
Introduction
Staphylococcus aureus make up a large proportion of human infections worldwide, causing various diseases that range from acute skin infections to life-threatening systemic toxic shock syndromes. The rise of methicillin-resistant S. aureus (MRSA) and other antibiotic-resistant strains has sparked the need for new treatment strategies in both immunodeficient and immunocompetent individuals [1-5]. Neutrophils are part of the innate immune system and are critical for clearing S. aureus infections. They are the first responders to invading bacteria and kill microbes using reactive oxygen species (ROS) produced by the NADPH oxidase complex [1, 6-10]. During ingestion, neutrophils first form a phagosomal cup, which then becomes a fully internalized phagosome where microorganisms are isolated and exposed to toxic levels of superoxide (O2-) and other reactive oxygen species (ROS) [11]. The NADPH oxidase complex is located within the phagosome membrane and it is made up of membrane-bound gp91phox and p22phox; and cytosolic p47phox, p67phox, p40phox, and Rac2 [12, 13]. Individuals with chronic granulomatous disease (CGD) demonstrate the importance of NADPH oxidase function for human health, as these patients lack a functional NADPH oxidase complex, and thus suffer from recurrent and severe bacterial and fungal infections [10, 14, 15].
A variety of receptors on neutrophil membranes contribute to the recognition of opsonized material. Two of the most important receptors are Fcγ receptors (FcγRs), which bind to IgG-coated pathogens, and complement receptors (CRs), which bind C3b on complement-coated pathogens [16-18]. FcγR or CR binding causes the cytosolic components of NADPH oxidase to translocate to gp91phox/p22phox at the membrane. Downstream of these receptors are many signaling molecules that regulate NADPH oxidase assembly and activation, including Class IA phosphoinositide 3-kinase (PI3K). PI3K is a heterodimer consisting of a regulatory subunit (p85α, p55α, p50α, or p85β) and a catalytic subunit (p110α, p110β, or p110δ) and phosphorylates the lipid PI(4,5)P2 to produce PI(3,4,5)P3 [12, 13]. Furthermore, pharmacologic inhibition and genetic ablation of the catalytic subunits have been shown to decrease neutrophil ROS production in response to IgG-zymosan and Aspergillus fumigatus hyphae [16, 19]. However, the specific role of p85α, the most abundant regulatory subunit of Class IA PI3K, has not been fully studied.
We previously found that a functional binding site on p47phox for Class IA PI3K-derived phospho-lipids, PI(3,4)P2 and PI(3,4,5)P3, is needed for extracellular ROS production, but is dispensable for intracellular ROS production during early phagocytosis [20]. This finding is consistent with the observation that PI(3,4)P2 and PI(3,4,5)P3
are found on the early phagosomal cup at the location of and during the time of extracellular ROS production, but are not detected on the mature, internalized phagosome. Notably, p85α, the regulatory subunit of Class IA PI3K and thus necessary for PI(3,4,5)P3 production, remains associated with the phagosome membrane even when PI(3,4,5)P3 is no longer present [21]. These observations led us to hypothesize that p85α differentially influences extracellular and intracellular NADPH oxidase activity and performs a function on the internalized, sealed phagosome independent of PI(3,4,5)P3 production.
To test this hypothesis, we used neutrophils lacking p85α, p55α, and p50α (encoded by Pik3r1), and distinguished production of extra- and intracellular ROS. We found that the PI3K regulatory subunits are not necessary for formation of the early phagosome cup or for production of extracellular ROS. However, we show that the loss of p85α leads to enhanced intracellular ROS, which also contributed to improved killing of methicillin-susceptible S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA).
Our work provides a novel target in the regulation of enhancing neutrophil intracellular ROS, which has been shown to cooperate with anti-microbial agents to increase bacterial killing [22, 23]. This is an improvement over indiscriminately increasing global ROS production, which could lead to inflammation-induced tissue injury. Using intracellular ROS to augment anti-microbial therapies may provide a novel strategy in the treatment of antibiotic-resistant pathogens.
Results and Discussion
Loss of regulatory subunits p85α, p55α, and p50α decreases extracellular ROS production, but increases intracellular ROS production
As global knockout of Pik3r1-/- leads to perinatal lethality [24], timed matings were performed between Pik3r1+/- mice for the isolation of fetal liver-derived hematopoietic cells at 14.5 days post-conception. WT (Pik3r1+/+), Pik3r1+/-, and Pik3r1-/- (lacking expression of p85α, p55α, and p50α) [24, 25] fetal liver mononuclear cells were differentiated in vitro to neutrophils [20], and each population demonstrated similar differentiation as assessed by morphology and Mac-1/Gr-1 staining (Figure 1A and 1B). Consistent with previous studies reporting increased PI3K catalytic subunit degradation upon loss of the stabilizing regulatory subunits [24, 26], p110α, p110β, and p110δ protein expression was substantially reduced in the Pik3r1-/- neutrophils (Figure 1C). Notably, however, the levels of the NADPH oxidase subunits (p67phox, p47phox, and p40phox) were unchanged (Figure 1C).
