
Introduction
Drugs that target genomically defined vulnerabilities in human tumors have been effective cancer therapies for decades [1]. The specificity of cancer drugs has progressively improved, from general cytotoxic agents such as nitrogen mustard in the 1940s [2] to natural-product anticancer drugs such as Vinca alkaloids and anthracyclines in the 1960s [3], to specific monoclonal antibodies [4], immunotoxins [5], and small molecules targeting cell surface receptors and growth-promoting signal transduction pathways [6]. Increased specificity has improved patient response rates while reducing the side effects of anticancer treatment. However, the rapid acquisition of resistance to drug treatments remains a substantial challenge to the clinical management of advanced cancers. Resistance to single drugs can be overcome by combinatorial treatment with drugs acting via different mechanisms, but cancer cells often evolve simultaneous resistance to different structurally and functionally unrelated drugs, a phenomenon known as multidrug resistance (MDR) [7, 8]. Resistance to anticancer drugs arises by various mechanisms and especially by the genetic instability of tumor cells driving heterogeneity. While therapies have become more targeted and effective, acquired resistance has remained the principal basis for treatment failure [9, 10].
One common reason for resistance to multiple anticancer drugs is the increased expression of one or more energy-dependent transporters that result in efflux of the drugs from cells [11, 12]. The first identification of a molecular mechanism of multidrug resistance was the identification of an energy-dependent drug efflux pump, known as P-glycoprotein (P-gp) or MDR1, the multidrug transporter [13, 14]. The product of the human MDR1 gene [15] and the products of two different but related mouse genes, Mdr1a and Mdr1b [16, 17], were among the first described members of a large family of ATP-dependent transporters known as the ATP-binding cassette (ABC) family [18]. From the 48 known ABC transporters [19], members of three subfamilies are important for drug efflux from cells: (i) MDR1 P-glycoprotein (ABCB1) from the “B” subfamily, which was the first identified ABC drug efflux transporter and has been the most completely characterized [11]; (ii) several multidrug resistance related protein (MRP) transporters from the “C” subfamily (ABCC1, ABCC2, ABCC3, ABCC4, ABCC5, ABCC10, ABCC11) [20-22] and (iii) ABCG2/BCRP from the “G” subfamily [23].
The SWI/SNF enzymes control gene expression through ATP-dependent remodeling of chromatin. Mammalian SWI/SNF complexes contain mutually exclusive ATPase subunits, either BRM (SMARCA2), or BRG1 (SMARCA4) [24-26]. SWI/SNF complexes containing BRG1 control cell proliferation, cell lineage differentiation and maintain cell pluripotency during early embryonic development [27-33]. A growing body of evidence suggests that BRG1 exhibits both tumor suppressing and tumor promoting functions, depending on the type of cancer [32]. Results published by us and by others demonstrate that the SWI/SNF ATPases BRG1 and BRM are up-regulated in primary breast cancer and are required for cancer cell proliferation in vitro and in vivo [27, 33]. These results suggest that BRG1, as a driver of proliferation, could be a drugable target in certain cancer types. In addition, BRG1 promotes chemoresistance in lung cancer cells [34], where BRG1 wildtype tumors upregulate BRG1 in response to EZH2 inhibitor and become more resistant to TOPOII inhibitor. In pancreatic tumors, BRG1 knockdown effectively reverses chemoresistance to gemcitabine [35].
Breast cancer is the most common cancer in women and one of the leading causes of cancer death for women, with triple negative breast cancer being the most invasive and life threatening [36-39]. Triple negative breast cancer has been shown to be highly glycolytic, metastatic, and chemotherapy resistant; currently there are no standard of care effective targeted therapies to combat triple negative breast cancer. Therefore, both early stage and advanced triple negative breast cancer tumors are treated with predominantly cytotoxic chemotherapy. We previously reported that reduction of BRG1 results in slow proliferation in triple negative breast cancer cells in vitro and in xenografts [33]. We report here that depletion of BRG1 or an inhibitor targeting the BRG1 ATPase domain sensitized triple negative breast cancer cells to chemotherapeutic drugs. BRG1 inhibition prevented chemotherapy drug-mediated induction of genes encoding specific ABC transporter proteins. We conclude that targeting the ATPase domain of BRG1, in combination with other chemotherapy drugs, is a promising strategy for breast cancer treatment.
Results
Breast tumors are heterogeneous with subtypes defined by pathology [40] and gene expression profiles [41]. Since we were studying chemotherapy drug efflux,we chose to focus on the subtype with the most resistance to those drugs, the most treatment failures, and the worst prognosis for patients [36]. These ‘triple negative’ tumors lack estrogen receptor α, lack the progesterone receptor, and do not have HER2 upregulation.
BRG1 depletion sensitized triple negative breast cancer cells to chemotherapy drugs
We tested the efficacy of six chemotherapy drugs on MDA-MB-231 cells expressing doxycyline-inducible shRNA targeting BRG1. Doxorubicin, 5-fluorouracil (5-FU), gemcitabine, cisplatin, cyclophosphamide, and paclitaxel are used clinically for treating breast cancer patients. Doxorubicin induces DNA double-strand breaks. 5-FU and gemcitabine are nucleoside analogs. Cisplatin crosslinks DNA, while cyclophosphamide alkylates and crosslinks DNA. Paclitaxel prevents normal breakdown of microtubules during mitosis. These drugs are structurally unrelated and have different mechanisms of action. Reduction of BRG1 expression significantly improved the efficacy of each of these drugs (Figure 1A-1F, Supplemental Figure 1A) as judged by cell viability in MTS assays [42]. The IC50 values were decreased 4 to 10 fold, supporting the concept that BRG1 reduction or inhibition might be effective as an adjunct to currently used chemotherapies.
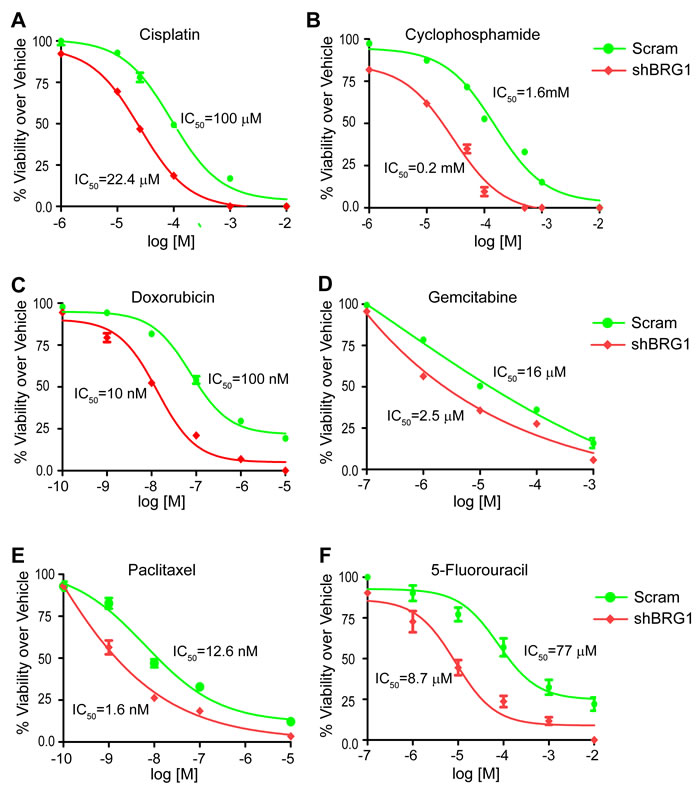
Figure 1: Reduction of BRG1 levels increased the chemosensitivity of triple-negative breast cancer cells. BRG1 knockdown was induced by Doxycycline in MDA-MB-231 cells engineered as previously described [33]. Cells were plated in 96-well plates with increasing doses of cisplatin A., cyclophosphamide B., doxorubicin C., gemcitabine D., paclitaxel E., or 5-fluorouracil F. for 72 hours. Cell viability was measured by MTS assay. Fold change over vehicle treated cells was calculated. IC50 values were determined by nonlinear regression analysis using a log (inhibitor) vs response (three parameters) model in GraphPad Prism 6.0. The results are presented as the mean of three independent experiments performed in triplicate. Error bars are standard deviations.
