
INTRODUCTION
Breast cancer (BC) is the leading cause of cancer death in females with an incidence growing worldwide [1,2]. Like other cancers, BC is considered as a complex disease in which gene aberrations, cellular context and environmental influences concur to tumor initiation and progression. Germline and somatic gene mutations play a crucial role in BC, being involved in inherited cancer syndromes [3,4], associated with either specific morphological stages [5] or response to therapy and poor disease outcome [6–8]. The therapeutic approach for women with BC includes a combination of chemotherapy, targeted and hormonal therapy. The latter is limited to patients with tumors expressing steroid hormone receptors, such as estrogen receptors (ER) and progesterone receptors (PR), which are about 65% of total cases [9–11]. The use of selective estrogen receptor modulators (SERMs), such as tamoxifen or aromatase inhibitors (AI), has led to remarkable improvements in cure efficacy for ER-positive (ER+) BC [12]. Nevertheless de novo and acquired resistance often limits the success of this therapeutic strategy in BC patients [13–15]. Understanding the molecular basis of such resistance may help us to develop alternative therapeutic strategies, thus improving response to treatment and preventing cancer deaths.
In this scenario, the receptor tyrosine kinase RET (REarranged during Transfection) has emerged as a new player in ER+ cancer development as well as a potential target to enhance sensitivity of BC to tamoxifen therapy and to avoid tamoxifen resistance [16]. RET activation is secondary to the formation of a multi-protein complex, including one of four soluble ligands, namely Glial cell Derived Neurotrophic Factor (GDNF), Neurturin (NRTN), Artemin (ARTN) or Persephin (PSPN), and one of four GPI-linked co-receptors (GFRα1-4), that leads to RET dimerization and autophosphorylation of tyrosine residues in the intracellular tyrosine kinase domain [17,18]. Alternative splicing of transcripts encoding the RET receptor leads to the isoforms RET9 and RET51 (either 9 or 51 carboxy-terminal aminoacids, respectively) with distinct biological functions [19–21].
Germline mutations of the RET gene are responsible for two different genetic disorders, Hirschsprung’s disease (HSCR) and Multiple Endocrine Neoplasia type 2 (MEN2), due to loss-of-function and gain-of-function mutations, respectively. Somatic mutations of the RET gene are also responsible for sporadic Medullary Thyroid Carcinoma (MTC), while gene rearrangements have been found in papillary thyroid carcinoma (PTC) and recently identified in lung adenocarcinoma [22]. Several studies have investigated the role of RET common SNPs in RET-related diseases [23–26]. Interestingly, one SNP (rs2435357) located in an intronic enhancer, was shown to reduce RET expression and therefore inversely associate with HSCR disease and sporadic MTC [26,27].
Several independent studies recently identified RET as a key player in BC pathogenesis [16,22]. In particular, RET and Gfra1 were over-expressed in a subset of ER+ tumors [28–30] and elevated RET levels correlated with decreased metastasis-free survival [31,32].
Estrogens induce transcription of RET and other ER+ dependent genes [33,34], in ER+ BC cell lines such as MCF7 and T47D [16,35,36], by activating the estrogen receptor. Regulatory elements responsive to this latter receptor are located at -50kb and + 32kb from the RET gene [35,36]. Moreover, additional target elements are located in the RET locus [37,38]. RET expression is also dependent on chromatin structure [39] and histone deacetylase (HDAC) inhibitors, such as Tricostatin A, Sodium Butyrate (Nabut), and 5 aza-2’ deoxycytidine (deAza), have opposite effects on estrogen mediated transcription in BC cell lines, thus affecting anti-estrogen therapy [40]. In particular, in ER-negative (ER-) cell lines, treatment with HDAC inhibitors reactivated ER expression and cellular response to hormone therapy [41,42] while in ER+ cell lines the same drugs have been associated with transcriptional down regulation of ER mRNA and its responsive genes [43,44]. Both RET and ER are tightly connected and drive proliferation and cell survival in luminal BC, thus demonstrating the potentiality of targeting both pathways [38]. Indeed, RET inhibition was shown to increase the efficacy of antiestrogen drugs, and combined therapy of tamoxifen with vandetanib was proven as a potential treatment strategy for RET positive luminal breast cancers [45].
Besides the expression of RET in ER+ BC, data suggest that RET can be expressed at low levels also in ER-ve and triple negative tumors. TFAP2C has been shown to induce ER independent RET expression, with important implications for the ER-ve breast cancers, such as MDA-MB-453 [38].
Finally, Morandi et al. [46] reported that the RET-GDNF pathway is a key determinant of response and resistance to AI in ER+ tumors.
Based on these lines of evidence, RET over-expression in BC might be due to transcriptional mechanisms involving estrogen receptors and chromatin conformation, so that targeting the RET pathway may lead to the development of new therapies in ER+ BC. To this end, we have investigated the molecular mechanisms (either genomic, transcriptional or post transcriptional) which underlie RET over-expression and its possible modulation in BC. Looking for RET genetic variants responsible for RET expression modulation, as well as BC progression and outcome, we also carried out a pilot association study in 93 ER+ BC patients.
RESULTS
Differential expression of RET, co-receptors and ligands in MCF7 and T47D
We have quantified RET mRNA levels in MCF7 and T47D in order to confirm that these cell lines, originated from ER+ BC, express different amounts of the RET gene. In agreement with literature data, we found that MCF7 showed significantly higher levels of RET compared to T47D (Figure 1A) [30,36]. Since the two isoforms RET9 and RET51 exert different oncogenic properties and differ for their C-terminals, we quantified and compared their relative amounts. To discriminate between RET9, RET51 and total RET, we set up two amplicons with a same forward primer, located upstream of the C-tail, and two alternative distal reverse primers, each specific for one isoform, in addition to an internal control amplimer in a 5’ region common to both isoforms. We found that MCF7 express a greater amount of the two isoforms than T47D (compare Y-axis scale in Figure 1B). Both cell lines express more RET9 than RET51, and in MCF7 this difference is statistically significant (Figure 1B). In addition to RET, we also investigated the mRNA levels of RET ligands (GDNF, NRTN, ARTN and PSPN) and co-receptors (GFRa1-3), except GFRa4 which seems to be expressed only in thyrocytes [18]. The results obtained are shown in Table 1. Consistent with literature data, MCF7 cells express very low levels of GDNF, which is undetectable in T47D [29]. The other genes display similar pattern of expression in the two cell lines, with the only exception of GFRa1, which is mainly express in MCF7 but not in T47D, and GFRA2 which is predominant in T47D, but poorly expressed in MCF7.