To examine the function of the PI3K regulatory subunits in neutrophil ROS production, we examined both extra- and intracellular ROS production in response to various stimuli. Pik3r1-/- neutrophils had significantly reduced FcγR (hIgG-latex)-stimulated extracellular ROS (50% of WT, Figure 1D), but similar amounts of CR (SOZ)-stimulated extracellular ROS (Figure 1D). Together with the lower expression of the PI3K catalytic subunits, this finding supports the notion that PI3K activity is required for extracellular ROS mediated by FcγR stimulation, and is consistent with our previous findings [20]. In contrast to that observed with extracellular ROS, loss of p85α/p55α/p50α led to significantly increased FcγR- and a trend of enhanced CR-stimulated intracellular ROS production (Figure 1E). Both extra- and intracellular ROS production from heterozygous Pik3r1+/- neutrophils was similar to that of WT.
Immunoblots confirmed that activated Akt was reduced in Pik3r1-/- neutrophils; however, myeloperoxidase (MPO) levels were equal in WT and Pik3r1-/- cells (Figure 1F), demonstrating that the increased intracellular ROS production in Pik3r1-/- neutrophils is due to a regulatory effect of p85α/p55α/p50α, rather than due to reduced MPO expression and diminished ROS consumption. Furthermore, by immunostaining with anti-F-actin and anti-Rac to visualize the phagosomes at 10min and 30min post-SOZ stimulation, we found a similar phagocytic index in WT and Pik3r1-/- neutrophils (Figure 1G and 1H), indicating that increased FcγR- and CR-stimulated intracellular ROS levels in Pik3r1-/- neutrophils is not merely due to enhanced phagocytosis, but to a regulatory role of p85α/p55α/p50α on NADPH oxidase activity.
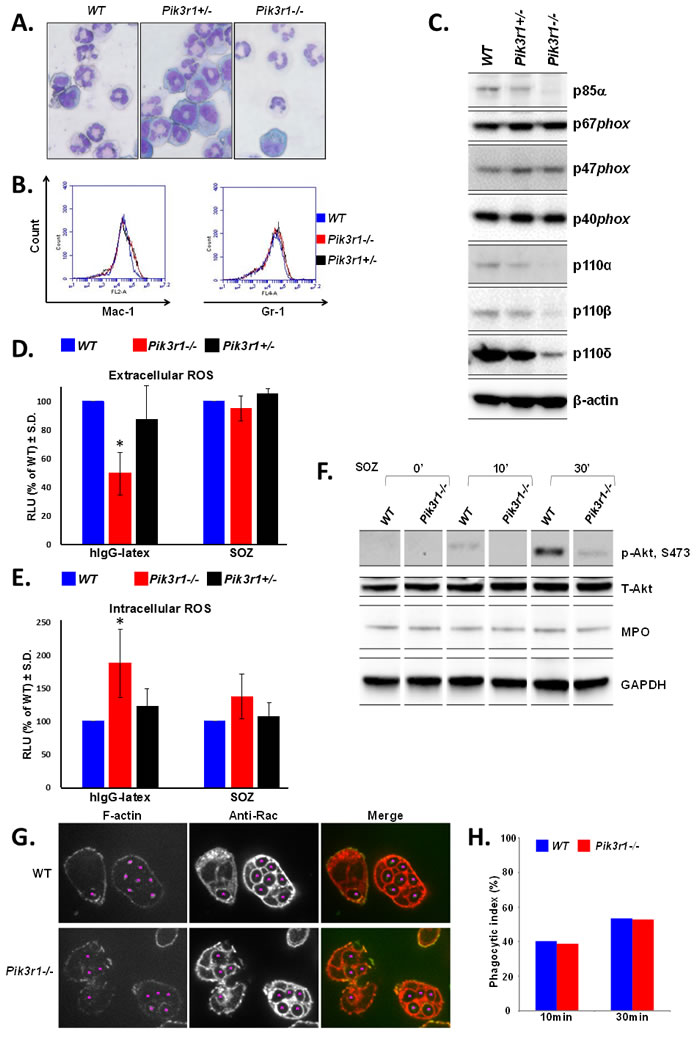
Figure 1: Characterization of WT, Pik3r1+/-, and Pik3r1-/- fetal liver-derived neutrophils. Differentiation of fetal liver cells taken from WT, Pik3r1+/-, and Pik3r1-/-embryos was assessed based on A. morphology (images taken with 40X objective) and B. Mac-1/Gr-1 staining; C. Protein expression of PI3K catalytic subunits and NADPH oxidase subunits as measured by immunoblot; D. Extracellular and E. intracellular ROS production was measured in WT, Pik3r1+/-, and Pik3r1-/- fetal liver-derived neutrophils stimulated with hIgG-latex (FcγR simulation) and SOZ (CR stimulation), n=10, *p<0.0001 comparing extracellular ROS production from Pik3r1-/- to WT in response to hIgG-latex, and n=10, p=0.0009 comparing intracellular ROS production from Pik3r1-/- to WT in response to hIgG-latex, statistical analyses performed by two-tailed, one-sample Student’s t-test; F. Immunoblotting for phospho-Akt and MPO in WT and Pik3r1-/- fetal liver-derived neutrophils, stimulated for 0min, 10min, and 30min with SOZ; G. WT and Pik3r1-/- fetal liver-derived neutrophils were stimulated with SOZ and immunostained with anti-F-actin and anti-Rac to visualize phagosomes and quantitate phagocytic index, images taken with 100× objective; H. 10min and 30min after SOZ stimulation, phagocytic index (PI) was calculated as PI = (% of phagocytic cells containing ≥ 1 particle) × (mean number of particles/phagocytic cell containing particles).
