
INTRODUCTION
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is a genetically and clinically heterogeneous malignancy characterized by the clonal expansion of tumor B-cell precursors [1]. In the past, several distinct genetic/molecular subtypes of BCP-ALL have been identified which show an age-associated distribution with prognostic impact [2, 3]. Chromosomal alterations from such BCP-ALL-associated genetic subgroups appear to be associated with the development of the disease and they frequently are a hallmark of the tumor. Despite this, evidences exist about the existence of a genetic background that might involve the whole hematopoiesis and favor the development of BCP-ALL, e.g. in subjects with Down syndrome (DS) and a 30 fold increased risk for BCP-ALL [4], in individuals with germline mutations of the RUNX1, PAX5, ETV6, GATA1, HOXA11, ANKRD26, MPL, TP53 and RMB8A genes [5–8] and in children with RUNX1 amplification associated to t(15;21)(q10;q10) and a 3 × 103 fold increase in the frequency of BCP-ALL [9]. In addition, around 2% of children with BCP-ALL present with a transient (preleukemic) aplastic phase, 2 to 9 months before initial diagnosis [10, 11]. In the past decades, efforts devoted to the analysis of BCP-ALL tumor cells have concentrated in the characterization of the oncogenetic steps that occur within the BCP-ALL blast cell compartment [2, 12]; in contrast, fewer studies have investigated the features of residual hematopoietic cells at diagnosis.
In addition to the above referred genetic predisposition to childhood BCP-ALL, other recent studies also provide further evidence about the potential involvement of the residual hematopoiesis in BCP-ALL; thus, the presence of genetic alterations which are typical of leukemic blast cells (e.g. MLL and BCR-ABL1 gene rearrangements) have been also detected in mesenchymal stem cells [13] and residual neutrophils [14]. Altogether, these findings suggest that bone marrow (BM) hematopoiesis could be altered in at least a fraction of childhood BCP-ALL patients. Thereby, detailed characterization of residual hematopoietic BM and peripheral blood (PB) cells may contribute to a better identification of such patients.
In the past decade, evidences have accumulated which indicate that altered myeloid and lymphoid cell phenotypes are a surrogate marker for an altered hematopoiesis [15, 16], particularly in the elderly [17, 18]. Thus, phenotypic alterations of residual hematopoietic BM cells, such as those involving maturing neutrophils, monocytic and/or erythroid cells, as well as CD34+ myeloid precursors, have been associated with clonal hematopoiesis in patients with myeloid malignancies including (adult) acute myeloid leukemia (AML), myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN) [15, 16, 19]. Similarly, immunophenotypic alterations have also been reported in systemic mastocytosis in association with multilineage KIT mutations [20, 21] and a greater risk of progression to aggressive disease e.g. AML and MDS. More recently, altered MDS-like phenotypes involving maturing and mature neutrophils, monocytic and/or erythroid precursors, have also been found in adult patients with mature lymphoid malignancies such as multiple myeloma and monoclonal gammopathy of undetermined significance (MGUS) cases; of note, these patients frequently showed also MDS-related genetic changes in the phenotypically altered BM myeloid cells and CD34+ precursors [16]. Whether such phenotypic alterations are also present in BM cells from BCP-ALL patients remains to be investigated.
Here we investigated the presence and the frequency of altered patterns of expression of neutrophil-associated proteins in BM- and PB-derived maturing/mature neutrophils from children with BCP-ALL studied at diagnosis, and their potential relationship with tumor cytogenetics and the clinical behavior of the disease.
RESULTS
Altered neutrophil immunopheno types in the bone marrow and peripheral blood of children with BCP-ALL
Altered maturing/mature neutrophil immunophenotypes were detected at diagnosis in the majority (85/110; 77%) of childhood BCP-ALL patients; in contrast, they were systematically absent in normal/reactive BM samples (0/12 cases; 0%) (p < 0.001), Figure 1. The most frequently altered marker was CD10, which was absent on BM and PB neutrophils of around half of the cases (62/118 patients; 53%) (p < 0.001). Absence of CD33 (36/107 patients; 34%) (p = 0.02), decreased expression of CD13 (17/111; 15%) (p = 0.14) heterogeneously low reactivity for CD15/CD65 (11/111; 10%) (p = 0.25) and overexpression of CD123 (8/109; 7%) (p = 0.33) (Figure 2A) were also found at variable frequencies. In more than one third of the patients (42/101; 42%) with altered neutrophil phenotypes these involved one marker; whereas two, three and four altered markers were found in 24/101 (24%), 9/101 (9%) and 1/101 (1%) BCP-ALL patients, respectively (Figure 2B). The most frequent combination of altered markers (11/101 cases; 11%) involved the absence of both CD10 and CD33 (Figure 2B).