A BRG1 ATPase domain inhibitor decreased breast cancer cell proliferation
Only two BRG1 inhibitors have been reported. PFI-3, or (2E)-1-(2-hydroxyphenyl)-3-[(1R,4R)-5-(pyridin-2-yl)-2,5-diazabicyclo[2.2.1]heptan-2-yl]prop-2-en-1-one, a small molecule inhibitor that specifically targets the bromo domains of BRG1, BRM, and PB1, is a Pfizer/ Structural Genomics Consortium candidate with in vitro potency in isothermal titration calorimetry at < 100 nM dose (http://www.thesgc.org/chemical-probes/PFI-3; [43, 44]). Previously, we reported that depletion of BRG1 in triple negative breast cancer cell lines reduced cell proliferation [33]. We treated three triple negative breast cancer cell lines, MDA-MB-231, MDA-MB-468, and HDQ-P1, with PFI-3 at different doses. No inhibition of cell proliferation was observed, even at the 10 μM dose as measured by MTT assay [45] (Figure 2A). This is consistent with recent results demonstrating that PFI-3 did not affect the proliferation rate of other cancer cell lines [44].
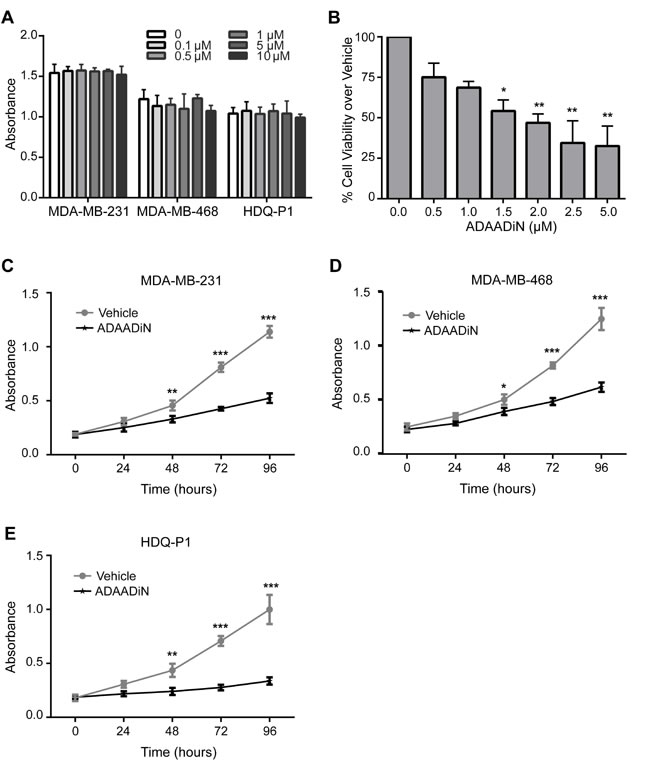
Figure 2: Effects of BRG1 inhibitors on breast cancer cell proliferation and viability. A. Cell proliferation was measured by MTT assay in three triple negative breast cancer cell lines (MDA-MB-231, MDA-MB-468 and HDQ-P1) with increasing doses of PFI-3 treatment for 72 hours. B. Cell viability in MDA-MB-231 cells treated with increasing doses of ADAADi was examined by MTT assay for 48 hours. MTT assays were performed on MDA-MB-231 C., MDA-MB-468 D. and HDQ-P1 E. cells treated with 2 μM ADAADi. Each data point represents the mean of 3 independent experiments performed in triplicate. Error bars are standard deviations. *P < 0.05, **P < 0.01, ***P < 0.001.
The bacterial APH (3’)-III enzyme that encodes for aminoglycoside resistance generates a minor product that can be chromatographically separated from the known 3’-phosphoaminoglycoside product as well as from the parental aminoglycoside. This product, named ADAADi (Active DNA-dependent ATPase A Domain inhibitor), inhibits the ATPase activity of the SWI2/SNF2 family of ATPases [46, 47]. Enzymes from other families of DNA-dependent ATPases showed no or greatly reduced sensitivity to ADAADi, and DNA-independent or RNA-dependent ATPases were not affected [47]. Prior studies indicated that ADAADi inhibited BRG1 nucleosome remodeling activity in vitro [47]. ADAADi derived from different aminoglycosides behaves similarly in all tested assays [46]; here we utilized ADAADi derived from neomycin (ADAADiN). We tested the ADAADiN inhibitor on three triple negative lines: MDA-MB-231, MDA-MB-468 and HDQ-P1. ADAADiN significantly decreased cell proliferation in these cell lines (Figure 2B-2E). However, ADAADiN failed to decrease cell proliferation significantly in cells with reduced BRG1 expression (Figure 3A-3B; Supplemental Figure 1B). This observation strongly suggests that ADAADiN targeted BRG1 in these cells by interfering with its ATPase function.
Brahma (BRM) is highly homologous to BRG1 [25, 48] and can also function as the catalytic subunit of mammalian SWI/SNF enzymes in a manner mutually exclusive of BRG1 [26]. ADAADiN decreased cell proliferation to roughly the same extent as shRNA mediated knockdown of BRM (Supplemental Figure 1 and 2). However, the combination of ADAADiN and shRNA targeting BRM further decreased proliferation in a manner that is statistically significant and additive (Supplemental Figure 2). This finding is in contrast to the results obtained for treatment of cells with a combination of ADAADiN and shRNA targeting BRG1 (Figure 3) and suggests that ADAADiN specifically targets BRG1 in these cells.
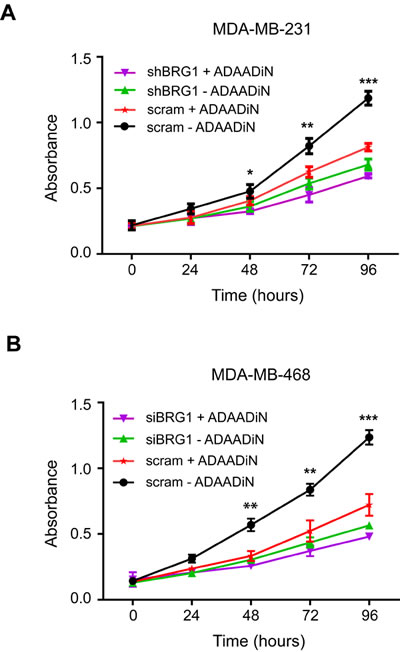
Figure 3: ADAADiN-mediated inhibition of triple negative breast cancer cell proliferation and viability is due to inhibition of BRG1. A. Cell proliferation was measured by MTT assay after ADAADi treatment, inducible BRG1 knockdown, or both in MDA-MB-231 cells. B. Cell proliferation was measured by MTT assay after ADAADi treatment, BRG1 knockdown by siRNA, or both in MDA-MB-468 cells. Each data point is the mean from 3 independent experiments performed in triplicate; error bars are standard deviations. *P < 0.05, **P < 0.01, ***P < 0.001.
ADAADiN treatment increased breast cancer cell sensitivity to chemotherapeutic drugs
Since ADAADiN inhibited breast cancer cell proliferation, we asked if it could also sensitize cells to chemotherapeutic drugs, just as BRG1 knockdown does. Following pretreatment with ADAADiN, cells were exposed to different doses of the same chemotherapy drugs, and cell viability was assayed by MTT assay. ADAADiN significantly increased the chemotherapeutic sensitivity of MDA-MB-231 and MDA-MB-468 cells from 3- to well over 10-fold (Table 1). These data establish the concept that chemical inhibition of the BRG1 ATPase domain might be used to target BRG1 mediated pro-survival pathways in breast cancer cells.
Table 1: ADAADiN significantly increased the sensitivity of MDA-MB-231 and MDA-MB-468 to chemotherapy drugs.