Re-introduction of p85α corrects the levels of extra- and intracellular ROS production in Pik3r1-/- neutrophils
We next examined the effect of re-introducing p85α (Figure 2A) on extra- and intracellular ROS production from Pik3r1-/- fetal liver-derived neutrophils [27]. We found comparable neutrophil differentiation (Mac-1, Gr-1) between Pik3r1-/- neutrophils and Pik3r1-/- neutrophils upon re-introduction of p85α (Figure 2B). Protein levels of PI3K catalytic subunit p110δ were increased upon re-introduction of p85α, and concordantly, Akt phosphorylation was normalized (Figure 2C). Moreover, p85α restored hIgG-latex-stimulated extracellular ROS levels and inhibited hIgG-latex-stimulated intracellular ROS compared to Pik3r1-/- neutrophils (Figures 2D, 2E, and 2F). Consistent with a dispensable role of p85 α, p55α, and p50α on SOZ-stimulated extracellular ROS (Figure 1D), re-introduction of p85α did not alter SOZ-stimulated extracellular ROS production from Pik3r1-/- neutrophils (Figure 2G); however, SOZ-stimulated intracellular ROS was inhibited by re-introduction of p85α. These findings demonstrate that of the PI3K regulatory subunits, p85α uniquely is able to negatively regulate hIgG-latex- and SOZ-stimulated intracellular ROS production. Furthermore, as the C-terminal portion of p85α (nSH2, iSH2, and cSH2 domains, the shared domains between the p85α, p55α, and p50α regulatory proteins) is critical for promoting PI3K activity, these findings suggest that the N-terminus of p85α (SH3 and BH domains) functions to negatively regulate intracellular ROS.
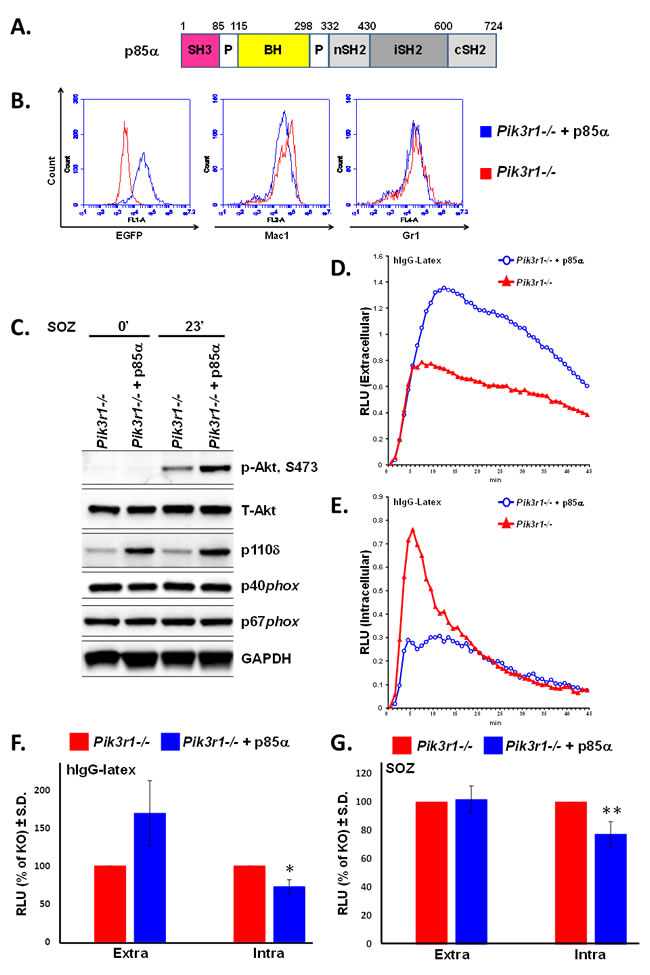
Figure 2: Re-introduction of p85α in Pik3r1-/- fetal liver-derived neutrophils normalizes hIgG-latex-stimulated extra- and intracellular ROS. A. Domain structure of p85α construct, BH: Bcr homology domain; B. Representative flow cytometry of fetal liver-derived neutrophils upon transduction with p85α (either in tandem with EGFP or tagged with YFP); C. Immunoblotting for Akt, PI3K catalytic subunit p110δ, and NADPH oxidase subunits in Pik3r1-/- and Pik3r1-/- + p85α neutrophils, stimulated for 0 and 23min with SOZ; D. Representative extracellular and E. representative intracellular ROS production measured in Pik3r1-/- and Pik3r1-/- + p85α neutrophils stimulated with hIgG-latex; F. Quantitative assessment of hIgG-latex-stimulated extra- and intracellular ROS production, n = 6, *p = 0.04 comparing intracellular ROS production from Pik3r1-/- to Pik3r1-/- + p85α; G. Quantitative assessment of SOZ-stimulated extra- and intracellular ROS production, n = 7, **p = 0.05 comparing intracellular ROS production from Pik3r1-/- to Pik3r1-/- + p85α; statistical analyses performed by two-tailed, one-sample Student’s t-test.