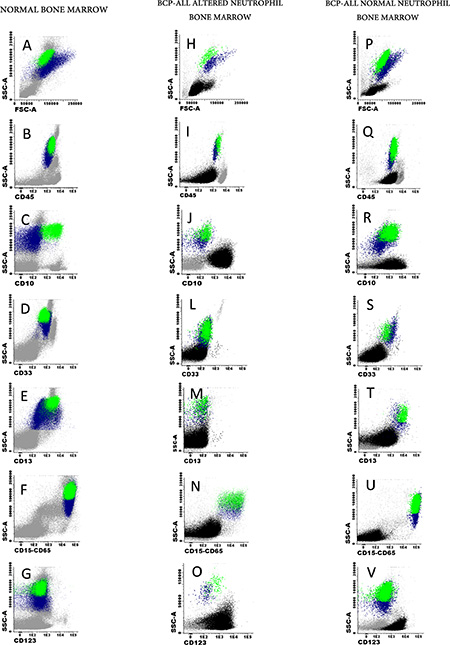
Figure 1: Illustrating examples of normal (A–G) and childhood BCP-ALL (H–V) BM mature/maturing neutrophil phenotypes for CD10 (C, J and R), CD33 (D, L and S), CD13 (E, M and T), CD15 plus CD65 (F, N and U) and CD123 (G, O and V). Mature neutrophils are depicted in green while neutrophil precursors are shown in blue. Blast cells and other BM cell compartmens are depicted as black and grey events, respectively.
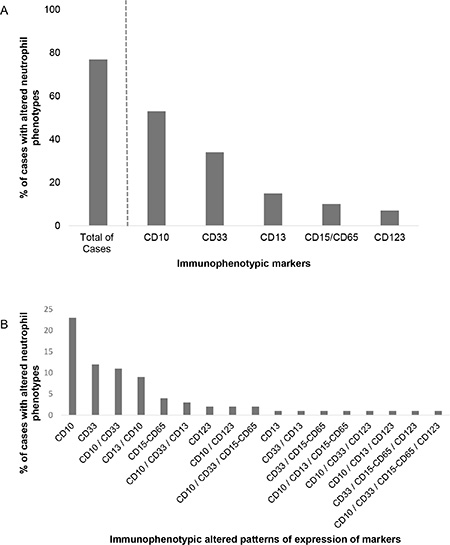
Figure 2: Frequency of BCP-ALL cases showing altered patterns of expression for individual (A) or multiple (B) markers on maturing/mature neutrophils.
Interestingly, no statistically significant differences were found (p > 0.05) between BM and PB samples, as regards the frequency of altered neutrophil phenotypes (79% in BM vs 58% in PB, p > 0.05), except for CD10 that was more frequently absent in BM than PB of childhood BCP-ALL (57% vs. 15%, p = 0.004). Of note, altered expression of CD10 on neutrophils did not correlate with the pattern of expression of CD10 on blast cells (p > 0.05).
In only 30% of cases (35/118) sufficient events (> 20) were measured that would allow analysis of the immuno phenotypic profile of CD34+/MPO+ neutrophil precursors. In all 35 cases, normal CD117 and CD13 expression patterns in the absence of CD7 were observed on CD34+/MPO+ precursors. In contrast, CD33 was expressed at abnormally low levels (i.e. negative) in CD34+/MPO+ precursors from 26% (9/35) of patients. Cases with an altered CD34+/MPO+/CD33–/lo phenotype systematically showed also an altered maturing/mature neutrophil phenotypes (100% (9/9) vs. 65% (17/26), p = 0.03). Thereby, they more frequently displayed abnormal expression of both CD33–66% (6/9) vs 13% (3/23), p = 0.01 and CD15/CD65–100% (4/4) vs 13% (5/30), p = 0.003 - on maturing/mature neutrophils. Furthermore, the proportion of cases which had an altered phenotype on CD34+/MPO+ cells progressively increased in parallel to the number of aberrant markers observed in maturing/mature neutrophils, from 3/14 cases (21%) to 4/8 patients (50%) and 2/2 cases (100%) for BCP-ALL patients presenting with 1, 2 and ≥ 3 aberrant markers on mature/maturing neutrophils (p = 0.02).
After therapy, most patients showed (normal) recovery of CD10 expression on mature neutrophils. Thus, decreased or negative CD10 expression was observed in PBof 8/26 cases (31%) at day8 of prednisone therapy, and in the BM of 25/81 (30%), 12/96 (12%) and 6/87 (7%) of patients studied at day 15, 33 and 78 of therapy, respectively. Expression of other markers such as CD33, CD15/CD65, CD13 and CD123, on PB or BM neutrophils was not evaluated during follow up.
Relationship between the presence of altered neutrophil phenotypes and the distribution of residual non-blast cell BM populations in childhood BCP-ALL
Children with BCP-ALL and altered neutrophil phenotypes showed a lower percentage of maturing neutrophils (19% vs. 28%, p = 0.004) and a greater proportion of lymphocytes (51% vs. 37%, p = 0.04), including mature B cells (8.8% vs. 7.1%, p = 0.03), than cases with normal neutrophil phenotypes (Table 1). In addition, the former cases also showed a significantly higher proportion of CD34+ myeloid precursors within the CD34+ (HPC) BM compartment (0.6% vs. 0.3%, p = 0.03) (Table 1). No statistically significant differences (p > 0.05) were found between both patient groups as regards the distribution of nucleated red blood cells (NRBC), monocytes, eosinophils, basophils, T lymphocytes and NK cells (Table 1).