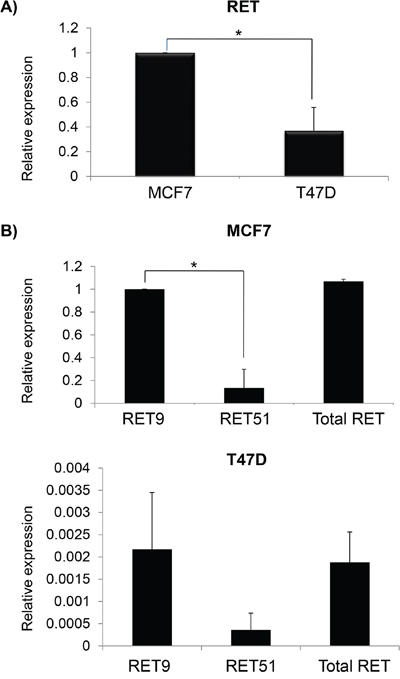
Figure 1: Differential expression of total RET and its isoforms in breast cancer cell lines. A. Relative basal RET mRNA levels in MCF7 (equal to 1) and T47D. B. mRNA quantification of RET9 and RET51 isoforms in MCF7 (top) and in T47D (bottom). Values are the mean ±SD of at least two independent experiments performed in triplicate; asterisks (*) indicate statistically significant differences (*p<0.05; **p<0.01).
Table 1: Expression of members of the RET pathway in breast cancer cell lines: qualitative analysis of RET, its ligands and co-receptors in MCF7 and T47D
Genes |
MCF7 |
T47D |
|---|---|---|
RET |
+++ |
+ |
GDNF |
+/- |
- |
NRTN |
++ |
++ |
ARTN |
++ |
+++ |
PSPN |
++ |
+++ |
GFRA1 |
+ |
- |
GFRA2 |
- |
+ |
GFRA3 |
+/- |
+/- |
RET over-expression in MCF7 is due to transcriptional mechanisms
We investigated the mechanisms potentially underlying the different expression of RET in MCF7 and T47D cell lines. First of all, to exclude duplications in MCF7 or deletions in T47D genomes, possibly accounting for their different RET mRNA levels, we quantified the relative RET locus dosage in the two BC cell lines. To this end, we developed a real time qPCR from genomic DNA, designing two amplimers in the 5’ and 3’ end of the RET gene respectively, and setting the threshold for RET amount compared to a reference gene at <0.5 for deletions and >1.5 for duplications. Consistent with known data, the results clearly showed a comparable dose of RET DNA in MCF7 and T47D (Figure 2A). Then, we sought to see whether the difference observed in RET mRNA levels between the two cell lines was due to transcriptional or post-transcriptional mechanisms. To this end, we investigated RET mRNA half-life in the two cell lines. Since preliminary experiments had suggested that RET mRNA is very stable, we decided to quantify RET mRNA amounts at 0, 2, 4 and 6 hours after transcriptional inhibition with DRB treatment. As shown in Figure 2B, RET half-life has turned out to last more than 6 hours in both cell lines, and even longer in the T47D cells that, having less RET transcript than MCF7, and presumably a less stable mRNA, were treated with DRB for up to 12 hours without observing any significant difference in the RET mRNA level (Supplementary Figure S1). These data prompted us to hypothesize that the differences observed in RET mRNA levels in MCF7 and T47D might be due mainly to transcriptional mechanisms.
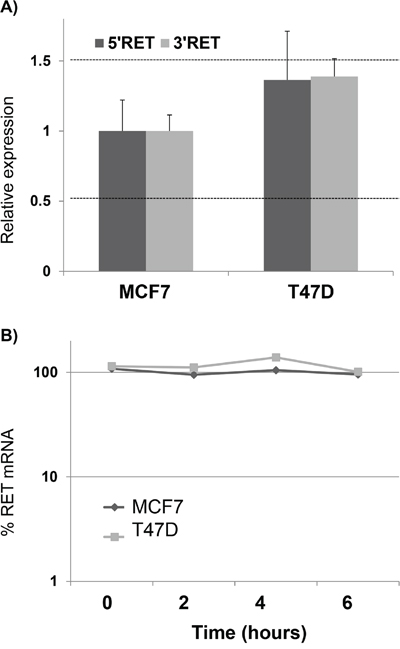
Figure 2: RET over-expression in MCF7 is due to transcriptional mechanisms. A. Quantification of RET genomic content in MCF7 and T47D by using two amplimers designed in the 5' and 3' UTRs respectively. Thresholds to detect DNA deletion or duplication were settled to 0.5 and 1.5 of % genomic DNA compared to a reference genome, respectively. B. Quantification of RET mRNA half-life by DRB pulse chase experiments in MCF7 and T47D. Log-linear regression curves are shown. Values are the mean ±SD of at least two independent experiments performed in triplicate.
Estrogen enhanced expression of RET and Artemin in breast cancer cells
To evaluate whether and how the expression of the RET gene and its co-receptors and ligands depends on estrogen levels in BC cell lines, we treated both MCF7 and T47D with 17-β-estradiol for 6 and 24 hours and performed a realtime PCR for RET, GFRa1, ARTN, and NRTN genes. We found that after 6 hours only RET and ARTN showed statistically significant differences in mRNA levels between treated and untreated samples (Figures 3A and 3B). This is consistent with previous observations showing that not only RET but also ARTN is an estrogen inducible gene and plays a role in BC pathogenesis [35,36,47]. At 6 hours, β-estradiol increased RET expression both in MCF7 and T47D, while its effect disappeared in T47D after 48 hours (Figures 3C and 3D). Interestingly, the amount of RET9 and RET51 mRNA, quantified in the two cell lines after the estrogen treatment, displayed a greater fold induction for RET51 than RET9. These data are in agreement with an already reported different response to estrogens of the two isoforms (Figure 3E) [37]. To confirm the effect of estrogens on RET expression, we compared levels of RET mRNA induced in MCF7 and T47D cell lines by two different cell culture media: standard DMEM and estrogens-depleted DMEM media, this latter lacking of phenol-red. As shown in Figure 3F, estrogens, provided either specifically or by means of standard DMEM medium, did activate RET expression. In order to assess whether RET transcript levels do correlate with ER amount, we then compared RET expression levels with Estrogen Receptor alpha (ESR1) mRNA levels in MCF7 and T47D. After growing cells in either normal DMEM medium or estrogens depleted DMEM medium, no correlation was demonstrated between RET and ESR1 expression (data not shown).