Elevated intracellular ROS in Pik3r1-/- neutrophils enhances bacterial killing
Based on our observation that Pik3r1-/- fetal liver-derived neutrophils have increased intracellular ROS production in response to Fcγ receptor and CR stimulation, we predicted that Pik3r1-/- neutrophils would demonstrate enhanced S. aureus-stimulated ROS production and enhanced bacterial killing compared to WT neutrophils. Consistent with our hypothesis, Pik3r1-/- fetal liver-derived neutrophils produce more intracellular ROS in response to serum-opsonized MSSA (Wood 46), while the extracellular ROS levels were not affected (Figures 3A and 3B).
To determine the antimicrobial function of Pik3r1-/- neutrophils, we performed a bacterial killing assay. WT and Pik3r1-/- fetal liver-derived neutrophils were incubated with serum-opsonized methicillin-sensitive S. aureus (MSSA) (Wood 46) over 60 min, and at various time points, neutrophil samples were quenched in ice-cold LB-saponin, sonicated to liberate ingested bacteria, and surviving bacteria were enumerated by plating on LB-agar. Surviving MSSA was reduced when incubated with Pik3r1-/- neutrophils compared to WT neutrophils at all time points, with a trend toward statistical significance at 60 minutes (Figure 3C).
Given the promising results using fetal liver-derived neutrophils and Wood 46 MSSA, we turned our attention to a more relevant pathogen, MRSA. Hypervirulent MRSA, such as LAC MRSA USA300 strain, has become an important public health problem due to the sheer number of infections and widespread antibiotic resistance [3]. Consistent with results observed using MSSA, we observed enhanced intracellular ROS in fetal liver-derived Pik3r1-/- neutrophils compared to WT neutrophils in response to serum-opsonized MRSA (USA300), and no difference in extracellular ROS production (Figures 3D and 3E). We next conducted bacterial killing assays using a more physiologic source of neutrophils directly isolated from the bone marrow using a percoll gradient. Since global knockout of Pik3r1 induces perinatal lethality, we utilized a murine model bearing a conditionally targeted Pik3r1 allele (Pik3r1flox/flox) crossed with Mx1-Cre. Pik3r1flox/flox;Mx1Cre+ and Pik3r1flox/flox;Mx1Cre- littermate controls were treated with polyI:polyC to induce recombination of the Pik3r1 allele. Animals were permitted to recover from polyI:polyC treatment for at least 12 weeks prior to isolation of bone marrow neutrophils. Phenotypically and morphologically, bone marrow neutrophils isolated directly from the Pik3r1flox/flox;Mx1Cre+ and Pik3r1flox/flox;Mx1Cre- bone marrow were similar (data not shown). To control for phagocytosis, neutrophils were incubated with GFP-expressing MRSA for 2 hours, followed by quenching of extracellular GFP using trypan blue. To measure bacterial killing, a second plate was incubated for an additional 2 hours and then quenched with trypan blue. Following quenching, intracellular GFP levels were read immediately on a fluorometer to measure phagocytosis and bacterial killing, respectively. GFP-expressing MRSA were phagocytized equally (Figure 3F); however, significantly reduced GFP levels, previously shown to correlate with bacterial survival [28], was observed in Pik3r1flox/flox;Mx1Cre+ neutrophils compared to Pik3r1flox/flox;Mx1Cre- neutrophils (Figure 3G). Collectively, these findings suggest that increased intracellular ROS production may provide enhanced bacterial killing, in particular of MSSA and MRSA.
Our findings demonstrate that genetic disruption of Pi3kr1 differentially regulates NADPH oxidase activity on the plasma membrane (extracellular ROS production) v. the phagosome membrane (intracellular ROS production). These novel findings are consistent with other studies demonstrating that the regulation of NADPH oxidase activity differs between the plasma and phagosome membranes, and highlights the varied environments of these two compartments. An example relevant to the current work is the Class III PI3K product, PI3P, which is a strong positive regulator of intracellular ROS production but plays no role in extracellular ROS production. A key molecule increasing Class III PI3K activity and increased PI3P production on the phagosome is the Rab GTPase, Rab5. Down-regulation of Rab5 reduces the capacity of S. aureus-containing phagosomes to fuse with endocytic organelles resulting in poorer bacterial killing [29]. Notably, p85α is known to interact with Rab5 and to bear GTPase Activating Protein (GAP) activity towards Rab5-GTP, which is localized to the p85α BH domain (Figure 2A). These considerations suggest that a feasible mechanism underlying the negative regulatory effect of p85 α on intracellular ROS may be downregulating Rab5a-GTP levels via its GAP function [30, 31], resulting in reduced Class III PI3K-derived PI3P. Thus, while our current work cannot exclude the possibility that p85α negatively regulates intracellular ROS production in an indirect manner due to altered expression of the Class IA PI3K catalytic subunits, a thought-provoking consideration is that p85 α functions in a Class IA PI3K catalytic subunit-independent manner to regulate NADPH oxidase activity on the phagosome membrane.