Table 1: Distribution of non-blast cell subsets in the BM of children with BCP-ALL at diagnosis according to the presence vs. absence of altered immunophenotypes on residual maturing and mature BM neutrophils
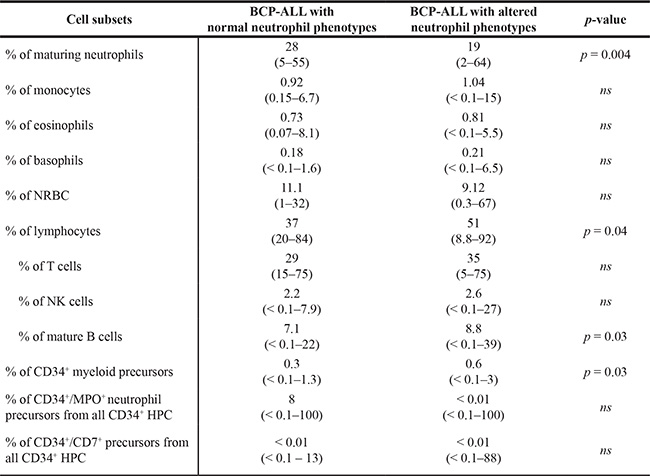
Results expressed as median (range). BCP-ALL, B-cell precursor acute lymphoblastic leukemia; BM, bone marrow; NRBC, nucleated red blood cells; HPC, hematopoietic progenitor cells.
Relationship between the presence of phenotypically altered (mature/maturing) neutrophils and the clinical features of the disease
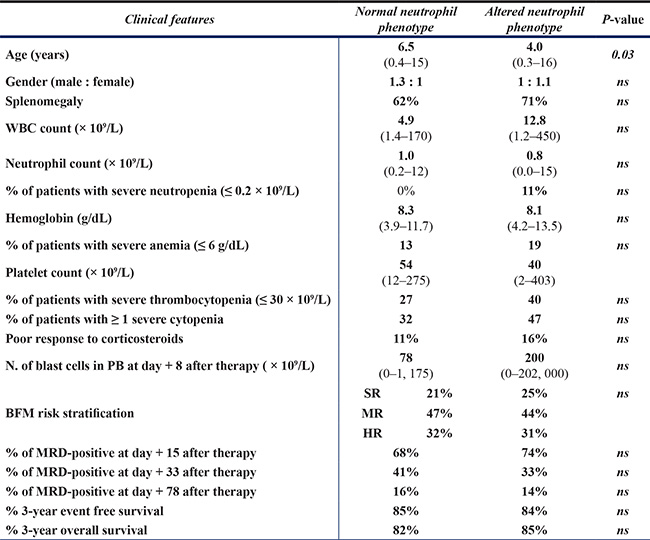
Interestingly, the frequency of cases with altered neutrophil phenotypes varied according to age; thus, BCP-ALL children with altered neutrophil phenotypes were younger than children who had normal phenotypes: median age at diagnosis of 4.0 vs. 6.5 years (p = 0.03) (Figure 3A–3B and Table 3). Thus, presence of at least one altered marker was detected in 62/82 (86%) BCP-ALL children younger than 6 years vs. 22/37 (60%) children older than 6 years (aged 6–16 years) (p = 0.002) (Figure 3A–3B). Overall, similar frequencies of altered neutrophil phenotypes were detected for the different markers investigated among BCP-ALL patients carrying distinct cytogenetic subtypes of the disease, except for a slightly lower frequency of cases showing (mature/maturing) neutrophils with a CD10– and CD33low phenotype among patients with MLL gene rearrangements - 27% vs 58% (p = 0.06), and 9% vs. 33% (p = 0.05) respectively - (Table 2). Regarding other features of the disease (Table 3), BCP-ALL children with altered patterns of expression of phenotypic markers on mature/maturing neutrophils showed similar PB cell counts, comparable frequencies of organomegalies, and overlapping rates of response to therapy, to those observed for cases who had normal neutrophil phenotypes.
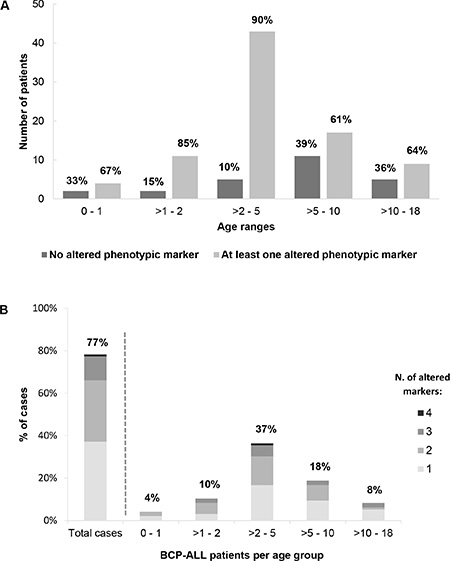
Figure 3: Frequency of childhood BCP-ALL cases with immunophenotypically altered maturing/mature neutrophils. (A) The distribution of patients with immunophenotypically normal vs. altered neutrophils is represented by bars whose height represents the number of cases per age group. The relative distribution of patients with normal (dark gray bars) vs. at least one altered phenotypic marker (light gray bars) within each age group is shown in percentage numbers above the bar. In (B) bars indicate the relative distribution of cases (i.e. percentages) within each age group according the number of altered markers on neutrophils. The overall percentage of cases with an altered neutrophil immunophenotype is shown per age group, as numbers above the corresponding bar.
