish legal regulations with the permission of ethical committees in Gothenburg (230-2014).
ACKNOWLEDGMENTS AND FUNDING
Work in the author’s laboratories has been supported by grants from the Swedish Cancer Society (BH 15-0323, RHP 15-0391), the Children’s Cancer Foundation (BH 14/150, 2015/80, RHP 15/0096), the Swedish Research Council (RHP 2015-04466, BH 2012-2831), and the Swedish Foundation for Strategic Research (RB13-0204). KP was supported by Carl Tryggers foundation (CTS KF15:15).
CONFLICTS OF INTEREST
V.M.R. is full-time employee of ARIAD Pharmaceuticals and holds ARIAD stock. There are no other relationships, conditions or circumstances that present a potential conflicts of interest to disclose.
REFERENCES
1. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994; 263:1281-1284.
2. Marino-Enriquez A, Dal Cin P. ALK as a paradigm of oncogenic promiscuity: different mechanisms of activation and different fusion partners drive tumors of different lineages. Cancer Genet. 2013; 206:357-373.
3. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013; 13:685-700.
4. (FDA). U.F.a.D.A. FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer. http://wwwfdagov/NewsEvents/Newsroom/PressAnnouncements/ucm269856htm. 2011.
5. (Roche). Japan becomes first country to approve Roche’s alectinib for people with a specific form of advanced lung cancer. http://wwwrochecom/media/store/releases/med-cor-2014-07-04htm. 2014.
6. Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011; 108:7535-7540.
7. Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J. 2011; 440:405-413.
8. Maris JM. Recent advances in neuroblastoma. The New England journal of medicine. 2010; 362:2202-2211.
9. Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KK, Hogarty M, Committee COGN. Children’s Oncology Group’s 2013 blueprint for research: neuroblastoma. Pediatric blood & cancer. 2013; 60:985-993.
10. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003; 3:203-216.
11. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008; 455:930-935.
12. George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008; 455:975-978.
13. Caren H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. 2008; 416:153-159.
14. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008; 455:971-974.
15. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008; 455:967-970.
16. Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, Ingle AM, Ahern C, Adamson PC, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013; 14:472-480.
17. Bown N, Lastowska M, Cotterill S, O’Neill S, Ellershaw C, Roberts P, Lewis I, Pearson AD, Group UKCC and the UKCsCSG. 17q gain in neuroblastoma predicts adverse clinical outcome. U.K. Cancer Cytogenetics Group and the U.K. Children’s Cancer Study Group. Med Pediatr Oncol. 2001; 36:14-19.
18. Guimier A, Ferrand S, Pierron G, Couturier J, Janoueix-Lerosey I, Combaret V, Mosseri V, Thebaud E, Gambart M, Plantaz D, Marabelle A, Coze C, Rialland X, et al. Clinical characteristics and outcome of patients with neuroblastoma presenting genomic amplification of loci other than MYCN. PLoS One. 2014; 9:e101990.
19. Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, Vermeulen J, Couturier J, Peuchmaur M, Valent A, Plantaz D, Rubie H, Valteau-Couanet D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol. 2009; 27:1026-1033.
20. Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, Klijanienko J, Couturier J, Pierron G, Mosseri V, Valent A, Auger N, Plantaz D, Rubie H, Valteau-Couanet D, Bourdeaut F, et al. Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol. 2010; 28:3122-3130.
21. Fransson S, Hansson M, Ruuth K, Djos A, Berbegall A, Javanmardi N, Abrahamsson J, Palmer RH, Noguera R, Hallberg B, Kogner P, Martinsson T. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes, chromosomes & cancer. 2015; 54:99-109.
22. Okubo J, Takita J, Chen Y, Oki K, Nishimura R, Kato M, Sanada M, Hiwatari M, Hayashi Y, Igarashi T, Ogawa S. Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene. 2012; 31:4667-4676.
23. Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, Pierron G, Lapouble E, Combaret V, Speleman F, de Wilde B, Djos A, Ora I, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. 2014; 32:2727-2734.
24. Schramm A, Koster J, Assenov Y, Althoff K, Peifer M, Mahlow E, Odersky A, Beisser D, Ernst C, Henssen AG, Stephan H, Schroder C, Heukamp L, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet. 2015; 47:872-877.
25. Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, Schild L, Bentahar NB, Bellini A, Chicard M, Lapouble E, Combaret V, Legoix-Ne P, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015; 47:864-871.
26. De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, Lakeman A, Vandesompele J, Hoebeeck J, Van Maerken T, De Paepe A, Laureys G, Schulte JH, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. 2010; 16:4353-4362.
27. Guan J, Umapathy G, Yamazaki Y, Wolfstetter G, Mendoza P, Pfeifer K, Mohammed A, Hugosson F, Zhang H, Hsu AW, Halenbeck R, Hallberg B, Palmer RH. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife. 2015; 4.
28. Reshetnyak AV, Murray PB, Shi X, Mo ES, Mohanty J, Tome F, Bai H, Gunel M, Lax I, Schlessinger J. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc Natl Acad Sci U S A. 2015.