MDA-MB-231 |
MDA-MB-468 |
|||
Vehicle |
ADAADi |
Vehicle |
ADAADi |
|
5-fluorouracil |
16.9 ± 4.6 μM |
4.2 ± 1.1 μM |
27.15 ± 5.45 μM |
5.22 ± 0.63 μM |
Cisplatin |
5.4 ± 0.8 μM |
0.14 ± 0.03 μM |
1.94 ± 0.27 μM |
0.37 ± 0.08 μM |
Cyclophosphamide |
1.45 ± 0.15 mM |
0.061± 0.005 mM |
0.4 ± 0.02 mM |
0.028 ± 0.003 mM |
Doxorubicin |
68.13 ± 5.11 nM |
7.16 ± 1.23 nM |
60.3± 4.9 nM |
8.12± 3.7 nM |
Gemcitabine |
166 ± 10.8 nM |
59.9 ± 7.2 nM |
2.21 ± 0.33 μM |
0.42 ± 0.015 μM |
Paclitaxel |
29.25 ± 7.49 nM |
1.35 ± 0.52 nM |
20.55 ± 1.95 nM |
1.21 ± 0.19 nM |
Shown are IC50 doses for each drug and cell line.
ADAADiN blocked induction of drug transporter gene expression in response to drug treatment
ABC transporters mediate the efflux of anti-cancer drugs and are critically involved in multidrug resistance [21, 49-51]; the expression of ABC transporters is up-regulated in patients after neoadjuvant therapy [52]. We first surveyed nine ABC transporter genes to determine whether BRG1 contributed to their expression in MDA-MB-231 cells. The results show that BRG1 contributed to the endogenous level of transporter gene expression for seven of the genes (Supplemental Figure 3).
Since ADAADiN sensitized breast cancer cells to chemotherapeutic drugs, we hypothesized that ADAADiN treatment might inhibit the transcriptional activation of the transporter genes upon chemotherapy drug treatment. From the literature, we identified six instances where ABC transporter genes are transcriptionally activated in response to one or more of the chemotherapeutic drugs used in our study. Each of the triple negative breast cancer cell lines were treated with vehicle alone or with one of the chemotherapy drugs at the IC50, dose and specific transporter mRNA levels were compared to levels present in cells exposed to drug plus ADAADiN.
ABCC11 was previously identified as a 5-FU efflux transporter that directly confers resistance to 5-FU [53, 54]. We observed up-regulation of ABCC11 expression in each of the three triple negative cell lines when treated with 5-FU, and this activation was attenuated in the presence of ADAADiN (Figure 4A).
Minegaki et al [55] reported that ABCC2 mRNA levels increased in a dose-dependent manner when treated with 5-FU. ABCC2 expression was increased more than 2-fold in all three cell lines when treated with an IC50 dose of 5-FU. When co-treated with ADAADiN, ABCC2 mRNA levels were significantly decreased (Figure 4B). ABCC2 also mediates cisplatin resistance and this is correlated with clinical outcome [56, 57]. In our study, cisplatin up-regulated ABCC2 expression by 4-fold in MDA-MB-468 cells. Activation in MDA-MB-231 and HDQ-P1 cells was less than 2-fold but was nevertheless statistically significant. However, the up-regulation of ABCC2 in each of the three cell lines was inhibited by co-treatment with ADAADiN (Figure 4C).
The expression of ABCG1 is increased in breast cancer patients during neoadjuvant therapy with 5-fluorouracil-doxorubicin-cyclophosphamide, and increased levels of ABCG1 predict poor prognosis [58]. ABCG1 expression increased in each of the cell lines when treated with cyclophosphamide, while the presence of ADAADiN suppressed ABCG1 up-regulation (Figure 4D).
Targeting ABCC2 by antisense RNA reduces the doxorubicin IC50 value by 12-fold in hepatocellular carcinoma cells, whereas overexpression of ABCC2 in HEK293 cells enhances doxorubicin resistance around 8-fold [56, 59]. These results suggest that ABCC2 is the efflux transporter for doxorubicin. When the three triple negative breast cancer lines were treated with the IC50 dose of doxorubicin, it caused a greater than 3-fold induction of ABCC2 in MDA-MB-231 and HDQ-P1 cells, and a modest but statistically significant increase in MDA-MB-468 cells. The addition of ADAADiN blocked ABCC2 induction in each cell line (Figure 4E).
ABCB1 is overexpressed in gemcitabine resistant pancreatic cells and in side population cells with high gemcitabine efflux capacity [60, 61]. In non-small-cell lung cancer cells, ABCB1 mRNA levels can predict gemcitabine chemosensitivity [62]. Homology modeling and docking of ABCB1 showed gemcitabine to be a high-affinity substrate [63]. In the three triple negative lines tested, ABCB1 was strongly induced by gemcitabine treatment, but ADAADiN treatment effectively blocked its induction (Figure 4F).
EGFR-mediated overexpression of ABCG2 is associated with paclitaxel resistance in drug resistant MCF-7 cells [64]. Silencing ABCB1 and ABCG2 by nanoparticle-facilitated siRNA in MCF-7 cells increases chemosensitivity to paclitaxel [65, 66]. As shown in Figure 4G-4H, the expression of ABCB1 and ABCG2 was up-regulated in each of the three cell lines treated with paclitaxel. However, the presence of ADAADiN significantly blocked the activation of these genes. In summary, ADAADiN treatment blocked the transcriptional induction of drug transporters by chemotherapeutic drugs, which may contribute to the increase chemosensivity in ADAADiN treated cells.
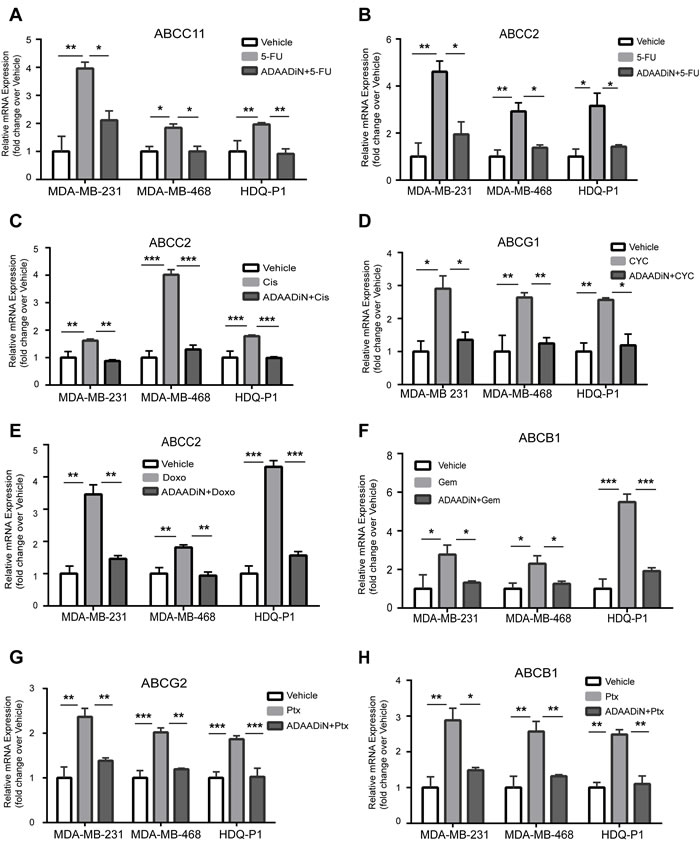
Figure 4: ADAADiN blocked drug-induced ABC transporter gene expression in triple negative breast cancer cell lines. Bar graphs present relative mRNA expression of specific ABC transporter genes in response to vehicle, the indicated chemotherapeutic drug, and the indicated chemotherapeutic drug in the presence of ADAADiN in each of the indicated breast cancer cell lines. Each bar presents the mean of 3 independent experiments performed in duplicate; error bars are standard deviations. *P < 0.05, **P < 0.01, ***P < 0.001. A.-B. 5-FU mediated induction of ABCC11 and ABCC2. C. Cisplatin (Cis) mediated induction of ABCC2. D. Cyclophosphamide (CYC) mediated induction of ABCG1. E. Doxorubicin (Doxo) mediated induction of ABCC2. F. Gemcitabine (Gem) mediated induction of ABCB1. G.-H. Paclitaxel (Ptx) mediated induction of ABCG2 and ABCB1.