Table 2: Distribution of aberrant patterns of antigen expression on bone marrow maturing and mature neutrophils from pediatric BCP-ALL patients studied at diagnosis: relationship with the genetic subtype of the disease
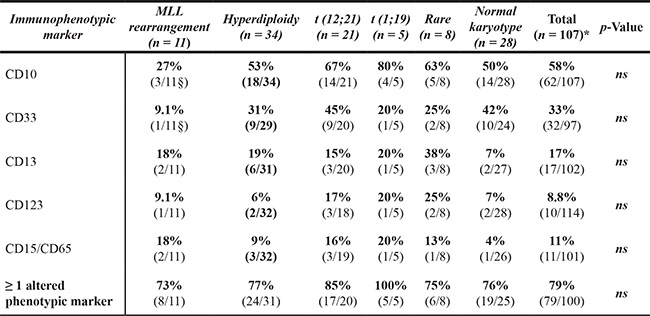
The denominator in each fraction is the total number of patients with the genetic alteration evaluated for each specific marker.
*The total of patients was 107 because there were 11 cases with missing data concerning genetic alterations. § p-value < 0.05, vs. patients without MLL gene rearrangements.
Table 3: Relationship between the number of immunophenotypically altered markers on neutrophils and the clinical features of the disease
DISCUSSION
By definition, childhood BCP-ALL is a relatively heterogeneous group of de novo acute leukemias, which is characterized by an abnormal expansion of genetically altered B-cell precursors blocked at relatively early stages of B-lymphoid maturation [3]. Despite this, accumulating evidences suggest that a genetic predisposition might exist which favors the development of childhood BCP-ALL [4–9, 11]. Such genetic predisposition might translate into uniquely altered phenotypes of maturing residual hematopoietic cells that can mimic those observed in MDS, MPN and clonal hematopoiesis of indeterminate potential (CHIP) [22–25]. Here we investigated for the first time the presence of altered neutrophil phenotypes in a relatively large cohort of childhood BCP-ALL. Interestingly, three quarters of all childhood BCP-ALL cases studied showed altered phenotypes on BM and/or PB maturing/mature neutrophils. Abnormally low-to-negative expression levels of CD10, CD33, CD13 and CD15/CD65, together with overexpression of CD123, were recurrently detected at decreasing frequencies. These findings are in line with previous observations pointing out the existence of an altered neutrophil function in these patients at diagnosis, including decreased chemotaxis, phagocytosis and oxidative burst [26–28].
Of note, the presence of altered neutrophil phenotypes was associated with lower percentages of maturing neutrophils and higher lymphocyte counts in the BM, as well as with a younger age at diagnosis. In contrast, no significant associations were found between the presence of altered neutrophil phenotypes and PB cell counts, tumor cytogenetics and other clinical features of the disease, except for a lower frequency of altered CD10 and CD33 expression in patients with MLL gene rearrangement. This later association could not be due to technical artifacts such as antibody consumption due to high numbers of CD10hi blast cells, among cases who show no MLL rearrangements (vs. MLL cases), since CD10 levels on blast cells did not show an association with CD10 expression levels on residual neutrophils. In addition, usage of different antibody clones in a subset of altered cases (n = 3) showed similar results, which would rule out also a reagent specific bias.
In recent years, several groups have independently reported the presence of clonal hematopoiesis in adults diagnosed with both de novo AML and mature lymphoid neoplasias (e.g. MM and MGUS), as well as in otherwise healthy adults [17, 22, 29–31]; of note, the frequency of leukemia and cancer cases increased with age among later group [23–26]. Based on these observations, it has been hypothesized that the presence of clonal hematopoiesis in the absence of clear cut criteria for a hematological disorder might precede (or even favor) the development of both myeloid and lymphoid malignancies, at least in a fraction of cases. Despite this, no study has been reported so far in which the presence of cytogenetic and/or phenotypic markers associated with clonal hematopoiesis have been systematically investigated in residual BM myeloid cells from children with BCP-ALL.
Here we report a high frequency of altered (e.g. MPN/MDS-like) phenotypes on BM and PB neutrophils of children with BCP-ALL; these findings would support the existence of an altered hematopoiesis also in childhood BCP-ALL. In line with this, patients with altered neutrophil phenotypes also showed reduced BM neutrophil counts vs. cases displaying normal neutrophil phenotypes, independently of the genetic subtype of BCP-ALL. These findings suggest a more pronounced impairment of neutrophil production among the former group of patients. Despite this, no statistically significant differences were observed between BCP-ALL children with normal vs. altered neutrophil phenotypes, as regards PB neutrophil counts. Such lack of association could be due to the fact that PB neutrophil counts also depend on factors other than a clonal hematopoiesis, such as the degree and the pattern of BM involvement by blast cells, the BM microenvironment and/or the local production of growth factors and cytokines, in addition to the specific underlying alteration of residual (potentially clonal) hematopoiesis. In fact, disruption of the normal HPC BM niches caused by leukemic cell growth has recurrently been reported [32, 33] and deserves further (ongoing) investigations.