29. Camidge DR, Bazhenova L, Salgia R, Langer CJ, Gold KA, Rosell R, Shaw AT, Weiss GJ, Narasimhan NI, Dorer DJ, Rivera VM, Clackson TP, Conlan MG, et al. Safety and efficacy of brigatinib (AP26113) in advanced malignancies, including ALK plus non-small cell lung cancer (NSCLC). Journal of Clinical Oncology. 2015; 33.
30. Ceccon M, Mologni L, Giudici G, Piazza R, Pirola A, Fontana D, Gambacorti-Passerini C. Treatment Efficacy and Resistance Mechanisms Using the Second-Generation Alk Inhibitor Ap26113 in Human Npm-Alk-Positive Anaplastic Large Cell Lymphoma. Mol Cancer Res. 2014.
31. Schleiermacher G, Janoueix-Lerosey I, Combaret V, Derre J, Couturier J, Aurias A, Delattre O. Combined 24-color karyotyping and comparative genomic hybridization analysis indicates predominant rearrangements of early replicating chromosome regions in neuroblastoma. Cancer genetics and cytogenetics. 2003; 141:32-42.
32. Cazes A, Louis-Brennetot C, Mazot P, Dingli F, Lombard B, Boeva V, Daveau R, Cappo J, Combaret V, Schleiermacher G, Jouannet S, Ferrand S, Pierron G, et al. Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res. 2013; 73:195-204.
33. Kohl NE, Gee CE, Alt FW. Activated expression of the N-myc gene in human neuroblastomas and related tumors. Science. 1984; 226:1335-1337.
34. Schonherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, Djos A, Martinsson T, Christensen JG, Palmer RH, Hallberg B. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012; 31:5193-5200.
35. Umapathy G, El Wakil A, Witek B, Chesler L, Danielson L, Deng X, Gray NS, Johansson M, Kvarnbrink S, Ruuth K, Schonherr C, Palmer RH and Hallberg B. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Science signaling. 2014; 7:ra102.
36. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012; 18:1472-1482.
37. Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994; 77:841-852.
38. Chand D, Yamazaki Y, Ruuth K, Schonherr C, Martinsson T, Kogner P, Attiyeh EF, Maris J, Morozova O, Marra MA, Ohira M, Nakagawara A, Sandstrom PE, et al. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis Model Mech. 2013; 6:373-382.
39. Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993; 118:401-415.
40. Martinsson T, Eriksson T, Abrahamsson J, Caren H, Hansson M, Kogner P, Kamaraj S, Schonherr C, Weinmar J, Ruuth K, Palmer RH, Hallberg B. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res. 2011; 71:98-105.
41. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, Ahmad Z, Barker K, Moreau L, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012; 22:117-130.
42. Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. 2014; 12:429-439.
43. Mano H. Second-generation ALK inhibitors. Clin Adv Hematol Oncol. 2015; 13:416-417.
44. UFaD. A. Breakthrough Therapy Designation, Case study Brigatinib. http://wwwfdagov/downloads/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/ucm447165pdf. 2014.
45. Fontana D, Ceccon M, Gambacorti-Passerini C, Mologni L. Activity of second-generation ALK inhibitors against crizotinib-resistant mutants in an NPM-ALK model compared to EML4-ALK. Cancer Med. 2015; 4:953-965.
46. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, Hogarty MD, London WB, Radhakrishnan R, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014; 26:682-694.
47. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013; 368:2385-2394.
48. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Iyer S, Reisman A, Wilner KD, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. The New England journal of medicine. 2014; 371:2167-2177.
49. Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, Morcos PN, Lee RM, Garcia L, Yu L, Boisserie F, Di Laurenzio L, Golding S, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014; 15:1119-1128.
50. Mok T, Spigel D, Felip E, deMarinis F, Ahn M-J, Groen HJM, Wakelee HA, Hida T, Crino L, Nishio M, Scagliotti GV, Branle F, Emeremni C, et al. ASCEND-2: A single-arm, open-label, multicenter phase II study of ceritinib in adult patients (pts) with ALK-rearranged (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (CRZ). ASCO Meeting Abstracts. 2015; 33:8059.
51. Ou S-HI, Ahn JS, De Petris L, Govindan R, Yang JC-H, Hughes BGM, Lena H, Moro-Sibilot D, Bearz A, Ramirez SV, Mekhail T, Spira AI, Zeaiter AH, et al. Efficacy and safety of the ALK inhibitor alectinib in ALK+ non-small-cell lung cancer (NSCLC) patients who have failed prior crizotinib: An open-label, single-arm, global phase 2 study (NP28673). ASCO Meeting Abstracts. 2015; 33:8008.
52. Toyokawa G, Seto T, Takenoyama M, Ichinose Y. Insights into brain metastasis in patients with ALK+ lung cancer: is the brain truly a sanctuaryΠCancer Metastasis Rev. 2015; 34:797-805.
53. Zhu J, Wang J, Zhen ZJ, Lu SY, Zhang F, Sun FF, Li PF, Huang JT, Cai RQ, Sun XF. Brain metastasis in children with stage 4 neuroblastoma after multidisciplinary treatment. Chin J Cancer. 2015; 34:531-537.