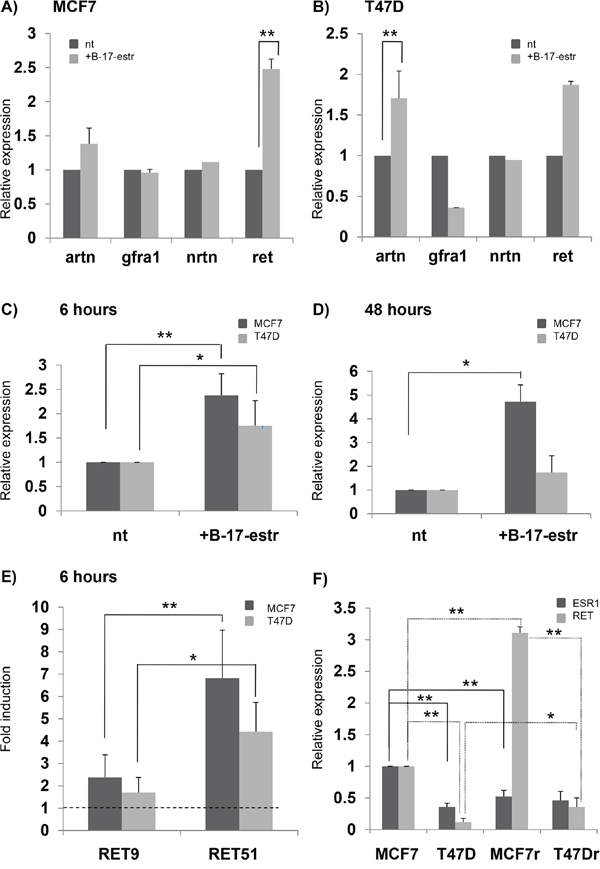
Figure 3: Effect of estrogens on RET-pathway gene expression. mRNA quantification of RET, GFRa1, NRTN and ARTN after 6 hours of 17β-estradiol (B-17-estr) treatment in MCF7 A. and T47D B. RET expression as assessed after 6 hours C. and 48 hours D. of B-17-estr treatment in both the MCF7 and T47D cell lines. E. RET9 and RET51 mRNA fold induction after 6 hours of B-17-estr administration in MCF7 and T47D. F. Levels of RET and estrogen receptor (ESR1) mRNAs induced by either estrogens depleted DMEM media (MCF7 and T47D) or standard DMEM (MCF7r and T47Dr). Values are the mean ±SD of at least two independent experiments performed in triplicate; asterisks (*) indicate statistically significant differences (*p<0.05; **p<0.01).
Inflammatory stimuli which may affect RET transcription
Considering the well-known connection between tumors and immunity, and the involvement of the RET gene in the immune system development and function [48], we treated BC cell lines for 6 and 24 hours with inflammatory stimuli which could modulate RET expression, namely the RET ligand GDNF, IL8, and TNFα. In particular, we tested the expression levels of both ARTN and RET, two genes demonstrated above to have an estrogen-dependent expression and considered to be oncogenic in BC cells. As shown in Figure 4, these treatments induced different effects in MCF7 and T47D cells. A significant decrease in RET expression with all the treatments (GDNF/GRFRA1, IL8, TNFα), was observed at both 6 and 24 hours in MCF7. Conversely, ARTN significantly decreased after 6 hours with GDNF treatment (Figure 4A), and after 24 hours with IL8 and TNFα treatments (Figure 4B). In T47D, RET was down-regulated by IL8, after both 6 and 24 hours of treatment, and by GDNF, after 24 hours. No change in ARTN levels was detected after 6 hours in any condition, while ARTN levels decreased after 24 hours of TNFα treatment (Figures 4C and 4D). Overall these data suggest that IL8, and at lesser extent TNFα, is able to induce a down-regulation of RET expression. In line with previous observations about IL-8 upregulation in the presence of GDNF+GFRα1 in SK-N-MC neuroectodermal tumor cells, stably transfected with the human RET gene, as well as in TT medullary thyroid carcinoma cells and PTC-1 papillary thyroid carcinoma cells [49,50], we sought to verify whether RET could induce IL-8 also in BC cell lines. We found the mRNA levels of IL-8 were significantly increased after 6 hours incubation of MCF7 but not of T47D with GDNF plus GFRα1, while incubation of 24 hours did not induce any change in both these BC cell lines (Supplementary file and Supplementary Figure S2).
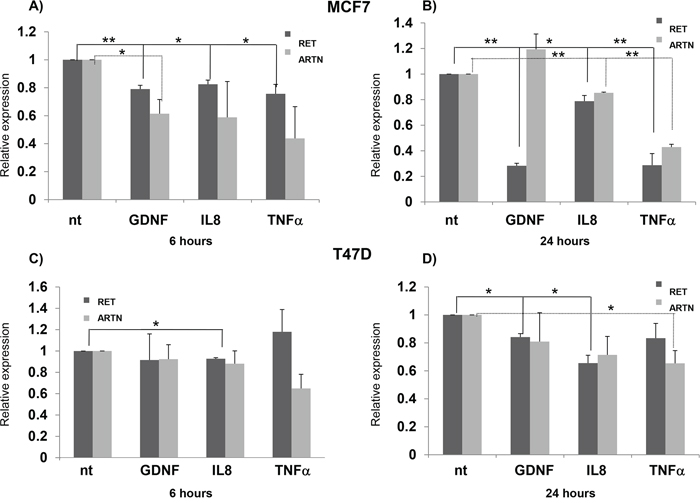
Figure 4: Inflammatory stimuli and RET transcription. RET and ARTN expression in MCF7 cells treated for 6 hours A. and 24 hours B. with GDNF/gfra1, IL8 and TNFalpha. Same experiments repeated in T47D cells are reported in panels C. and D. Values are the mean ±SD of at least two independent experiments performed in triplicate; asterisks (*) indicate statistically significant differences (*p<0.05; **p<0.01).
Chromatin structure and RET expression
In order to verify whether and how RET transcription is modulated by HDAC inhibitors in BC, we treated the above cell lines with Nabut at different concentrations (1, 2 and 5 nM) and investigated, 48 hrs later, the levels of RET and ESR1 mRNA thus induced. As shown in Figure 5A (left panel), in MCF7 we observed a Nabut dose-dependent, statistically significant down-regulation of RET and ESR1 compared to untreated cells, with the trend of the two genes clearly correlated (R=0.847). Surprisingly, T47D cells were not affected by the drug (Figure 5B, left panel). Similar results were observed after treatments with 5-Aza-2'-deoxycytidine (DAC): in MCF7 both RET and ESR1 decreased, while in T47D we observed an increase in RET mRNA levels which is statistically significant (Figures 5A and 5B, right panels). Overall, these data suggest that in our cellular model RET expression is affected by the chromatin structure.