Targeting BRG1 led to an increase in drug retention
Altering drug efflux transporter expression might change the intracellular concentration of the chemotherapeutic drugs. Because of the limited availability of radiolabeled versions of the drugs used in this study, we used only 14C-5-FU and 3H-paclitaxel to test this hypothesis. The uptake of the radiolabeled drugs was identical in cell cultures expressing control shRNA or shRNA targeting BRG1 (Figure 5A-5B). Subsequent analysis of cell cultures that were pulse-chased with the drugs showed that BRG1 knockdown resulted in increased intracellular retention of the drugs (Figure 5C-5D). We conclude that BRG1-dependent induction of drug transporter gene expression by 5-FU and Paclitaxel results in increased intracellular concentrations of the drugs. This may explain the increased chemosensitivity of cells that express reduced levels of BRG1 or that are treated with a BRG1 inhibitor.
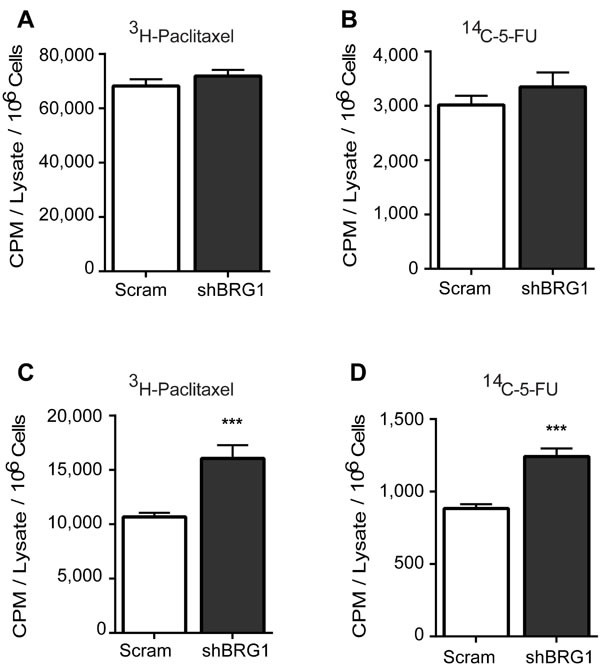
Figure 5: Targeting BRG1 results in increased retention of chemotherapeutic drugs. Radiolabeled paclitaxel or 5-FU were incubated with MDA-MB-231 cells expressing a control (Scram) shRNA or shRNA targeting BRG1. A.-B. Cellular uptake was determined by scintillation counting after harvested cells were washed repeatedly, counted, and lysed. Results were normalized by cell count. C.-D. Drug retention was determined by scintillation counting after cells were pulse-chased and prepared as described in Materials and Methods. Each bar presents the mean of 3 independent experiments performed in triplicate; error bars are standard deviations. ***P < 0.001.
Drug treatment increased BRG1 binding at drug transporter promoters
SWI/SNF enzymes regulate gene expression by altering chromatin structure, and BRG1 binds to chromatin at many genes that are actively transcribed [30, 32]. We asked whether BRG1 is directly involved in the drug-induced transcriptional activation of the tested transporters. Binding of BRG1 at transporter genes was examined by chromatin immunoprecipitation (ChIP) in MDA-MB-231 cells treated with vehicle or with individual chemotherapeutic drugs at the IC50 dose, and these results were compared to results from cells treated with ADAADiN prior to and during induction with the chemotherapeutic drug. BRG1 binding sites at transporter genes promoters were predicted from BRG1 ChIP-seq data deposited for HeLa and K562 cells [67, 68]. 5-FU treatment enhanced BRG1 binding at ABCC2 by 3-fold and ABCC11 by 2-fold, and ADAADiN had no effect on BRG1 binding at these genes (Figure 6A). This result is consistent with the idea that ADAADiN inhibits ATPase activity but has no effect on the ability of the enzyme to bind to chromatin [46]. Cisplatin increased BRG1 enrichment at ABCC2 more than 3-fold, and co-treatment with ADAADiN did not change BRG1 binding (Figure 6B). Similar results were seen in cyclophosphamide, doxorubicine, gemcitabine and paclitaxel treated cells, where these drugs significantly increased BRG1 binding at target drug transporter genes and ADAADiN showed no effect on BRG1 binding (Figure 6C-6F). We were unable to examine BRG1 binding at the ABCG2 locus in paclitaxel treated cells, as there were no BRG1 binding sites identified at this locus in reported ChIP-seq data sets in different cell contexts. Overall, chemotherapeutic drugs increased BRG1 binding to drug transporter genes, and these binding events were not affected by the ADAADiN BRG1 ATPase inhibitor.
PFI-3, a compound targeting the bromodomain of BRG1, BRM, and PB1 [44] had no effect on proliferation of any of the breast cancer cell lines tested (Figure 2A). This is consistent with recent work of others using different cancer cell lines [44] showing that PFI-3 was unable to dislodge BRM from chromatin, either globally or at specific gene loci. We performed ChIP assays to address whether BRG1 remained bound in the presence of PFI-3. Eight transporter genes were examined for the binding of BRG1 in proliferating MDA-MB-231 cells. Four of the eight transporter genes tested had BRG1 binding. PFI-3 had no effect on the extent of BRG1 binding observed at any of the loci (Supplemental Figure 4). These data are consistent with the prior observation that PFI-3 could not displace BRM from chromatin [44] and extend those findings by showing that PFI-3 also cannot dislodge BRG1 from chromatin.
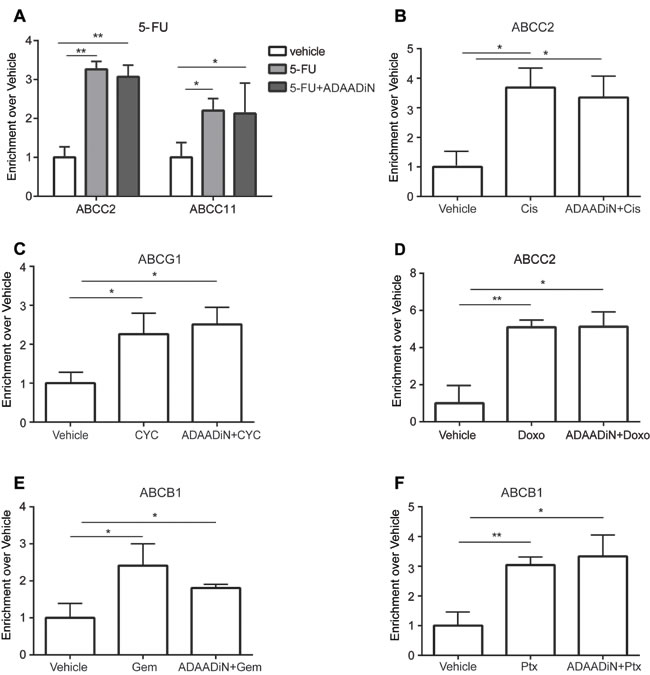
Figure 6: Chemotherapeutic drugs increased BRG1 binding at ABC transporter genes in a manner independent of ADAADiN. ChIP was performed in MDA-MB-231 cells treated with vehicle, chemotherapy drug alone or chemotherapy drug in combination with ADAADi. BRG1 binding at transporter genes was measured by quantitative PCR using primers listed in Supp. Table 2. The bar graphs represent the ratio (enrichment) of BRG1 binding to sequences near the indicated transporter gene in cells treated with the indicated chemotherapeutic drug alone or in combination with ADAADi relative to binding in vehicle treated cells. Each bar presents the mean of 3 independent experiments performed in duplicate; error bars are standard deviations. *P < 0.05, **P < 0.01, ***P < 0.001. A. BRG1 binding at the ABCC2 and ABCC11 genes was analyzed in vehicle, 5-FU and ADAADi plus 5-FU treated cells. B. BRG1 binding at ABCC2 was analyzed in vehicle, cisplatin (Cis) and ADAADi plus Cis treated cells. C. BRG1 binding at ABCG1 was analyzed in vehicle, cyclophosphamide (CYC) and ADAADi plus CYC treated cells. D. BRG1 binding at ABCC2 was analyzed in vehicle, Doxorubicin (Doxo) and ADAADi plus Doxo treated cells. E. BRG1 binding at ABCB1 was analyzed in vehicle, gemcitabine (Gem) and ADAADi plus Gem treated cells. F. BRG1 binding at ABCB1 was analyzed in vehicle, paclitaxel (Ptx) and ADAADi plus Ptx treated cells.