Collectively, our findings show that neutrophils lacking the PI3K regulatory subunit p85α produce significantly more intracellular ROS without affecting phagocytosis. Furthermore, this correlates to significantly increased killing of both MSSA and MRSA. These results suggest a new strategy for combating the growing threat of resistant microorganisms.
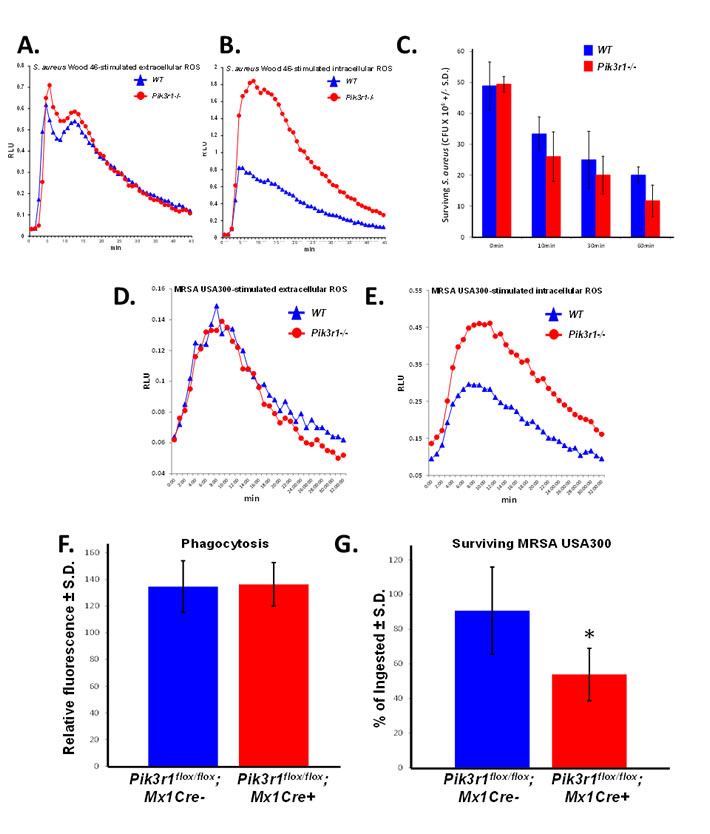
Figure 3: Pik3r1-/- neutrophils demonstrate superior killing of MSSA (Wood 46) and MRSA (USA300). A. Extracellular and B. intracellular ROS production from WT and Pik3r1-/- fetal liver-derived neutrophils in response to serum-opsonized MSSA (Wood 46), representative of 2 independent experiments; C. WT and Pik3r1-/- fetal liver-derived neutrophil killing of MSSA (Wood 46) was measured by counting the number of surviving bacteria after 0, 10, 30, and 60min incubation, n = 3, p = 0.09 comparing Pik3r1-/- to WT at 60min, statistical analysis performed by unpaired, two-tailed Student’s t-test; D. Extracellular and E. intracellular ROS production was measured in WT and Pik3r1-/- neutrophils stimulated with serum-opsonized MRSA (USA300), experiment performed on one occasion. F. Phagocytosis and G. killing of MRSA (USA300) by Pik3r1flox/flox;Mx1Cre- and Pik3r1flox/flox;Mx1Cre+ bone marrow neutrophils was measured by fluorescence remaining inside cells after washing and quenching extracellular fluorescence, n = 30, *p < 0.001 comparing Pik3r1flox/flox;Mx1Cre- to Pik3r1flox/flox;Mx1Cre+, statistical analysis by unpaired, two-tailed Student’s t-test, experiment conducted on two independent occasions.
Materials and Methods
Preparation of particulate stimuli
Human IgG-opsonized latex beads (1.98 µm or 2.94µm), serum opsonized-zymosan A particles (Sigma, St. Louis, MO, USA), and serum-opsonized MSSA (Wood 46) were prepared as previously described [32-34]. MRSA USA300 (LAC) expressing superfolded GFP was grown overnight at 37°C with shaking, OD600 was measured to identify mid-logarithmic growth, and bacteria were washed with PBS before adding to PMNs [35]. Final concentrations were 5:1 IgG-beads/PMNs; 400 µg/ml SOZ; 40:1 S. aureus/PMNs; 60 µg/ml hyphae; 50:1 MRSA/PMNs.