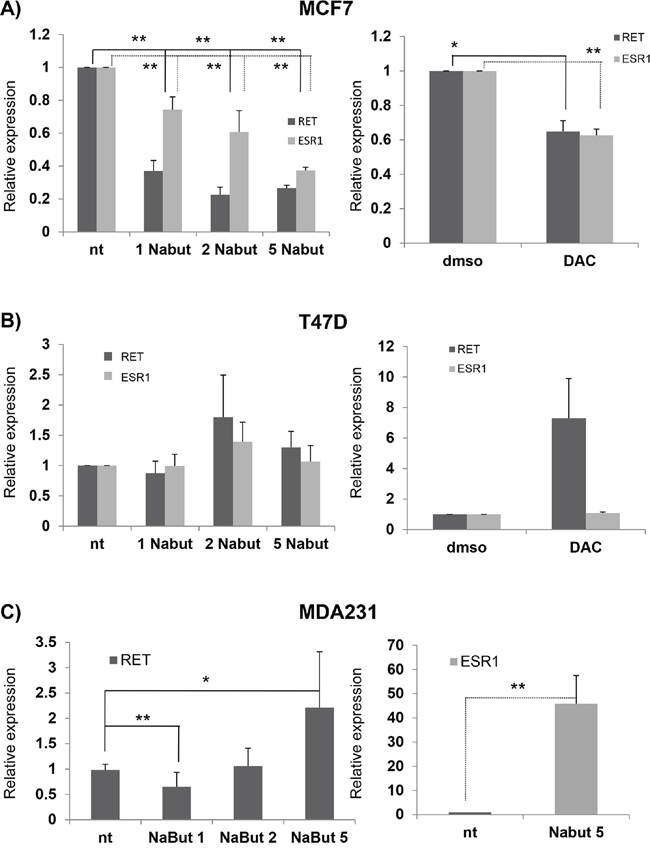
Figure 5: HDAC inhibitors and RET expression in breast cancer cell lines. mRNA expression of RET and ESR1 in MCF7 A. and T47D B. cells after treatment with 1, 2, 5 nM Nabut (left panel, normalised to untreated cells) and 10uM 5-Aza-2'-deoxycytidine (DAC) (right panel, normalised to DMSO treated cells). RET and ESR1 expression levels in MDA231 upon Nabut treatments are reported in the left and right C panels respectively. Values are the mean ±SD of at least two independent experiments performed in triplicate; asterisks (*) indicate statistically significant differences (*p<0.05; **p<0.01).
A similar experiment has already been carried out in the ER- cell line MDA231, physiologically expressing very low levels of RET [36]. After Nabut treatments, and compared to untreated cells, we observed a dose-dependent increase of RET expression (Figure 5C, left panel) while the ESR1 mRNA levels resulted significantly enhanced after Nabut 5nM, but undetectable for lower concentrations (Figure 5C, right panel). In order to identify the treatment most effectively affecting RET expression, we treated both MCF7 and T47D with 17-β-estradiol, Nabut and the two drugs together for 48 hours. These treatments confirmed the opposite effects of the two compounds on MCF7 and T47D (Figure 6). Moreover, when we treated the cells with Nabut and β-estradiol together, we observed a RET expression more comparable to the effect of Nabut alone than β-estradiol in both MCF7 (Figure 6, top panel) and T47D (Figure 6, bottom panel) cell lines, demonstrating the upstream effect of Nabut with respect to estrogens.
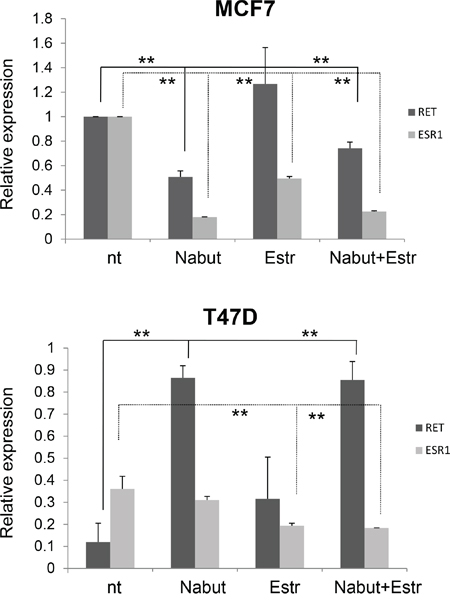
Figure 6: Combined effect of HDAC inhibitors and estrogens. Effect of 48 hours treatment with Nabut and 17-β-estradiol on RET and ERS1 transcript levels in MCF7 (top panel) and T47D (bottom panel) cell lines. A different scale has been used to optimize visualization of the fold change of RET expression in the two cell lines. Values are the mean ±SD of at least two independent experiments performed in triplicate; asterisks(*) indicate statistically significant differences (*p<0.05; **p<0.01).
RET genotyping and statistical analysis and clinical study
The different RET expression modulation assessed in the MCF7 and T47D cell lines in response to Nabut and estrogens may be accounted for by their RET gene sequence, including functional elements lying in the RET regulatory regions. As two ER-responsive enhancers (ERE) have already been located at -50 kb from the transcription start and in intron 6 of the RET gene [35], we looked up the UCSC genome browser and performed a bioinformatic analysis of the RET locus. Results thus obtained and data already known were used to achieve a refined map of ER-sensitive loci at the RET locus (Figure 7). We sequenced genomic DNA samples from MCF7 and T47D cells for all the above mentioned putative ER-responsive regions, as well as a conserved region in intron 1, which is known to act as a RET enhancer during development [24,51]. Only variants at two single nucleotide polymorphisms, rs12247450 and rs2435357, resulted to differ between the two cell lines MCF7 and T47D (rs12247456: AA vs GG; rs2435357: CC vs TT), possibly accounting for their different RET expression patterns. SNP rs2435357 (also known as RET+3 polymorphism) is located at +5 kb inside intron 1 and shown to be associated with reduced expression of the gene, as in vitro assays have demonstrated that the T variant disrupts a SOX10 binding site within a highly conserved sequence (MCS+9.7) thus compromising RET transactivation [23,27]. Consistent with these data, T47D cells carry the genotype associated with lower expression of the RET gene, an observation which might explain both the reduced expression of the RET gene and the remarkable response to Nabut, different from the MCF7 response.