BRG1 expression level correlated with breast cancer patient survival
To determine the correlation between BRG1 expression and breast cancer patient survival, we retrieved 7 microarray datasets of human breast cancer (GSE1456, GSE2034, GSE2990, GSE3494, GSE12093, GSE11121 and Chin et al., combined N = 1339) profiled on Affymetrix HG-U133A platform [69-75] from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) and European Bioinformatics Institute (http://www.ebi.ac.uk/arrayexpress for Chin et al, 2006 [75]). We normalized these datasets using MAS 5.0 software in GeneSpring 12.0 (Agilent Technologies, Santa Clara, CA), scaled to a mean target intensity of 600, and log2 transformed. 88 samples from GSE2990 were also used in GSE3494, and thus excluded from GSE2990. 23 samples without survival information and 7 samples with distance metastasis-free survival of 0 were excluded, which left 1221 non-redundant samples for survival analysis. There was a negative correlation between BRG1 expression level and patient prognosis, in which high levels of BRG1 were associated with poor prognosis (Figure 7). This result provides independent corroboration of a prior study [27] linking BRG1 levels with poor clinical outcomes of breast cancer patients. That conclusion, like our analysis of patient data here, was not limited to triple negative tumors, which make up only about 15% of patient tumors. Future studies will need to determine whether specific breast tumor subtypes have a negative BRG1 level correlation with prognosis.
Our results show that BRG1 knockdown or inhibition increases chemosensitivity and decreases drug-induced increases in ABC transporter gene expression. This suggests that BRG1 overexpression that is normally observed in primary breast tumors [27, 33] may result in elevated ABC transporter gene expression and possible chemoresistance. We might therefore expect that transporter gene expression would correlate with BRG1 expression. The microarray datasets used for correlating high BRG1 expression with decreased survival were interrogated for expression levels of the transporter genes that were stimulated by chemotherapeutic drugs in a BRG1-dependent manner (Figure 4). Three of these genes (ABCB1, ABCC2, ABCG2) showed increased levels of expression that correlated with BRG1 expression, whereas one of the genes (ABCG1) showed an inverse correlation (Supplemental Figure 5). The fifth gene, ABCC11, was not present in the microarray datasets. It is important to remember, however, that levels of specific transporter gene expression were generated from the combined dataset representing patients with a spectrum of breast cancers and that the majority of samples were isolated upon biopsy prior to any treatment. Nevertheless, this analysis provides support for the idea that ABC transporter gene expression is linked to BRG1 expression.
Discussion
Small molecule inhibition of the BRG1 ATPase domain is a promising therapeutic strategy
Previous work by us and others indicates that BRG1 is overexpressed in most breast tumors regardless of classification and that BRG1 knockdown in triple negative breast cancer cells caused a slow proliferation phenotype [27, 33]. Here we report that BRG1 knockdown cells have increased sensitivity to chemotherapeutic drugs currently used to treat breast cancer (Figure 1), suggesting that targeting BRG1 may be a viable approach to augmenting current therapeutic regimens.
Delivery to and continued expression of knockdown vectors in most tissues and specifically in tumors presents significant challenges [76]. Identification of small molecule inhibitors, in contrast, has been an effective therapeutic approach for decades. PFI-3 is a cell-permeable small molecule inhibitor that specifically targets the bromodomains of BRG1, BRM, and a third mammalian SWI/SNF subunit, Polybromo (BAF180) via tight interaction [44, 77]. Bromodomains bind to acetylated chromatin and therefore have a targetable molecular function [78-80]. A recent study showed that PFI-3 treated embryonic stem cells lost stemness and deregulated lineage specification. Exposure of trophoblast stem cells to PFI-3 markedly enhanced differentiation [43]. These results emphasize a key function of the BRG1 bromodomain in stem cell maintenance and differentiation. However, PFI-3, unlike BRG1 knockdown, did not change the proliferation rate of triple negative breast cancer cells at any concentration (Figure 2A), nor did it displace BRG1 from chromatin at specific gene loci (Supplemental Figure 4). These results are consistent with a recent study indicating that PFI-3 failed to alter proliferation in multiple tumor cell types in which BRG1 or BRM was mutated or deficient. In that report, the drug was unable to displace endogenous BRM from chromatin, suggesting at least one reason for its lack of effect [44]. The data are also consistent with studies showing that the C-terminal portion of BRG1, which includes the bromodomain, was dispensable for glucocorticoid receptor-mediated gene induction [81] and that the BRG1 ATPase domain, not the bromo domain, is required for leukemia cell proliferation [82].
In contrast, ADAADiN [46, 47] targets the BRG1 ATPase domain and in our study decreased breast cancer cell proliferation (Figure 2B-2E). The specificity of ADAADi for BRG1 in the cells tested was demonstrated by two experiments. First, the combination of ADAADiN treatment and BRG1 knockdown resulted in an inhibition of cell proliferation that was not statistically different from either treatment alone. Second, whereas knockdown of the BRG1 homologue BRM and ADAADiN treatment provided similar inhibition of breast cancer cell proliferation, the combination of the two resulted in an apparent additive effect on proliferation. This suggests that ADAADiN does not or minimally targets BRM in these cells. Thus, one or more molecules that could simultaneously target BRG1 and BRM might have greater potential as a therapeutic agent. Furthermore, ADAADiN treatment, like BRG1 knockdown, increased the sensitivity of triple negative breast cancer cells to chemotherapeutic drugs that are used clinically (Table 1). These results are consistent with the observation that the ATPase domain of BRG1 is required for GR-mediated transactivation [83] and leukemia cell proliferation [82]. In addition, though the chemotherapeutic agents used in this study have diverse mechanisms of action, none directly target an epigenetic regulator, suggesting that combining epigenetic therapies with conventional chemotherapies has great potential in combinatorial drug approaches to treating cancer. Future work will be needed to further explore this possibility. Xenografts and animal models could be used to probe the efficacy of BRG1 inhibitors. Both in vitro and in vivo approaches to understanding differences in BRG1 functions in normal compared to cancer cells will help inform attempts to improve screening or design of new inhibitors.
The work presented here supports for the idea that targeting BRG1 in breast cancer and in other cancers such as melanoma and colorectal, gastric, and prostate cancer [84-88] could be therapeutic via mechanisms that reduce cell proliferation and increase chemosensitivity. Coupled with recent proposals on targeting the BRG1 homologue BRM in BRG1-deficient tumors [89-92], it is apparent that strategies for targeting the human SWI/SNF enzyme ATPases in context-dependent manners will be an expanding area of focus. Indeed, these advances are only part of a larger movement demonstrating that broad classes of epigenetic regulatory proteins are viable targets for novel cancer therapies [93].
Regulation of ABC transporter genes by BRG1
Reduction of BRG1 levels or interference with BRG1 catalytic function reduces breast cancer cell proliferation, and yet these slow proliferating cells are more sensitive to cytotoxic agents that preferentially target rapidly growing cells. This paradox prompted us to investigate BRG1 effects on the expression of transporters responsible for drug trafficking in cancer cells. Our data show that BRG1 is a regulator of ABC transporters that are implicated as efflux transporters for chemotherapy drugs [94]. ADAADiN inhibited drug-mediated up-regulation of specific transporter genes, indicating a functional role for BRG1. Demonstration that BRG1 was bound to sequences near each transporter gene’s transcription start site indicates a direct role for BRG1 during therapeutic drug mediated gene activation. Together these data suggest a possible mechanism for the increased sensitivity of breast cancer cells to chemotherapeutic drugs.