Western blots
Cell lysates were prepared from murine neutrophils using 1% Triton X-100, 15 µg lysate was subjected to SDS-PAGE separation and immunoblotting using a nitrocellulose membrane (Thermo Scientific). Blots were blocked with 5% BSA and then incubated overnight with anti-p85α, -p110δ (Santa Cruz);anti-p67phox, -p47phox, -p40phox (Millipore); anti-p110a, -p110b, -phospho-Akt (Ser473), or -MPO (Cell Signaling). Blots were then incubated with HRP-conjugated secondary antibody and signal was visualized with ECL detection (Pierce).
Confocal microscopy
SOZ-induced phagocytosis in WT and Pik3r1-/- fetal liver-derived neutrophils was imaged using a spinning-disk (CSU10) confocal system mounted on a Nikon TE-2000U inverted microscope with an Ixon air-cooled EMCCD camera (Andor Technology, South Windsor, CT) and a Nikon Plan Apo 100X 1.4 N.A. objective as described previously [16, 20, 21]. Images were analyzed with Metamorph software (Universal Imaging; Downington, PA).
Animal husbandry
Mice were housed and bred in accordance with the Institutional Animal Care and Use Committee of the Indiana University School of Medicine. Pik3r1+/- mice [24, 25] were subjected to timed matings, dams were euthanized at day 14 post-conception, and fetal livers were harvested for generation of Pik3r1+/+, Pik3r1+/-, and Pik3r1-/- neutrophils. Pik3r1flox/flox mice [36] were crossed with Mx1-Cre+ mice to produce Pik3r1flox/flox; Mx1-Cre+ mice. Cre expression was induced by 3 intraperitoneal injections of polyI:polyC (300ug), and recombination of Pik3r1 was confirmed by genotyping.
Neutrophil differentiation
Fetal liver cells were collected from embryos at day 14 of gestation, genotyped, and differentiated into neutrophils in α-minimum essential medium with 20% heat-inactivated FCS, 1% penicillin/streptomycin, 50ng/ml G-CSF and 50units/ml IL-3. Every 2 days, cells were counted and replated at a concentration of 0.5×106/ml in fresh differentiation medium. Activity was analyzed on days 6 and 7 of differentiation [20].
Reintroduction of p85α
p85α cDNA was cloned upstream of an internal entry site (IRES) and the enhanced green fluorescence protein (EGFP) in the bicistronic retroviral plasmid, pMIEG3 [37]. Alternatively, p85α was tagged on the C-terminal end with yellow fluorescent protein (YFP) and cloned into the retroviral plasmid, pMSCV (Clontech). Ecotropic retrovirus containing the vectors was used to transduce Pik3r1-/- fetal liver cells, cells were sorted for EGFP or YFP positivity, and differentiated into neutrophils. Data using both constructs are combined for statistical analyses, as the YFP tag did not alter the function of p85α (data not shown).
ROS detected by chemiluminescence in intact cells
Oxidant production was measured by chemiluminescence using 2×105 neutrophils during synchronized phagocytosis of particulate stimuli [20, 21, 38, 39]. Extracellular ROS was measured using 20µM isoluminol with 20U/ml horseradish peroxidase (HRP), and intracellular ROS was measured using 20µM luminol with 20U/ml HRP and 10µg/ml superoxide dismutase (SOD) [20, 21, 32]. An Lmax microplate luminometer (Molecular Devices, Sunnyvale, CA, USA) was used to record luminescence as previously described [20, 21].
Methicillin-susceptible S. aureus (Wood 46) killing
6×106 WT or Pik3r1-/- fetal liver-derived neutrophils were incubated with 1.5×106 serum-opsonized MSSA (strain Wood 46) in 300 µl PBS for 60 min. Samples were added to ice-cold Difco nutrient broth (BD, Franklin Lakes, NJ, USA) with 10% saponin and sonicated to liberate ingested bacteria [32]. Surviving bacteria were enumerated by plating on Columbia agar (Sigma, St. Louis, MO, USA).
Neutrophil isolation from bone marrow and MRSA (USA300 LAC) killing
Mice were sacrificed, bone marrow cells were collected from the pelvis, femur, and tibia, and neutrophils were isolated using a 62% and 55% percoll gradient. Neutrophils were washed in Hanks’ Balanced Salt solution (Sigma, St. Louis, MO, USA), resuspended in Iscove’s Modified Dulbecco’s Medium (Life Technologies, Carlsbad, CA, USA), and plated at 2x105 cells per well into a 96well plate coated with celltak (Corning, Corning, NY, USA). MRSA (USA300 strain LAC) was incubated with neutrophils for 2h at 37°C, and then plates were washed with warm PBS. For phagocytosis analysis, 50uL of 500mg/mL trypan blue was added immediately to PBS-washed plates to quench extracellular fluorescence. For bacterial survival analysis, PBS-washed plates were incubated for an additional 2h at 37°C followed by the addition of 50uL of 500mg/mL trypan blue. For both phagocytosis and bacterial killing analyses, plates were read on a fluorometer to measure intracellular GFP fluorescence promptly following the addition of trypan blue. Fluorescence intensity correlates to bacterial survival based on the previously defined correlation of HOCl-bleaching of superfolded GFP to bacterial viability.