Independently of whether or not the presence of altered neutrophil phenotypes might be due to an underlying genetically altered (clonal) hematopoiesis, it might be speculated that this would provide an environment that might favor an earlier onset of the disease. Thus, children with BCP-ALL and altered neutrophil phenotypes also showed a significantly lower age at disease onset. In contrast, no significant differences were found in the clinical behavior of the disease and patient outcome, depending on the presence vs. absence of altered neutrophil phenotypes. Altogether, these observations suggest that the course of BCP-ALL more likely depends on features of the disease related to the tumor cells, tumor burden and response to therapy, than to the presence of an underlying altered hematopoiesis, at least in the short-to-medium term, which is also supported by the progressive recovery of CD10 expression on neutrophils from treated BCP-ALL cases here reported. In turn, such lack of clinical correlation could also be due to the fact that we only investigated the presence of altered phenotypes on neutrophil lineage cells, but not on other relevant hematopoietic cell compartments (e.g. maturing monocytes and NRBC) which might also be abnormal, even in cases that showed normal neutrophil immunophenotypic patterns. Interestingly, our results showed a close association between the presence of altered phenotypes on CD34+/MPO+ neutrophil precursors and a greater number of aberrant phenotypes on maturing/mature neutrophils. These results might suggest that alteration of a single marker such as CD10 might not be as relevant as the coexistence of multiple phenotypic alterations involving also precursor cells, in line with what has been previously reported for MDS patients [24]. Further studies, in which the presence of altered phenotypes is investigated in BM/PB cell compartments other than neutrophils, are required to confirm this hypothesis.
Despite all the above, it might be speculated that the herein reported altered phenotypes might not be specific for an underlying clonal hematopoiesis, since they may also be found in other reactive non-clonal conditions. In this regard, absent-to-low expression of CD10 has been reported for mature neutrophils from septic shock patients [34] in association with a greater chemotactic response to activated complement (vs. CD10+ neutrophils) [35]. Similarly, decreased CD13 levels have been reported on neutrophils from HIV+ patients in parallel to the decrease in PB CD4+ T cell counts [36]; in these patients, greater CD13 levels have an inhibitory effect on TNFα induced apoptosis [37]. In fact, CD10 and CD13 can be downregulated by cytokines, and growth factors such as TNFα, and GM-CSF [28]. Thereby, the time span from disease onset to diagnosis could contribute to the decreased expression of CD10 and/or CD13, due to more prolonged time of exposure to an inflammatory microenvironment. Regarding altered expression of other markers here investigated in reactive conditions, low expression of CD15 on mature neutrophils has also been reported among individuals with delayed asthmatic response, suggesting the involvement of a cell-mediated hypersensitivity mechanism in bronchial asthma [38]. Concerning CD33, no clear association has been reported between decreased levels of expression of this molecule and other non-neoplastic conditions, except for a rare single nucleotide polymorphism that prevents binding of specific anti-CD33 antibody clones to CD33 [39]. In contrast, greater levels of CD123 on neutrophil lineage cells have been recurrently reported in association with clonal hematopoietic disorders [40, 41]. Altogether, these findings support the notion that, despite some of the herein described phenotypic alterations might occur in rare disease conditions or small groups of individuals, these could not explain the rather high frequency of phenotypically altered neutrophils observed among our BCP-ALL patients.
In summary, here we describe for the first time, the presence of altered phenotypes on neutrophils from children with BCP-ALL, in association with an earlier onset of the disease. Such findings support the existence of an altered residual hematopoiesis in most childhood BCP-ALL cases. Whether such altered phenotypes also reflect an underlying clonal hematopoiesis in a substantial fraction of childhood BCP-ALL patients or they are a consequence of massive leukemic infiltration of the BM microenvironment, deserves further investigations.
MATERIALS AND METHODS
Patients and samples
Overall, 105 BM and 13 PB samples from 118 children diagnosed with BCP-ALL by the WHO criteria (59 males and 59 females; mean age: 6 ± 4 years, ranging from 0.3 to 16 years), were studied. No paired BM and PB samples were analyzed. In parallel, normal and reactive BM samples from 12 children (6 males and 6 females; mean age of 6 ± 6 years, ranging from 0 to 15 years), were also studied as controls. Samples were obtained at three different institutions – Institute of Pediatrics and Puericulture Martagão Gesteira, Rio de Janeiro Federal University (IPPMG/UFRJ), Rio de Janeiro (Brazil); University Hospital of Salamanca, University of Salamanca, Salamanca (Spain), and; Medical University of Silesia, Zabrze (Poland) – after informed consent was given according to the Helsinki Declaration protocol. The study was approved by the local Ethics Committees. Patients were treated with the IC-ALL-BFM 2002 (32% - 38/118), IC-ALL-BFM 2009 (34% - 40/118), INTERFANT 2006 (5% - 6/118) and SEHOP-PETHEMA LAL 2013 (28% - 33/118) protocols. Of note, a high number of infants were studied since IPPMG/UFRJ is a reference center for infant ALL.