It has been shown that more than half of the ABC transporters are involved in drug resistance using the NCI60 cell line panel [95]. This redundancy in transporter function has limited therapeutic approaches that target specific transporters. For example, MDR1 inhibitors such as zosuquidar and tariquidar failed in clinical trials despite their high potency and specificity [96]. Our discovery that catalytic activity of BRG1 is required for the up-regulation of multiple ABC transporters in response to drug treatment pioneers a new pan-transporter approach to combating drug resistance by targeting BRG1.
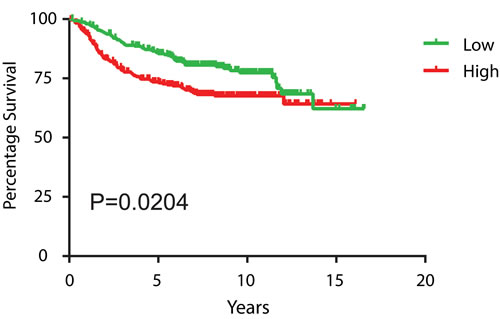
Figure 7: High BRG1 expression levels in breast tumors predicts poor patient prognosis. Kaplan-Meier curves of distance metastasis free survival of human breast cancer patients from 7 combined datasets were plotted according to the level of BRG1 expression, with the 1st quartile having the lowest BRG1 expression designated as “low” and 4th quartile having the highest BRG1 expression designated as “high”. The log-rank test was used for statistical analysis.
Materials and Methods
Cell culture
MDA-MB-231 cells were obtained from T. Guise [97]. MDA-MB-468 cells were obtained from ATCC. HDQ-P1 cells were purchased from DSMZ (Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Culture, 38124 Braunschweig, Germany. MDA-MB-231, MDA-MB-468 and HDQ-P1 cells were maintained in DMEM supplemented with 10% FBS and Penicillin/Streptomycin. BRG1 knockdown by doxycycline-inducible shRNA expression in MDA-MB-231 cells was performed as previously described [33]. siRNA mediated knockdown of BRG1 in MDA-MB-468 and HDQ-P1 cells was performed using reagents and methods previously described [33, 98]. The identities of all four triple negative breast tumor lines were authenticated by Short Tandem Repeat profiling at the Genetic Resources Core Facility, Johns Hopkins School of Medicine, Institute of Genetic Medicine.
Reagents
5-FU, cyclophosphamide, gemcitabine, doxorubicin, cisplatin, paclitaxel, and 3-(4,5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO). 3H-Paclitaxel and 14C-5-Fluorouracil were purchased from Moravek Biochemicals (Brea, CA). PFI-3 was purchased from Xcess Biosciences, Inc (San Diego, CA). ADAADiNN was prepared from neomycin as previously described [46]. CellTiter 96 AQueous One Solution was purchased from Promega (Madison, WI).
Drug treatment
Cells were plated and incubated overnight before treatment with increasing doses of drugs for 72 hours to determine the IC50 value. When combined with ADAADiN treatment, cells were pre-treated with 2 μM ADAADiN for 48 hours and then different drugs were added to culture medium at the IC50 value incubated for another 24 hours and collected for analysis.
Drug uptake and retention studies
MDA-MB-231 scram and shBRG1 cells were treated with doxycycline to induce BRG1 knockdown as described previously [33]. Cells were then treated with 0.1 μCi 3H-Paclitaxel or 14C-5-Fluorouracil for 1 hour or 6 hours, respectively. Uptake of radiolabeled drug was measured after washing the cells repeatedly, cell counting, and scintillation counting. For assessing drug retention, labeled cells were washed 3 times with PBS to remove residual labeling medium and replaced with growth medium containing doxycycline and 100 μM paclitaxel or 1 mM 5-FU for an additional 2 hours before harvest. All cells, including any floating cells, were collected, counted and lysed by addition of 0.5 N NaOH. Cell lysates were analyzed by scintillation counting. Readouts were normalized by cell number.
MTS assay
Cells were seeded in 96-well plates (5,000 cells/well) overnight prior to drug treatment, and were then maintained in the presence of vehicle or drug for 72 hours before addition of 20 μL CellTiter 96 AQueous One Solution per well. Plates were incubated for 2 hours before absorbances at 490 nm were measured with a Synergy H4 Hybrid microplate reader (Bio Tek, Winooski, VT).
MTT assay
Cells were seeded in 96-well plates (5,000 cells/well) overnight prior to drug treatment, and were then maintained in presence of vehicle or drug for 72 hours before addition of MTT solution (5 ug/mL). Plates were incubated for 4 hours in MTT solution, then the media was removed and plates were air-dried. One hundred microliters of DMSO were added to each well and incubated at room temperature for 30 minutes with gentle shaking before the absorbances were measured as described above.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously [99] with the following modifications: cells were cooled to room temperature before being cross-linked with ice-cold growth medium containing 3.7% formaldehyde for 40 min at 4 oC. 50 μg of chromatin extract was used for each ChIP with 10 μL of BRG1 antisera or normal rabbit IgG (Millipore, Billerica, MA). BRG1 binding at sequences at transporter genes was measured by real time qPCR using primers listed in Supp. Table. 1.
RT-qPCR
Total RNA was extracted from one million cells using RNeasy Plus following manufacturer’s instruction (Qiagen Inc., Valencia, CA). cDNA was synthesized using SuperScript III kit (Invitrogen, San Diego, CA). Gene expression was measured by real time qPCR on a StepOne Plus realtime PCR System (Applied BioSystems, Grand Island, NY) using the primers listed in Supp. Table 2. Relative gene expression was normalized to beta actin in each sample in experiments comparing vehicle vs drug treatment. In experiments comparing scram and shBRG1 samples, expression was calculated relative to 45S pre-rRNA.
Survival analysis
Kaplan-Meier survival curves were plotted using GraphPad Prism 5.0 software and statistical significance was analyzed using the log-rank test.
Statistical analyses
All quantitative data points represent the mean of three independent experiments performed in duplicates or triplicates with standard deviation (S.D). Unless indicated, statistical analysis was performed using GraphPad Instat two-tail P value student test (Graphpad Software, Inc., La Jolla, CA). The significance of the correlation between BRG1 expression and the expression of each of the transporter genes was determined by calculating the Pearson coefficient. The lines present in the graphs shown in Supplemental Figure 5 were determined by linear regression.
Acknowledgments
We thank Tara Smith for a critical reading of the manuscript.
Grant Support
This work was supported by NIH grants P01 CA82834, R21 CA185926, and R01 EB014869.
Conflicts of Interest
No conflicts.
References
1. Barinaga M. From bench top to bedside. Science. 1997; 278:1036-1039.
2. Gilman A and Philips FS. The biological actions and therapeutic applications of the B-chloroethyl amines and sulfides. Science. 1946; 103:409-415.
3. Cutts JH, Beer CT and Noble RL. Biological properties of Vincaleukoblastine, an alkaloid in Vinca rosea Linn, with reference to its antitumor action. Cancer Res. 1960; 20:1023-1031.
4. Nabholtz JM and Slamon D. New adjuvant strategies for breast cancer: meeting the challenge of integrating chemotherapy and trastuzumab (Herceptin). Semin Oncol. 2001; 28:1-12.
5. Pastan II and Kreitman RJ. Immunotoxins for targeted cancer therapy. Adv Drug Deliv Rev. 1998; 31:53-88.
6. Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R and Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001; 344:1038-1042.
7. Persidis A. Cancer multidrug resistance. Nat Biotechnol. 1999; 17:94-95.
8. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C and Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006; 5:219-234.
9. Garraway LA and Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012; 2:214-226.
10. Lackner MR, Wilson TR and Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012; 8:999-1014.
11. Gottesman MM and Ling V. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett. 2006; 580:998-1009.
12. Holohan C, Van Schaeybroeck S, Longley DB and Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013; 13:714-726.
13. Juliano RL and Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976; 455:152-162.
14. Ueda K, Cardarelli C, Gottesman MM and Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci U S A. 1987; 84:3004-3008.
15. Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM and Roninson IB. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986; 47:381-389.
16. Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P and Housman DE. The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol. 1989; 9:1346-1350.