Abbreviations
CGD = chronic granulomatous disease, CR = complement receptor, FcγR = Fcγ receptor, hIgG = Human IgG-opsonized latex beads, HRP = horseradish peroxidase, MPO = myeloperoxidase, MRSA = methicillin-resistant S. aureus, MSSA = methicillin-susceptible S. aureus, PI3K = phosphoinositide 3-kinase, PMN = polymorphonuclear neutrophil, ROS = reactive oxygen species, S. aureus = Staphylococcus aureus, SOD = superoxide dismutase, SOZ = serum opsonized zymosan
Acknowledgments
The authors thank the Indiana University Simon Cancer Center Flow Cytometry Core and Indiana Center for Biological Microscopy. We gratefully acknowledge Dr. William Nauseef (Department of Medicine, University of Iowa School of Medicine, Iowa City, IA, USA) for providing the GFP-MRSA and Marilyn L. Wales for her administrative assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
Grant Support
This work was supported by the U.S. National Institutes of Health (RO1 CA134777 to RJC, T32 AI060519 (Blum) to SLB, T32 HL007910 (Broxmeyer) to LD, HL124159 and AI110990 to HS), the Indiana Center for Excellence in Molecular Hematology (NIDDK P30 DK090948), the Showalter Research Trust (XJL & RJC), and the Riley Children’s Foundation (RJC).
Authorship
XJL: designed and conducted experiments; analyzed data; prepared figures
LD: designed and conducted experiments; wrote the manuscript
SLB: designed and conducted experiments
CBG: designed and conducted experiments
PM: designed and conducted experiments
ZY: designed and conducted experiments
RSM: designed and conducted experiments
ZL: statistical analyses
RK: designed experiments
CHS: designed experiments
RJC: designed experiments, analyzed data, wrote the manuscript
References
1. Rigby KM and DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Seminars in Immunopathology. 2012; 34:237-259.
2. DeLeo FR, Otto M, Kreiswirth BN and Chambers HF. Community-associated methicillin-resistant Staphylococcus aureus. Lancet. 2010; 375:1157-1568.
3. Krishna S and Miller LS. Innate and adaptive immune responses against Staphylococcus aureus skin infections. Seminars in Immunopathology. 2012; 34:261-280.
4. Miller LS and Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nature Reviews Immunology. 2011; 11:505-518.
5. Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB and Talan DA. Methicillin-Resistant S. aureus Infections among Patients in the Emergency Department. The New England Journal of Medicine. 2006; 355:666-674.
6. Babior BM, Kipnes RS and Curnutte JT. Biological Defense Mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. The Journal of Clinical Investigation. 1973; 52:741-744.
7. Dinauer MC. Chronic Granulomatous Disease and Other Disorders of Phagocyte Function. American Society of Hematology Education Book. 2005; 2005:89-95.
8. Sbarra AJ and Karnovsky ML. The Biochemical Basis of Phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. The Journal of Biological Chemistry. 1959; 234:1355-1362.
9. Nauseef WM. The NADPH-dependent oxidase of phagocytosis. Proceedings of the Association of American Physicians. 1999; 111:373-382.
10. Stasia MJ and Li XJ. Genetics and immunopathology of chronic granulomatous disease. Seminars in Immunopathology. 2008; 30:209-235.
11. Swanson JA. Shaping cups into phagosomes and macropinosomes. Nature Reviews Molecular Cell Biology. 2010; 9:639-649.
12. Hawkins PT, Davidson K and Stephens L. The role of PI3Ks in the regulation of the neutrophil NADPH oxidase. Biochemical Society Symposium. 2007; 74:59-67.
13. Cadwallader KA, Condliffe AM, McGregor A, Walker TR, White JF, Stephens LR and Chilvers ER. Regulation of Phosphatidylinositol 3-Kinase Activity and Phosphatidylinositol 3,4,5-Trisphosphate Accumulation by Neutrophil Priming Agents. The Journal of Immunology. 2002; 169:3336-3344.
14. Heyworth PG, Cross AR and Curnutte JT. Chronic granulomatous disease. Current Opinion in Immunology. 2003; 15:578-584.
15. Holland SM. Chronic Granulomatous Disease. Clinical Reviews in Allergy & Immunology. 2010; 38:3-10.
16. Tian W, Li XJ, Stull ND, Ming W, Suh C-I, Bissonnette SA, Yaffe MB, Grinstein S, Atkinson SJ and Dinauer MC. FcγR-stimulated activation of the NADPH oxidase: phosphoinositide-binding protein p40phox regulates NADPH oxidase activity after enzyme assembly on the phagosome. Blood. 2008; 112:3867-3877.
17. Underhill DM and Ozinsky A. Phagocytosis of microbes: complexity in action. Annual Reviews Immunology. 2002; 20:825-852.
18. Joshi T, Butchar J and Tridandapani S. Fcgamma receptor signaling in phagocytes. International Journal of Hematology. 2006; 84:210-216.