Multiparameter flow cytometry immunophenotypic studies
Individual BM and PB samples were obtained at diagnosis prior to the administration of any therapy, and were stained with the EuroFlow 8-colour – fluorescein isothiocyanate (FITC)/phycoerythrin (PE)/PE-cyanin7(PECy7)/peridinin chlorophyll protein-Cy5.5 (PerCP-Cy5.5)/allophycocyanin (APC)/APC-hilite7 (APC- H7)/pacific blue (PacB)/pacific orange (PacO) – acute leukemia orientation tube (ALOT) plus the BCP-ALL panel [42]: i) cytoplasmic (Cy) MPO/CyCD79a/CD19/CD34/CD7/surface membrane (Sm) CD3/CyCD3/CD45; ii) CD58/CD66c/CD19/CD34/CD10/CD38/CD20/CD45; iii) CyIgM/CD33/CD19/CD34/CD117-SmIgM/SmIgλ/ SmIgκ/CD45; iv) nuclear (Nu) TdT/CD13/CD19/CD34/CD22/CD24/CD9/CD45; v) CD15-CD65/NG2/CD19/CD34/CD123/CD81/CD21/CD45. Staining of BM and PB cells was performed as previously described in detail, according to the standard operating procedures proposed by the EuroFlow Consortium [43]. Immediately after staining, cells were acquired in a FACSCanto II flow cytometer – Beckton/Dickinson Biosciences, San José, CA (BD), using the FACSDiVa software (BD). For data analysis, the INFINICYT™ software (Cytognos SL, Salamanca, Spain) was used. Mature neutrophils were identified as SSChi, FSCint and CD45hi cells; maturing neutrophil precursors were defined as SSCint-to-hi, FSCint-to-hi and CD45lo cells (Figure 1), both in normal (Figure 1A–1G) and BCP-ALL samples (Figure 1H–1O). Altered neutrophil phenotypes were defined by the absence or abnormally low (< 2 SD of normal BM neutrophils) expression levels of CD10, CD33 and CD15/CD65 and/or by aberrantly high expression levels of CD123 [44].
Cytogenetic and fluorescence in situ hybridization FISH studies
Cytogenetic studies were performed on short-term (2–72 h) cultured BM or PB cells collected at diagnosis and processed by standard procedures [45]. Conventional cytogenetic analysis was performed by GTG or QFQ banding using the Band View 5.5 software (Applied Spectral Imaging, Vista, CA, USA) or the IKAROS 5.4 software (MetaSystems, Altlusshein, Germany). Karyotypes were reported following International System for Human Cytogenetic Nomenclature (ISCN 2013).
FISH analyses were performed on methanol/acetic 3/1 (v/v) fixed cells. Slides were prepared, hybridized and analyzed using commercially available probes following the manufacturer’s recommendations. Locus-specific DNA probes (LSI Abbott VYSIS, Des Plaines, IL, USA) were used to detect ETV6-RUNX1 (Dual-color, dual fusion), MLL(Dual-color, break apart) and BCR-ABL (Dual-color, dual fusion) gene rearrangements. Fluorescence signals were detected using a BX-51 epifluorescence microscope (Olympus, Miami, FL, USA) equipped with the ISS 5.4 software (MetaSystems) and the Fish View 5.5 software (Applied Spectral Imaging). The number of mitosis and interphase nuclei investigated by classical cytogenetics and FISH was of 20 metaphases (whenever available) and > 200 interphase nuclei, respectively. Multicolor banding FISH was performed in cases with complex karyotypes [46] and fluorescence signals were detected using an AXIO- Image D2 epifluorescence microscope (ZEISS, Thornwood, NY, USA) equipped with the MetaArquive 5.2 software (MetaSystems).
Statistical methods
In order to establish the statistical significance of differences observed between groups, the Kruskall-Wallis and either the Mann-Whitney U tests (for continuous variables) or the Chi square test (for categorical variables) were used (SPSS software package, version 18.0, SPSS Inc., Chicago, IL, USA). P-values < 0.05 were considered to be associated with statistical significance.
ACKNOWLEDGMENTS
The authors would like to thank all clinicians and technicians from the Institute of Pediatrics and Puericulture Martagão Gesteira, (Rio de Janeiro Federal University, Rio de Janeiro, Brazil) for their support in data collection.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by the following grants: Bilateral Cooperation Program between Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES (Brasília/Brazil) and Dirección General de Políticas Universitárias – Ministério de Educación, Cultura y Deportes - DPGU (Madrid/Spain) (311/15) and RD12/0036/0048 from RETICS, (Instituto de Salud Carlos III, Ministerio de Economia y Competitividad, Madrid, Spain and Fondos FEDER); Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro of Brazil (E26/110.105/2014; E26/102.191/2013); Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPQ of Brazil (400194/2014–7). EO and DNRG were both supported by grants from CAPES (Brazil). NLD was partly supported by a grant from CNPQ (Brazil). TS and LS were supported by the internal grants from the Medical University of Silesia (Katowice, Poland). The funders had no role in the design of the study, data collection and analysis, decision to publishing nor in the preparation of the manuscript.