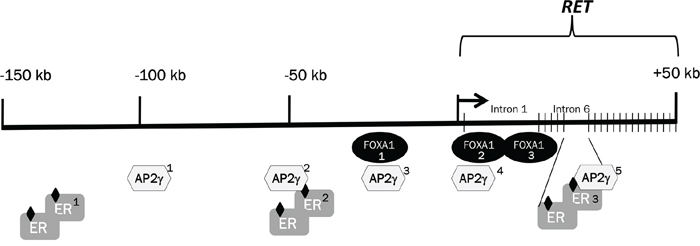
Figure 7: Selected transcription factors and positional map of their binding sites at the RET locus. Schematic representation of the 150 kb upstream of the RET gene followed by the 50 kb of the RET genomic region. The localization of ER and FOXA1 candidate binding regions has been obtained from the UCSC browser, while the five TFAp2C positions were published by Tan et al. [37]
To verify whether the different alleles have effects in vivo, the rs2435357C>T SNP was genotyped in a cohort of 93 ER + BC patients (see Table 2 for clinical description of the patients). Intriguingly enough, the presence of at least one variant allele (CT or TT) was associated with an increased Overall Survival (OS) compared to patients carrying the CC wild type alleles. In particular, patients carrying the CC genotype displayed a median OS of 145.1 months (95% CI: 115.6 - 338.3) while those carrying the CT/TT genotype had a median OS of 180.6 months (95% CI: 159.7 - not estimable). The association was confirmed in multivariate analyses, where nodal status, grading, HER2 status and Ki67 were adjusted for (HR=0.243, 95%; CI=0.088-0.675; P=0,007) (Figure 8). This result is fully consistent with the observation that RET over-expression is associated with poor prognosis in ER+ BC and strongly suggests this SNP as prognostic factor in this subset of patients.
Table 2: Clinical parameters of 93 patients affected by breast cancer
N (%) |
|
|---|---|
Age (years) |
70 |
Adjuvant CT |
44 (47.3) |
Adjuvant HT |
89 (95.7) |
Type of HT (N=89) |
13 (14.6) |
Nodal Status |
50 (53.8) |
HER2 status |
10 (10.8) |
Grading |
32 (34.4) |
Ki-67 |
52 (55.9) |
Abbreviations: CT= Chemotherapy. HT= Hormone therapy. TAM= Tamoxifen. AI= Aromatase Inhibitors
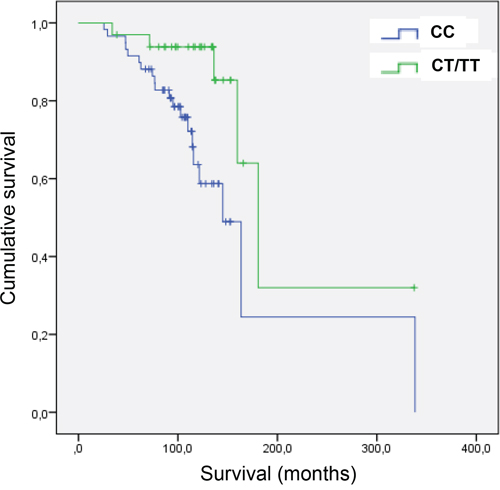
Figure 8: Overall survival (OS) in BC patients carrying the variant allele of SNP rs2435357. The two ROC curves show increased overall survival (OS) in carriers of the variant T allele of SNP rs2435357, among a pilot cohort of 93 ER+ breast cancer patients.
To further deepen into such an association, we sought to verify whether tumors assessed were RET-expressing and to compare RET levels in the breast cancer tissues of the different genotype groups. To this end, we collected 80 paraffin-embedded tumor tissues from BC patients already genotyped at the SNP rs2435357 locus. RNA extraction was followed by real-time PCR for all of them, as reported in the Supplementary Information file. Unfortunately, we could estimate the RET level only in 21 samples, namely those that returned assessable and reliable levels of the beta-2-microglobulin housekeeping, besides RET. Despite this was not enough to draw a genotype-phenotype correlation, we could obtain RET expression data which overall confirmed the expression of this gene in the BC tissues collected (Supplementary Information file and Figure S3).
DISCUSSION
The RET gene is a receptor tyrosine kinase which mediates the activation of intracellular pathways involved in proliferation, migration and differentiation of neural crest derived cells. It is regarded as the major oncogene involved in thyroid cancer, since RET rearrangements are detected in radiation-induced papillary thyroid carcinoma and gain-of-function mutations do segregate in inherited and sporadic medullary thyroid carcinoma [22].
More recently, RET has been involved in other forms of neoplasia including lung, pancreatic and breast cancers, thus demonstrating expanded oncogenic potentialities [22]. In particular, many studies have highlighted the role played by RET in estrogen receptor positive BC and its direct involvement in endocrine resistance [16]. Based on evidence collected so far, it is clear that RET involvement in BC is not due to mutations but to its over-expression. Moreover, RET is an estrogen inducible gene whose activation directly induces the estrogen receptor pathway, which mediates resistance to endocrine treatment. Such an autoregulatory loop is intriguing and has to be taken into consideration for the development of new targeted therapies in BC [45].
After confirming the expression of RET and of its ligands and coreceptors in two BC cell lines, MCF7 and T47D, here we have sought to investigate the possible molecular mechanisms underlying the different RET levels of these cells. We firstly established that the differences observed are mainly due to transcriptional events and not to DNA rearrangements. Given that the mean estimated half life of human transcripts is 3 hrs, and mRNA species with short half-life are enriched among genes with regulatory functions [52], we would have expected RET mRNA to be much more unstable and to start to decay earlier than 6-12 hrs. Therefore, the results obtained in our experiments are enough to conclude that the peculiar RET transcript stability observed in the BC cell lines tested might be the underlying cause of their sustained RET expression level. Thus, we investigated the effect of estrogens on RET expression, confirming that in both cell lines RET is estrogen inducible and interestingly finding that estrogens do modulate the expression of the two gene isoforms, RET9 and RET51. Several studies have highlighted a different role for RET9 and RET51, leading to the conclusion that RET51 activates a specific oncogenic pathway [21]. Here we have shown that, consistently with previous observations, estrogens selectively up-regulate RET51 compared to RET9 [37] thus raising questions, deserving further investigations, about function of the two isoforms.
RET is also known to be expressed in subsets of immune cells and can orchestrate and activate the immune response in the gut [53,54]. In previous studies, we investigated the relationship between RET and the immune system finding that the activation of RET-GDNF pathway specifically modulates the expression of inflammatory cytokines and that RET expression is associated with Interleukin 8 (CXCl8 or IL8) transcript levels [48,55]. Here we report that RET expression in MCF7 and T47D is responsive to treatment with several inflammatory stimuli, besides GDNF/gfra1 [48]. In BC cell lines, treatments with GDNF for 6 and 24 hours significantly reduced the amount of RET (negative loop) in MCF7 and at lesser extent in T47D, as previous studies had already shown [29]. The same reduction was observed in MCF7 after treatment with IL8 and TNFα, and a similar trend was also observed in T47D. These data suggest that RET-expressing BC cells still maintain sensitivity to cytokines, such as IL8 and TNFα, which are physiologically produced by the innate system to contrast tumor development. Moreover, a significant upregulation of IL-8 after 6 hrs of treatment with GDNF/gfra1 in the MCF7 cells could be shown, a circumstance which suggests that RET activation mediates a fast IL-8 production in these cells, giving rise to a loop where RET activation seems to enhance the inflammatory effects of cytokine secretion.