17. Lothstein L, Hsu SI, Horwitz SB and Greenberger LM. Alternate overexpression of two P-glycoprotein [corrected] genes is associated with changes in multidrug resistance in a J774.2 cell line. J Biol Chem. 1989; 264:16054-16058.
18. Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992; 8:67-113.
19. Moitra K and Dean M. Evolution of ABC transporters by gene duplication and their role in human disease. Biol Chem. 2011; 392:29-37.
20. Keppler D. (2011). Multidrug Resistance Proteins (MRPs, ABCCs): Importance for Pathophysiology and Drug Therapy. In: Fromm MF and Kim RB, eds. Drug Transporters: Springer Berlin Heidelberg), pp. 299-323.
21. Nies AT and Lang T. (2014). Multidrug Resistance Proteins of the ABCC Subfamily. In: You G and Morris ME, eds. Drug Transporters: Molecular characterization and role in drug disposition. (Hoboken, NJ: John Wiley & Sons, Inc.), pp. 161-185.
22. Slot AJ, Molinski SV and Cole SP. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011; 50:179-207.
23. Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M and Bates SE. ABCG2: a perspective. Adv Drug Deliv Rev. 2009; 61:3-13.
24. Khavari PA, Peterson CL, Tamkun JW, Mendel DB and Crabtree GR. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature. 1993; 366:170-174.
25. Muchardt C and Yaniv M. A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J. 1993; 12:4279-4290.
26. Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL and Crabtree GR. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996; 15:5370-5382.
27. Bai J, Mei P, Zhang C, Chen F, Li C, Pan Z, Liu H and Zheng J. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS One. 2013; 8:e59772.
28. Cohet N, Stewart KM, Mudhasani R, Asirvatham AJ, Mallappa C, Imbalzano KM, Weaver VM, Imbalzano AN and Nickerson JA. SWI/SNF chromatin remodeling enzyme ATPases promote cell proliferation in normal mammary epithelial cells. J Cell Physiol. 2010; 223:667-678.
29. de la Serna IL, Ohkawa Y, Higashi C, Dutta C, Osias J, Kommajosyula N, Tachibana T and Imbalzano AN. The microphthalmia-associated transcription factor requires SWI/SNF enzymes to activate melanocyte-specific genes. J Biol Chem. 2006; 281:20233-20241.
30. Ho L and Crabtree GR. Chromatin remodelling during development. Nature. 2010; 463:474-484.
31. Naidu SR, Love IM, Imbalzano AN, Grossman SR and Androphy EJ. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene. 2009; 28:2492-2501.
32. Wu JI. Diverse functions of ATP-dependent chromatin remodeling complexes in development and cancer. Acta Biochim Biophys Sin (Shanghai). 2012; 44:54-69.
33. Wu Q, Madany P, Akech J, Dobson JR, Douthwright S, Browne G, Colby JL, Winter GE, Bradner JE, Pratap J, Sluder G, Bhargava R, Chiosea SI, van Wijnen AJ, Stein JL, Stein GS, et al. The SWI/SNF ATPases Are Required for Triple Negative Breast Cancer Cell Proliferation. J Cell Physiol. 2015; 230:2683-2694.
34. Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK and Kim CF. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015; 520:239-242.
35. Liu X, Tian X, Wang F, Ma Y, Kornmann M and Yang Y. BRG1 promotes chemoresistance of pancreatic cancer cells through crosstalking with Akt signalling. Eur J Cancer. 2014; 50:2251-2262.
36. Oakman C, Viale G and Di Leo A. Management of triple negative breast cancer. Breast. 2010; 19:312-321.
37. Brenton JD, Carey LA, Ahmed AA and Caldas C. Molecular classification and molecular forecasting of breast cancer: ready for clinical application? Journal of clinical oncology. 2005; 23:7350-7360.
38. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research. 2007; 13:4429-4434.
39. Dreyer G, Vandorpe T, Smeets A, Forceville K, Brouwers B, Neven P, Janssens H, Deraedt K, Moerman P, Van Calster B, Christiaens MR, Paridaens R and Wildiers H. Triple negative breast cancer: clinical characteristics in the different histological subtypes. Breast. 2013; 22:761-766.
40. Livasy CA. Triple-Negative Breast Carcinoma. Surg Pathol Clin. 2009; 2:247-261.
41. The-Cancer-Genome-Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61-70.
42. Cory AH, Owen TC, Barltrop JA and Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991; 3:207-212.
43. Fedorov O, Castex J, Tallant C, Owen DR, Martin S, Aldeghi M, Monteiro O, Filippakopoulos P, Picaud S, Trzupek JD, Gerstenberger BS, Bountra C, Willmann D, Wells C, Philpott M, Rogers C, et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Science advances. 2015; 1:e1500723.
44. Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, Zhan Y, Leo E, Mahadeshwar HS, Protopopov A, Futreal A, Tieu TN, Peoples M, Heffernan TP, Marszalek JR, Toniatti C, et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF mutant cancers: Insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 2015; 18:3865-3878
45. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983; 65:55-63.
46. Dutta P, Tanti GK, Sharma S, Goswami SK, Komath SS, Mayo MW, Hockensmith JW and Muthuswami R. Global epigenetic changes induced by SWI2/SNF2 inhibitors characterize neomycin-resistant mammalian cells. PLoS One. 2012; 7:e49822.
47. Muthuswami R, Mesner LD, Wang D, Hill DA, Imbalzano AN and Hockensmith JW. Phosphoaminoglycosides inhibit SWI2/SNF2 family DNA-dependent molecular motor domains. Biochemistry. 2000; 39:4358-4365.
48. Chiba H, Muramatsu M, Nomoto A and Kato H. Two human homologues of Saccharomyces cerevisiae SWI2/SNF2 and Drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic acids research. 1994; 22:1815-1820.
49. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002; 53:615-627.
50. Gottesman MM, Fojo T and Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002; 2:48-58.
51. Vtorushin SV, Khristenko KY, Zavyalova MV, Perelmuter VM, Litviakov NV, Denisov EV, Dulesova AY and Cherdyntseva NV. The phenomenon of multi-drug resistance in the treatment of malignant tumors. Exp Oncol. 2014; 36:144-156.
52. Hlavac V, Brynychova V, Vaclavikova R, Ehrlichova M, Vrana D, Pecha V, Kozevnikovova R, Trnkova M, Gatek J, Kopperova D, Gut I and Soucek P. The expression profile of ATP-binding cassette transporter genes in breast carcinoma. Pharmacogenomics. 2013; 14:515-529.
53. Guo Y, Kotova E, Chen ZS, Lee K, Hopper-Borge E, Belinsky MG and Kruh GD. MRP8, ATP-binding cassette C11 (ABCC11), is a cyclic nucleotide efflux pump and a resistance factor for fluoropyrimidines 2’,3’-dideoxycytidine and 9’-(2’-phosphonylmethoxyethyl)adenine. J Biol Chem. 2003; 278:29509-29514.
54. Oguri T, Bessho Y, Achiwa H, Ozasa H, Maeno K, Maeda H, Sato S and Ueda R. MRP8/ABCC11 directly confers resistance to 5-fluorouracil. Mol Cancer Ther. 2007; 6:122-127.
55. Minegaki T, Takara K, Hamaguchi R, Tsujimoto M and Nishiguchi K. Factors affecting the sensitivity of human-derived esophageal carcinoma cell lines to 5-fluorouracil and cisplatin. Oncol Lett. 2013; 5:427-434.
56. Cui Y, Konig J, Buchholz JK, Spring H, Leier I and Keppler D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol. 1999; 55:929-937.
57. Surowiak P, Materna V, Kaplenko I, Spaczynski M, Dolinska-Krajewska B, Gebarowska E, Dietel M, Zabel M and Lage H. ABCC2 (MRP2, cMOAT) can be localized in the nuclear membrane of ovarian carcinomas and correlates with resistance to cisplatin and clinical outcome. Clin Cancer Res. 2006; 12:7149-7158.