REFERENCES
1. Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, Vora A, Baruchel A, Silverman LB, Schmiegelow K, Escherich G, Horibe K, Benoit YC, et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J Clin Oncol. 2015; 33:2938–2948.
2. Bhojwani D, Yang JJ, Pui CH. Biology of childhood acute lymphoblastic leukemia. Pediatr Clin North Am. 2015; 62:47–60.
3. Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015; 12:344–357.
4. Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol. 2014; 167:587–599.
5. Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, Rajpurkar M, Jones K, Gowan K, Balduini CL, Pecci A, Gnan C, De Rocco D, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015; 47:535–538.
6. Stieglitz E, Loh ML. Genetic predispositions to childhood leukemia. Ther Adv Hematol. 2013; 4:270–290.
7. Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, Wechsler J, Yang J, Hayes J, Klein RJ, Zhang J, Wei L, Wu G, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013; 45:1226–1231.
8. Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A, Haferlach T, Haferlach C. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014; 124:251–258.
9. Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, Moorman AV, Dyer S, Borrow J, Griffiths M, Heerema NA, Carroll AJ, Talley P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014; 508:98–102.
10. Disperati P, Ichim CV, Tkachuk D, Chun K, Schuh AC, Wells RA. Progression of myelodysplasia to acute lymphoblastic leukaemia: implications for disease biology. Leuk Res. 2006; 30:233–239.
11. Horsley SW, Colman S, McKinley M, Bateman CM, Jenney M, Chaplin T, Young BD, Greaves M, Kearney L. Genetic lesions in a preleukemic aplasia phase in a child with acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2008; 47:333–340.
12. Mullighan CG. The molecular genetic makeup of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2012; 2012:389–396.
13. Menendez P, Catalina P, Rodriguez R, Melen GJ, Bueno C, Arriero M, Garcia-Sanchez F, Lassaletta A, Garcia-Sanz R, Garcia-Castro J. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J Exp Med. 2009; 206:3131–3141.
14. Primo D, Flores J, Quijano S, Sanchez ML, Sarasquete ME, del Pino-Montes J, Gaarder PI, Gonzalez M, Orfao A. Impact of BCR/ABL gene expression on the proliferative rate of different subpopulations of haematopoietic cells in chronic myeloid leukaemia. Br J Haematol. 2006; 135:43–51.
15. Fernandez C, Santos-Silva MC, Lopez A, Matarraz S, Jara-Acevedo M, Ciudad J, Gutierrez ML, Sanchez ML, Salvador-Osuna C, Berruezo MJ, Diaz-Arias JA, Palomo-Hernandez AM, et al. Newly diagnosed adult AML and MPAL patients frequently show clonal residual hematopoiesis. Leukemia. 2013; 27:2149–2156.
16. Matarraz S, Paiva B, Diez-Campelo M, Barrena S, Jara-Acevedo M, Gutierrez ML, Sayagues JM, Sanchez ML, Barcena P, Garrastazul MP, Berruezo MJ, Duran JM, et al. Immunophenotypic alterations of bone marrow myeloid cell compartments in multiple myeloma patients predict for myelodysplasia-associated cytogenetic alterations. Leukemia. 2014; 28:1747–1750.
17. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landen M, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014; 371:2477–2487.
18. Lichtman MA. Clonal hematopoiesis: a “CHIP” off the old block. Blood. 2015; 126:1–2.
19. van de Loosdrecht AA, Alhan C, Bene MC, Della Porta MG, Drager AM, Feuillard J, Font P, Germing U, Haase D, Homburg CH, Ireland R, Jansen JH, Kern W, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica. 2009; 94:1124–1134.
20. Teodosio C, Mayado A, Sanchez-Munoz L, Morgado JM, Jara-Acevedo M, Alvarez-Twose I, Garcia-Montero AC, Matito A, Caldas C, Escribano L, Orfao A. The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J Leukoc Biol. 2015; 97:49–59.
21. Garcia-Montero AC, Jara-Acevedo M, Alvarez-Twose I, Teodosio C, Sanchez-Munoz L, Muniz C, Munoz-Gonzalez JI, Mayado A, Matito A, Caldas C, Morgado JM, Escribano L, Orfao A. KIT D816V-mutated bone marrow mesenchymal stem cells in indolent systemic mastocytosis are associated with disease progression. Blood. 2016; 127:761–768.
22. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015; 126:9–16.
23. Primo D, Sanchez ML, Espinosa AB, Tabernero MD, Rasillo A, Sayagues JM, Gonzalez M, Hernandez JM, Orfao A. Lineage involvement in chronic myeloid leukaemia: comparison between MBCR/ABL and mBCR/ABL cases. Br J Haematol. 2006; 132:736–739.
24. Matarraz S, Lopez A, Barrena S, Fernandez C, Jensen E, Flores-Montero J, Rasillo A, Sayagues JM, Sanchez ML, Barcena P, Hernandez-Rivas JM, Salvador C, Fernandez-Mosteirin N, et al. Bone marrow cells from myelodysplastic syndromes show altered immunophenotypic profiles that may contribute to the diagnosis and prognostic stratification of the disease: a pilot study on a series of 56 patients. Cytometry B Clin Cytom. 2010; 78:154–168.