RET over-expression is considered as a negative prognostic factor in ER+ tumors and significantly associated with development of endocrine resistance. Looking for molecules able to modulate RET expression, we focused on deacetylase inhibitors such as Nabut and deAza, previously investigated by us and known to modulate RET expression [39]. Deacetylase inhibitors are known to have different effect on estrogen dependent genes in BC cell lines. In ER- cells this class of compounds reactivates ER and ER-dependent target gene expression. Conversely, in ER+ cells deacetylase inhibitors have the effect of decreasing the expression of estrogen receptor and of its target genes. In our previous work, we demonstrated for the first time that Nabut but not deAza can reactivate RET expression in cells expressing low levels of RET such as lymphoblasts and neuroblastoma SKNMC cells, while it had no effect on cells expressing high levels of RET such as the MTC-TT line [39]. Our present experiments have shown that the two estrogen receptor expressing BC cell lines, MCF7 and T47D, have opposite RET reactions when treated with increasing doses of Nabut. These results have prompted us to characterize the genotype of the two BC cell lines, looking for ERE candidate sequences and known RET enhancers. Interestingly, we have found that T47D cells, which express less RET mRNA than MCF7 cells, are homozygous for the T allele of SNP rs2435357, already described to be associated with a reduced expression of RET [27].
In the light of this observation, we have tested the hypothesis that the presence of the variant allele of SNP rs2435357, assuring low RET levels, can be found in association with either less severe oncogenic phenotypes or a set of more favorable prognostic parameters. Consistent with our expectation, the genetic analysis of 93 ER+ BC patients (4 patients never started adjuvant endocrine therapy) at the SNP rs2435357 (C>T) locus has shown that the presence of at least one variant allele (genotypes CT and TT) is associated with an increased OS compared to patients carrying the CC wild type allele. Such an association has been confirmed in multivariate analyses, where nodal status, grading, HER2 status and Ki67 were adjusted for.
The down-regulation of the RET gene achieved through the variant allele of SNP rs2435357 is clearly the reason for the observed OS increase. The same SNP is known to be associated with Hirschsprung disease (HSCR) through a similar mechanism: lack of adequate levels of RET expression during development does predispose to impaired enteric nervous system and therefore to colonic aganglionosis [18]. Very recently another RET SNP, rs2506030, located about 125 kb upstream of RET, has been reported as the second largest known genetic risk factor in HSCR [56]. These two RET SNPs, rs2435357 and rs2506030, have very low linkage disequilibrium in European-ancestry controls thereby suggesting two independent, and therefore additive, genetic effects at RET. Thus, it may be worthwhile to extend the genetic analysis to this second RET SNP in ER+ BC patients. Moreover, given the increasing number of cancers turned out to be associated with either rearranged or mutated forms of the RET receptor or RET gene over-expression, studies similar to that presented here might be fruitfully carried out in patient sets affected with different RET-related neoplasia.
The association reported here between RET SNP rs2435357 and OS in ER+ BC patients is of utmost importance and fully consistent with the observation that RET over-expression is associated with poor prognosis in ER+ BC and strongly candidate this SNP as prognostic factor in ER+ breast cancer. In addition, our findings suggest that RET and its downstream pathway can be proposed as a therapeutic target in order to improve response to endocrine therapy in selected BC patients. Indeed, targeting the RET tyrosine kinase activity is already an active line of research for the development of therapies against other RET-related tumors such as Medullary Thyroid Carcinoma [57].
MATERIALS AND METHODS
Cell lines and treatments
MCF7 and T47D cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FCS and 1% L-Glutamine 100X, 100U/ml penicillin and 100 μg/ml streptomycin. 17β-estradiol 1nM, Sodium Butyrate (Nabut) 1, 2, 5nM and 5 aza-2’ deoxycytidine (deAza) 10uM were used for treatments. Moreover, cells were also stimulated with either human GDNF and human GFRα1 at the final concentration of 100 ng/ml and 1μg/ml respectively (R&D Systems Minneapolis, MN, USA), TNFα (R&D System Minneapolis, MN, USA) 40 ng/ml, or IL8 (R&D System Minneapolis, MN, USA) 10 ng/ml. Cells were then incubated at 37°C in 5% CO2 and harvested at 6 and 24 hours after treatment.
RNA isolation and Real Time PCR
Total RNA from cells was isolated by a commercial RNA purification kit (RNeasyMini kit, Qiagen, GmbH, Germany) according to the manufacturer’s protocol. One μg of total RNA was reverse transcribed with iScript cDNA Syntesis kit (Bio-Rad Hercules, CA, USA) according to the manufacture’s protocol. Real-time quantitative PCR was performed using inventoried Assays-on-Demand™ provided by Applied Biosystem. Hs01120027_m1 was used to detect the RET gene and Hs99999903_m1 was used to detect the reference gene Beta-Actin. mRNA half-life was detected after treatment with 5,6-Dichloro-1-Beta-D-ribofuranosylbenzimidazole (DRB), an inhibitor of transcription [58]. To study ligands and coreceptors of RET we used Assays-on-Demand™ for GDNF (Hs00181185_m1), NRTN (Hs00177922_m1), ARTN (Hs00365083_m1), PSPN (Hs00358822_g1), GFRα1 (Hs00237133_m1), GFRα2 (Hs00176393_m1), GFRα3 (Hs00181751_m1). PCR reactions were performed using the iQ™5 Real Time thermal cycler (Bio-Rad Hercules, CA, USA). The expression of mRNA was evaluated using the relative Ct method (ΔΔCt) and Real Time PCR amplification was performed in triplicate and repeated at least twice. To detect the two RET9 and RET51 isoforms, the same forward primer 5’-CGT CCA CTC CAT CTG ACT CC-3’ was used together with reverse primers 5’-GAT AGT GCA AAG GGG ACA GC-3’ for RET9 and 5’TAG TGC CAT CAG CTC TCG TG-3’ for RET51. β2microglobulin was used as reference gene (Forward: 5’-AGG ACA AGA AGC CCT GAG CA-3’; Reverse 5’-GCCGTC TTC CCC TCC ATC-3’). The expression of mRNA was evaluated using the relative Ct method (ΔΔCt) and the Pfaffl method when the efficiencies of the amplicons were not similar. Genomic RET content was quantified by using primers in 5' and 3'UTR of the gene described in Griseri et al. [59].
Sequencing and patients analysis
DNA samples from 93 patients affected by ER+ BC were collected in the Laboratory of Cancer Genetics and Translational Oncology, in Cuneo. The study was approved by the Ethical Committee of the Croce & Carle Teaching Hospital in Cuneo, and patients recruited upon signing a specific Informed Consent. Genomic DNA was extracted from peripheral lymphocytes by a standard technique and subjected to RET SNPs screening by means of direct sequencing of the corresponding amplification products. PCR products were purified by ExoSAP IT (GE Healthcare) and directly sequenced using Big Dye v1.1 and a ABI3130 automated sequencer (Applied Biosystems, Foster City, CA, USA). Genetic screening for SNP rs2435357 (C>T) was performed by using primers F: 5’-AGAGGCACCAGGGTCAAAG-3’ and R: 5’-ATGCAAAGGAAACTGCCAAT-3’. Additional PCR details are available upon request.