58. Litviakov NV, Cherdyntseva NV, Tsyganov MM, Denisov EV, Garbukov EY, Merzliakova MK, Volkomorov VV, Vtorushin SV, Zavyalova MV, Slonimskaya EM and Perelmuter VM. Changing the expression vector of multidrug resistance genes is related to neoadjuvant chemotherapy response. Cancer Chemother Pharmacol. 2013; 71:153-163.
59. Folmer Y, Schneider M, Blum HE and Hafkemeyer P. Reversal of drug resistance of hepatocellular carcinoma cells by adenoviral delivery of anti-ABCC2 antisense constructs. Cancer Gene Ther. 2007; 14:875-884.
60. Yao J, Feng FY, Lin C, Zhang XY, Fu M, Liang X and Yang Y. [The mechanism of resistance to 2' 2'-difluorodeoxycytidine (gemcitabine) in a pancreatic cancer cell line]. Zhonghua Zhong Liu Za Zhi. 2005; 27:721-726.
61. Zhou J, Wang CY, Liu T, Wu B, Zhou F, Xiong JX, Wu HS, Tao J, Zhao G, Yang M and Gou SM. Persistence of side population cells with high drug efflux capacity in pancreatic cancer. World J Gastroenterol. 2008; 14:925-930.
62. Meng X, Wang G, Guan R, Jia X, Gao W, Wu J, Yu J, Liu P, Yu Y, Sun W, Dong H and Fu S. Predicting chemosensitivity to gemcitabine and cisplatin based on gene polymorphisms and mRNA expression in non-small-cell lung cancer cells. Pharmacogenomics. 2015; 16:23-34.
63. Subhani S, Jayaraman A and Jamil K. Homology modelling and molecular docking of MDR1 with chemotherapeutic agents in non-small cell lung cancer. Biomed Pharmacother. 2015; 71:37-45.
64. Xu JW, Li QQ, Tao LL, Cheng YY, Yu J, Chen Q, Liu XP and Xu ZD. Involvement of EGFR in the promotion of malignant properties in multidrug resistant breast cancer cells. Int J Oncol. 2011; 39:1501-1509.
65. Li YT, Chua MJ, Kunnath AP and Chowdhury EH. Reversing multidrug resistance in breast cancer cells by silencing ABC transporter genes with nanoparticle-facilitated delivery of target siRNAs. Int J Nanomedicine. 2012; 7:2473-2481.
66. Sprouse AA and Herbert BS. Resveratrol augments paclitaxel treatment in MDA-MB-231 and paclitaxel-resistant MDA-MB-231 breast cancer cells. Anticancer Res. 2014; 34:5363-5374.
67. Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J, Bhardwaj N, Gerstein MB and Snyder M. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet. 2011; 7:e1002008.
68. Rosenbloom KR, Dreszer TR, Pheasant M, Barber GP, Meyer LR, Pohl A, Raney BJ, Wang T, Hinrichs AS, Zweig AS, Fujita PA, Learned K, Rhead B, Smith KE, Kuhn RM, Karolchik D, et al. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010; 38:D620-625.
69. Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005; 7:R953-964.
70. Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D and Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005; 365:671-679.
71. Zhang Y, Sieuwerts AM, McGreevy M, Casey G, Cufer T, Paradiso A, Harbeck N, Span PN, Hicks DG, Crowe J, Tubbs RR, Budd GT, Lyons J, Sweep FC, Schmitt M, Schittulli F, et al. The 76-gene signature defines high-risk patients that benefit from adjuvant tamoxifen therapy. Breast Cancer Res Treat. 2009; 116:303-309.
72. Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET and Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005; 102:13550-13555.
73. Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, Desmedt C, Larsimont D, Cardoso F, Peterse H, Nuyten D, Buyse M, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006; 98:262-272.
74. Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kolbl H and Gehrmann M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008; 68:5405-5413.
75. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006; 10:529-541.
76. Thomas CE, Ehrhardt A and Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003; 4:346-358.
77. Fish PV, Filippakopoulos P, Bish G, Brennan PE, Bunnage ME, Cook AS, Federov O, Gerstenberger BS, Jones H, Knapp S, Marsden B, Nocka K, Owen DR, Philpott M, Picaud S, Primiano MJ, et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J Med Chem. 2012; 55:9831-9837.
78. Barbieri I, Cannizzaro E and Dawson MA. Bromodomains as therapeutic targets in cancer. Brief Funct Genomics. 2013; 12:219-230.
79. Horn PJ and Peterson CL. The bromodomain: a regulator of ATP-dependent chromatin remodeling? Front Biosci. 2001; 6:D1019-1023.
80. Mujtaba S, Zeng L and Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007; 26:5521-5527.
81. Trotter KW, Fan HY, Ivey ML, Kingston RE and Archer TK. The HSA domain of BRG1 mediates critical interactions required for glucocorticoid receptor-dependent transcriptional activation in vivo. Mol Cell Biol. 2008; 28:1413-1426.
82. Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, Campbell AE, Kawaoka S, Shareef S, Zhu Z, Kendall J, Muhar M, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes & development. 2013; 27:2648-2662.
83. Trotter KW and Archer TK. Reconstitution of glucocorticoid receptor-dependent transcription in vivo. Mol Cell Biol. 2004; 24:3347-3358.
84. Sentani K, Oue N, Kondo H, Kuraoka K, Motoshita J, Ito R, Yokozaki H and Yasui W. Increased expression but not genetic alteration of BRG1, a component of the SWI/SNF complex, is associated with the advanced stage of human gastric carcinomas. Pathobiology : journal of immunopathology, molecular and cellular biology. 2001; 69:315-320.
85. Sun A, Tawfik O, Gayed B, Thrasher JB, Hoestje S, Li C and Li B. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. The Prostate. 2007; 67:203-213.
86. Saladi SV, Keenen B, Marathe HG, Qi H, Chin KV and de la Serna IL. Modulation of extracellular matrix/adhesion molecule expression by BRG1 is associated with increased melanoma invasiveness. Molecular cancer. 2010; 9:280.
87. Watanabe T, Semba S and Yokozaki H. Regulation of PTEN expression by the SWI/SNF chromatin-remodelling protein BRG1 in human colorectal carcinoma cells. British journal of cancer. 2011; 104:146-154.
88. Lin H, Wong RP, Martinka M and Li G. BRG1 expression is increased in human cutaneous melanoma. The British journal of dermatology. 2010; 163:502-510.
89. Hohmann AF and Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends in genetics : TIG. 2014; 30:356-363.
90. Oike T, Ogiwara H, Amornwichet N, Nakano T and Kohno T. Chromatin-regulating proteins as targets for cancer therapy. Journal of radiation research. 2014; 55:613-628.
91. Marquez SB, Thompson KW, Lu L and Reisman D. Beyond Mutations: Additional Mechanisms and Implications of SWI/SNF Complex Inactivation. Frontiers in oncology. 2014; 4:372.
92. Helming KC, Wang X and Roberts CW. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer cell. 2014; 26:309-317.
93. Cai SF, Chen CW and Armstrong SA. Drugging Chromatin in Cancer: Recent Advances and Novel Approaches. Molecular cell. 2015; 60:561-570.
94. Nigam SK. What do drug transporters really do? Nat Rev Drug Discov. 2015; 14:29-44.
95. Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, Reinhold W, Guo Y, Kruh GD, Reimers M, Weinstein JN and Gottesman MM. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. 2004; 6:129-137.
96. Ruff P, Vorobiof DA, Jordaan JP, Demetriou GS, Moodley SD, Nosworthy AL, Werner ID, Raats J and Burgess LJ. A randomized, placebo-controlled, double-blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar (LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen. Cancer Chemother Pharmacol. 2009; 64:763-768.
97. Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, Yoneda T and Mundy GR. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996; 98:1544-1549.
98. Imbalzano KM, Cohet N, Wu Q, Underwood JM, Imbalzano AN and Nickerson JA. Nuclear shape changes are induced by knockdown of the SWI/SNF ATPase BRG1 and are independent of cytoskeletal connections. PLoS One. 2013; 8:e55628.
99. de la Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS, Tapscott SJ and Imbalzano AN. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol. 2005; 25:3997-4009.