19. Boyle KB, Gyori D, Sindrilaru A, Scharffetter-Kochanek K, Taylor PR, Mocsai A, Stephens LR and Hawkins PT. Class IA Phosphoinositide 3-Kinase β and δ Regulate Neutrophil Oxidase Activation in Response to Aspergillus fumigatus Hyphae. The Journal of Immunology. 2011; 186:2978-2989.
20. Li XJ, Marchal CC, Stull ND, Stahelin RV and Dinauer MC. p47phox Phox homology domain regulates plasma membrane but not phagosome neutrophil NADPH oxidase activation. The Journal of Biological Chemistry. 2010; 285:35169-35179.
21. Li XJ, Tian W, Stull ND, Grinstein S, Atkinson S and Dinauer MC. A fluorescently tagged C-terminal fragment of p47phox detects NADPH oxidase dynamics during phagocytosis. Molecular Biology of the Cell. 2009; 20:1520-1532.
22. Grant SS, Kaufmann BB, Chand NS, Haseley N and Hung DT. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:12147-12152.
23. Shatalin K, Shatalina E, Mironov A and Nudler E. H2S: A Universal Defense Against Antibiotics in Bacteria. Science. 2011; 334:986-990.
24. Fruman DA, Mauvais-Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT, Kahn CR and Cantley LC. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nature Genetics. 2000; 26:379-382.
25. Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW and Cantley LC. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999; 283:393-397.
26. Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr G and Backer J. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Molecular and Cell Biology. 1998; 18:1379-1387.
27. Hawkins PT, Anderson K, Davidson K and Stephens L. Signalling through Class I PI3Ks in mammalian cells. Biochemical Society Transactions. 2006; 34:647-662.
28. Schwartz J, Leidal KG, Femling JK, Weiss JP and Nauseef WM. Neutrophil Bleaching of GFP-Expressing Staphylococci: Probing the Intraphagosomal Fate of Individual Bacteria. The Journal of Immunology. 2009; 183:2632-2641.
29. Perskvist N, Roberg K, Kulyté A and Stendahl O. Rab5a GTPase regulates fusion between pathogen-containing phagosomes and cytoplasmic organelles in human neutrophils. Journal of Cell Science. 2002; 115:1321-1330.
30. Chamberlain MD, Berry TR, Pastor MC and Anderson DH. The p85alpha Subunit of Phosphatidylinositol 3’-Kinase Binds to and Stimulates the GTPase Activity of Rab Proteins. The Journal of Biological Chemistry. 2004; 279:48607-48614.
31. Chamberlain MD, Chan T, Oberg JC, Hawrysh AD, James KM, Saxena A, Xiang J and Anderson DH. Disrupted RabGAP Function of the p85 Subunit of Phosphatidylinositol 3-Kinase Results in Cell Transformation. The Journal of Biological Chemistry. 2008; 283:15861-15868.
32. Ellson C, Davidson K, Anderson K, Stephens LR and Hawkins PT. PtdIns3P binding to the PX domain of p40phox is a physiological signal in NADPH oxidase activation. The EMBO Journal. 2006; 25:4468-4478.
33. Matute JD, Arias AA, Wright NAM, Wrobel I, Waterhouse CCM, Li XJ, Marchal CC, Stull ND, Lewis DB, Steele M, Kellner JD, Yu W, Meroueh SO, Nauseef WM and Dinauer MC. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009; 114:3309-3315.
34. Suh C-I, Stull ND, Li XJ, Tian W, Price MO, Grinstein S, Yaffe MB, 4 SA and Dinauer MC. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcgammaIIA receptor-induced phagocytosis. The Journal of experimental medicine. 2006; 203:1915-1925.
35. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR and Nauseef WM. agr-Dependent Interactions of Staphylococcus aureus USA300 with Human Polymorphonuclear Neutrophils. Journal of Innate Immunity. 2010; 2:546-559.
36. Luo J, McMullen JR, Sobkiw CL, Zhang L, Dorfman AL, Sherwood MC, Logsdon MN, Horner JW, DePinho RA, Izumo S and Cantley LC. Class IA Phosphoinositide 3-Kinase Regulates Heart Size and Physiological Cardiac Hypertrophy. Molecular and Cell Biology. 2005; 25:9491-9502.
37. Krishnan S, Mali R, Ramdas B, Sims E, Ma P, Ghosh J, Munugalavadia V, Hanneman P, Beane J and Kapur R. p85β regulatory subunit of class IA PI3 kinase negatively regulates mast cell growth, maturation, and leukemogenesis. Blood. 2012; 119:3951-3961.
38. Dahlgren C and Karlsson A. Respiratory burst in human neutrophils. Journal of Immunological Methods. 1999; 232:3-14.
39. Li XJ, Grunwald D, Mathieu J, Morel F and Stasia M-J. Crucial role of two potential cytosolic regions of Nox2, 191TSSTKTIRRS200 and 484DESQANHFAVHHDEEKD500, on NADPH oxidase activation. The Journal of Biological Chemistry. 2005; 280:14962-14973.