Overall Survival (OS) was defined as the time between surgery and death, whatever the cause. Observation time of patients alive at the last follow-up was censored. Confidence intervals of median survival times were calculated according to the log–log method of Brookmeyer and Crowley. Hazard ratios and appropriate 95% CIs were calculated by means of the Cox proportional hazard model. A multivariate Cox regression model was fitted to evaluate the independent effect of rs2435357 on OS. Starting from a full model, including all covariates, non significant variables were progressively removed according to a backward stepwise procedure based on the Wald's chi-square test.
ACKNOWLEDGMENTS AND FUNDING
We would like to thank Luca Boni (Istituto Toscano Tumori, ITT) for his assistance and suggestions with statistical matters during the revision. This work has been performed thanks to the following supports: Young Investigator Fellowship awarded by the Fondazione Umberto Veronesi to PG, Italian Association for Cancer Research (GI-13217 to IC), Italian Ministry of Health (“Cinque per mille” and “Ricerca Corrente” to the Gaslini Institute).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69-90.
2. Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast Cancer: Epidemiology and Etiology. Cell Biochem Biophys. 2014; 28.
3. van der Groep P, van der Wall E, van Diest PJ. Pathology of hereditary breast cancer. Cell Oncol (Dordr). 2011; 34: 71-88.
4. Shiovitz S, Korde LA. Genetics of Breast Cancer: A Topic in Evolution. Ann Oncol. 2015; 26: 1291-1299.
5. Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013; 14: 703-718.
6. Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, Turpin E, Plassa LF, de Roquancourt A, Bourstyn E, de Cremoux P, Janin A, Giacchetti S, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013; Suppl 2: S27-29.
7. Goncalves R, Warner WA, Luo J, Ellis MJ. New concepts in breast cancer genomics and genetics. Breast Cancer Res. 2014; 16: 460.
8. Byler S, Goldgar S, Heerboth S, Leary M, Housman G, Moulton K, Sarkar S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014; 34: 1071-1077.
9. Livi L, Palar F, Saieva C, Simontacchi G, Nori J, Sanchez L, Santini R, Mangoni M, Fondelli S, Distante V, Bianchi S, Biti G. Breast cancer in the elderly: treatment of 1500 patients. Breast J. 2006; 12: 353-359.
10. Huang B, Warner M, Gustafsson JA. Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol Cell Endocrinol. 2014; 26.
11. Brufsky AM. Predictive and prognostic value of the 21-gene recurrence score in hormone receptor-positive, node-positive breast cancer. Am J Clin Oncol. 2014; 37: 404-410.
12. Jordan VC. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr Relat Cancer. 2014; 21: R235-246.
13. Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002; 2: 101–112.
14. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009; 9: 631–643.
15. Milani A, Geuna E, Mittica G, Valabrega G. Overcoming endocrine resistance in metastatic breast cancer: Current evidence and future directions. World J Clin Oncol. 2014; 5: 990-1001.
16. Morandi A, Plaza-Menacho I, Isacke CM. RET in breast cancer: functional and therapeutic implications. Trends Mol Med. 2011; 17: 149-157.
17. Airaksinen MS, TitievskyA, Saarma M. GDNF family neurotrophic factor signaling: four masters, one servant? Mol Cell Neurosci. 1999; 13: 313-325.
18. Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005; 16: 441-467.
19. de Graaff E, Srinivas S, Kilkenny C, D'Agati V, Mankoo BS, Costantini F, Pachnis V. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 2001; 15: 2433-2444.
20. Scott RP, Eketjäll S, Aineskog H, Ibáñez CF. Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J Biol Chem. 2005; 280: 13442-13449.
21. Hickey JG, Myers SM, Tian X, Zhu SJ, Shaw JL, Andrew SD, Richardson DS, Brettschneider J, Mulligan LM. RET-mediated gene expression pattern is affected by isoform but not oncogenic mutation. Genes Chromosomes Cancer. 2009; 48: 429-440.
22. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. 2014; 14: 173-186.
23. Griseri P, Bachetti T, Puppo F, Lantieri F, Ravazzolo R, Devoto M, Ceccherini I. A common haplotype at the 5' end of the RET proto-oncogene, overrepresented in Hirschsprung patients, is associated with reduced gene expression. Hum Mutat. 2005; 25: 189-195.
24. Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, Portnoy ME, Cutler DJ, Green ED, Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005; 434: 857-863.
25. Lantieri F, Griseri P, Puppo F, Campus R, Martucciello G, Ravazzolo R, Devoto M, Ceccherini I. Haplotypes of the human RET proto-oncogene associated with Hirschsprung disease in the Italian population derive from a single ancestral combination of alleles. Ann Hum Genet. 2006; 70: 12-26.
26. Borun P, Jerzy S, Ziemnicka K, Kubaszewski L, Lipinski D, Plawski A. Absence of the RET+3:T allele in the MTC patients. Hered Cancer Clin Pract. 2012; 10: 14.
27. Emison ES, Garcia-Barcelo M, Grice EA, Lantieri F, Amiel J, Burzynski G, Fernandez RM, Hao L, Kashuk C, West K, Miao X, Tam PK, Griseri P, et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet. 2010; 87: 60-74.
28. Tozlu S, Girault I, Vacher S, Vendrell J, Andrieu C, Spyratos F, Cohen P, Lidereau R, Bieche I. Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr Relat Cancer. 2006; 13: 1109-1120.
29. Esseghir S, Todd SK, Hunt T, Poulsom R, Plaza-Menacho I, Reis-Filho JS, Isacke CM. A role for glial cell derived neurotrophic factor induced expression by inflammatory cytokines and RET/GFR alpha 1 receptor up-regulation in breast cancer. Cancer Res. 2007; 67: 11732-11741.
30. Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, Wartmann M, Stumm M, Lane HA, Hynes NE. The Ret receptor tyrosine kinase pathway functionally interacts with the ERalpha pathway in breast cancer. Cancer Res. 2008; 68: 3743-3751.
31. Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, Martin LA, Isacke CM. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene. 2010; 29: 4648-4657.
32. Gattelli A, Nalvarte I, Boulay A, Roloff TC, Schreiber M, Carragher N, Macleod KK, Schlederer M, Lienhard S, Kenner L, Torres-Arzayus MI, Hynes NE. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol Med. 2013; 5: 1335-1350.