25. Matarraz S, Lopez A, Barrena S, Fernandez C, Jensen E, Flores J, Barcena P, Rasillo A, Sayagues JM, Sanchez ML, Hernandez-Campo P, Hernandez Rivas JM, Salvador C, et al. The immunophenotype of different immature, myeloid and B-cell lineage-committed CD34+ hematopoietic cells allows discrimination between normal/reactive and myelodysplastic syndrome precursors. Leukemia. 2008; 22:1175–1183.
26. Tanaka F, Goto H, Yokosuka T, Yanagimachi M, Kajiwara R, Naruto T, Nishimaki S, Yokota S. Suppressed neutrophil function in children with acute lymphoblastic leukemia. Int J Hematol. 2009; 90:311–317.
27. Hofmann WK, Stauch M, Hoffken K. Impaired granulocytic function in patients with acute leukaemia: only partial normalisation after successful remission-inducing treatment. J Cancer Res Clin Oncol. 1998; 124:113–116.
28. Shipp MA, Stefano GB, Switzer SN, Griffin JD, Reinherz EL. CD10 (CALLA)/neutral endopeptidase 24.11 modulates inflammatory peptide-induced changes in neutrophil morphology, migration, and adhesion proteins and is itself regulated by neutrophil activation. Blood. 1991; 78:1834–1841.
29. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, Hassimi M, Socci N, Bhatt PK, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012; 44:1179–1181.
30. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371:2488–2498.
31. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, McLeod JL, Doedens M, Medeiros JJ, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014; 506:328–333.
32. Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008; 322:1861–1865.
33. Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015; 126:2443–2451.
34. Martens A, Eppink GJ, Woittiez AJ, Eidhof H, de Leij LF. Neutrophil function capacity to express CD10 is decreased in patients with septic shock. Crit Care Med. 1999; 27:549–553.
35. McCormack RT, Nelson RD, Chenoweth DE, LeBien TW. Identification and characterization of a unique subpopulation (CALLA/CD10/negative) of human neutrophils manifesting a heightened chemotactic response to activated complement. Blood. 1987; 70:1624–1629.
36. Cloke T, Munder M, Bergin P, Herath S, Modolell M, Taylor G, Muller I, Kropf P. Phenotypic alteration of neutrophils in the blood of HIV seropositive patients. PLoS One. 2013; 8:e72034.
37. Cowburn AS, Sobolewski A, Reed BJ, Deighton J, Murray J, Cadwallader KA, Bradley JR, Chilvers ER. Aminopeptidase N (CD13) regulates tumor necrosis factor-alpha-induced apoptosis in human neutrophils. J Biol Chem. 2006; 281:12458–12467.
38. Pelikan Z. Expression of surface markers on the blood cells during the delayed asthmatic response to allergen challenge. Allergy Rhinol (Providence). 2014; 5:96–109.
39. Lamba JK, Pounds S, Cao X, Downing JR, Campana D, Ribeiro RC, Pui CH, Rubnitz JE. Coding polymorphisms in CD33 and response to gemtuzumab ozogamicin in pediatric patients with AML: a pilot study. Leukemia. 2009; 23:402–404.
40. Liu K, Zhu M, Huang Y, Wei S, Xie J, Xiao Y. CD123 and its potential clinical application in leukemias. Life Sci. 2015; 122:59–64.
41. Huang S, Chen Z, Yu JF, Young D, Bashey A, Ho AD, Law P. Correlation between IL-3 receptor expression and growth potential of human CD34+ hematopoietic cells from different tissues. Stem Cells. 1999; 17:265–272.
42. van Dongen JJ, Lhermitte L, Bottcher S, Almeida J, van der Velden VH, Flores-Montero J, Rawstron A, Asnafi V, Lecrevisse Q, Lucio P, Mejstrikova E, Szczepanski T, Kalina T, et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia. 2012; 26:1908–1975.
43. Kalina T, Flores-Montero J, van der Velden VH, Martin-Ayuso M, Bottcher S, Ritgen M, Almeida J, Lhermitte L, Asnafi V, Mendonca A, de Tute R, Cullen M, Sedek L, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012; 26:1986–2010.
44. Westers TM, Ireland R, Kern W, Alhan C, Balleisen JS, Bettelheim P, Burbury K, Cullen M, Cutler JA, Della Porta MG, Drager AM, Feuillard J, Font P, et al. Standardization of flow cytometry in myelodysplastic syndromes: a report from an international consortium and the European LeukemiaNet Working Group. Leukemia. 2012; 26:1730–1741.
45. Silva ML, Ornellas de Souza MH, Ribeiro RC, Land MG, Boulhosa de Azevedo AM, Vasconcelos F, Otero L, Vasconcelos Z, Bouzas LF, Abdelhay E. Cytogenetic analysis of 100 consecutive newly diagnosed cases of acute lymphoblastic leukemia in Rio de Janeiro. Cancer Genet Cytogenet. 2002; 137:85–90.
46. Liehr T, Heller A, Starke H, Rubtsov N, Trifonov V, Mrasek K, Weise A, Kuechler A, Claussen U. Microdissection based high resolution multicolor banding for all 24 human chromosomes. Int J Mol Med. 2002; 9:335–339.