33. Fiorito E, Katika MR, Hurtado A. Cooperating transcription factors mediate the function of estrogen receptor. Chromosoma. 2013; 122: 1-12.
34. Renoir JM, Marsaud V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol. 2013; 85: 449-465.
35. Stine ZE, McGaughey DM, Bessling SL, Li S, McCallion AS. Steroid hormone modulation of RET through two estrogen responsive enhancers in breast cancer. Hum Mol Genet. 2011; 20: 3746-3756.
36. Wang C, Mayer JA, Mazumdar A, Brown PH. The rearranged during transfection/papillary thyroid carcinoma tyrosine kinase is an estrogen-dependent gene required for the growth of estrogen receptor positive breast cancer cells. Breast Cancer Res Treat. 2012; 133: 487-500.
37. Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, Yong EL, Sung WK, Cheung E. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J. 2011; 30: 2569-2581.
38. Spanheimer PM, Woodfield GW, Cyr AR, Kulak MV, White-Baer LS, Bair TB, Weigel RJ. Expression of the RET proto-oncogene is regulated by TFAP2C in breast cancer independent of the estrogen receptor. Ann Surg Oncol. 2013; 20: 2204-2212.
39. Griseri P, Patrone G, Puppo F, Romeo G, Ravazzolo R, Ceccherini I. Rescue of human RET gene expression by sodium butyrate: a novel powerful tool for molecular studies in Hirschsprung disease. Gut. 2003; 52: 1154-1158.
40. Thomas S, Munster PN. Histone deacetylase inhibitor induced modulation of anti-estrogen therapy. Cancer Lett. 2009; 280: 184-191.
41. Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001; 61: 7025–7029.
42. Fan J, Yin WJ, Lu JS, Wang L, Wu J, Wu F, Di G, Shen Z, Shao ZM. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J Cancer Res Clin Oncol. 2008; 134: 883–890.
43. Travaglini L, Vian L, Billi M, Grignani F, Nervi C. Epigenetic reprogramming of breast cancer cells by valproic acid occurs regardless of estrogen receptor status. Int J Biochem Cell Biol. 2009; 41: 225-234.
44. Yi X, Wei W, Wang SY, Du ZY, Xu YJ, Yu XD. Histone deacetylase inhibitor SAHA induces ERalpha degradation in breast cancer MCF-7 cells by CHIP-mediated ubiquitin pathway and inhibits survival signaling, Biochem. Pharmacol. 2008; 75: 1697–1705.
45. Spanheimer PM, Park JM, Askeland RW, Kulak MV, Woodfield GW, De Andrade JP, Cyr AR, Sugg SL, Thomas A, Weigel RJ. Inhibition of RET increases the efficacy of antiestrogen and is a novel treatment strategy for luminal breast cancer. Clin Cancer Res. 2014; 20: 2115-2125.
46. Morandi A, Martin LA, Gao Q, Pancholi S, Mackay A, Robertson D, Zvelebil M, Dowsett M, Plaza-Menacho I, Isacke CM. GDNF-RET signaling in ER-positive breast cancers is a key determinant of response and resistance to aromatase inhibitors. Cancer Res. 2013; 73: 3783-3795.
47. Kang J, Qian PX, Pandey V, Perry JK, Miller LD, Liu ET, Zhu T, Liu DX, Lobie PE. Artemin is estrogen regulated and mediates antiestrogen resistance in mammary carcinoma. Oncogene. 2010; 29: 3228-3240.
48. Rusmini M, Griseri P, Lantieri F, Matera I, Hudspeth KL, Roberto A, Mikulak J, Avanzini S, Rossi V, Mattioli G, Jasonni V, Ravazzolo R, Pavan WJ, et al. Induction of RET dependent and independent pro-inflammatory programs in human peripheral blood mononuclear cells from Hirschsprung patients. PLoS One. 2013; 8: e59066.
49. Iwahashi N, Murakami H, Nimura Y, Takahashi M. Activation of RET tyrosine kinase regulates interleukin-8 production by multiple signaling pathways. Biochem Biophys Res Commun. 2002; 294: 642-649.
50. Borrello MG, Alberti L, Fischer A, Degl'innocenti D, Ferrario C, Gariboldi M, Marchesi F, Allavena P, Greco A, Collini P, Pilotti S, Cassinelli G, Bressan P, et al. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci U S A. 2005; 102: 14825-14830.
51. Grice EA, Rochelle ES, Green ED, Chakravarti A, McCallion AS. Evaluation of the RET regulatory landscape reveals the biological relevance of a HSCR-implicated enhancer. Hum Mol Genet. 2005; 14: 3837-3845.
52. Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MSH. Database for mRNA Half-Life of 19 977 Genes Obtained by DNA Microarray Analysis of Pluripotent and Differentiating Mouse Embryonic Stem Cells. DNA Research. 2009; 16: 45–58.
53. Veiga-Fernandes H, Coles MC, Foster KE, Patel A, Williams A, Natarajan D, Barlow A, Pachnis V, Kioussis D. Tyrosine kinase receptor RET is a key regulator of Peyer's patch organogenesis. Nature. 2007; 446: 547-551.
54. Patel A, Harker N, Moreira-Santos L, Ferreira M, Alden K, Timmis J, Foster K, Garefalaki A, Pachnis P, Andrews P, Enomoto H, Milbrandt J, Pachnis V, et al. Differential RET signaling pathways drive development of the enteric lymphoid and nervous systems. Sci Signal. 2012; 5: ra55.
55. Rusmini M, Griseri P, Matera I, Pontarini E, Ravazzolo R, Mavilio D, Ceccherini I. Expression variability and function of the RET gene in adult peripheral blood mononuclear cells. J Cell Physiol. 2014; 229: 2027-2037.
56. Jiang Q, Arnold S, Heanue T, Kilambi KP, Doan B, Kapoor A, Ling AY, Sosa MX, Guy M, Jiang Q, Burzynski G, West K, Bessling S, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability. Am J Hum Genet. 2015; 96: 581-596.
57. Maxwell JE, Sherman SK, O'Dorisio TM, Howe JR. Medical management of metastatic medullary thyroid cancer. Cancer. 2014; 120: 3287-301.
58. Griseri P, Bourcier C, Hieblot C, Essafi-Benkhadir K, Chamorey E, Touriol C, Pagès G. A synonymous polymorphism of the Tristetraprolin (TTP) gene, an AU-rich mRNA-binding protein, affects translation efficiency and response to Herceptin treatment in breast cancer patients. Hum Mol Genet. 2011; 20: 4556-4568.
59. Griseri P, Lantieri F, Puppo F, Bachetti T, Di Duca M, Ravazzolo R, Ceccherini I. A common variant located in the 3'UTR of the RET gene is associated with protection from Hirschsprung disease. Hum Mutat. 2007; 28: 168-76.