
INTRODUCTION
Monocyte chemotactic protein-1 (MCP-1) is a member of the chemokine family composed of 76 amino acids, and it is 13 kDa in size [1]. Monocyte chemotactic protein-1 is primarily identified as a potent chemotactic factor for attracting monocytes, macrophages and other inflammatory cells to the site of inflammation during tissue injury and infection [2]. Studies in recent years reveal that functions of MCP-1 are far beyond tissue repair; it participates in pathophysiological development of various diseases including cancer and obesity.
High levels of MCP-1 are associated with a poor outcome and a short disease-free survival after surgery because of the high incidence of metastasis in patients with cancer [3–5]. For example, high expression of MCP-1 is positively associated with advanced pathologic stages of prostate cancer [3] and with multiple liver metastases of colorectal cancer [5]. In breast cancer, it is a significant indicator of early relapse [4]. Animal studies indicate that MCP-1 participates in primary tumor growth and metastasis [6–8]. Treatment of mice with a neutralizing antibody to MCP-1 reduces the growth and metastasis of non-small-cell lung carcinoma [6]; stable knockdown of MCP-1 in MDA-MB-231 mammary carcinoma cells reduces their metastasis in vivo [7].
Obesity is a risk factor for cancer. Accumulation of adipose tissue in viscera is a strong indicator of detrimental health outcomes in obesity-associated diseases including cancer [9, 10]. A variety of cell types (e.g. hepatocytes, fibroblasts) produce MCP-1 [11]; among them adipocytes are recognized as an important source of MCP-1 [9, 10]. Adipose MCP-1 mRNA expression is correlated with adiposity and body mass index, and circulating MCP-1 is reduced after weight loss in obese subjects [12]. Being obese at the time of diagnosis of primary cancer can be predictive of poor prognosis. For example, breast cancer patients who are obese are at a greater risk of recurrence [13] with a shorter disease-free interval than those with normal body weight [14]. Similarly, obese or overweight prostate cancer patients are more likely to have prostate cancer recurrence after radical prostatectomy than those of normal weight [15, 16]. These observations suggest that pro-inflammatory adipokines may interact with cancer cells and play an active role in obesity-associated cancer progression.
Feeding laboratory rodents an obesogenic, high-fat diet results in increases in body fat mass and plasma concentration of MCP-1 [17–19]. In our studies on the roles of diet in metastasis using the spontaneous metastasis model of Lewis lung carcinoma (LLC), we found that high-fat diet enhances metastasis, which is accompanied by increases in plasma concentrations of adipokines including MCP-1 [20, 21]. We hypothesized that MCP-1 participates in metastatic development and growth and that adipose-produced MCP-1 contributes to the high-fat diet-enhanced metastasis. The present experiments were conducted to test the hypothesis in MCP-1 deficient (MCP-1-/-) mice using the spontaneous metastasis model of LLC.
RESULTS
Consumption of the high-fat diet, regardless of genotypes of mice, increased body weight compared to the AIN93G diet. The difference was statistically significant 3 weeks after initiation of experimental feeding (p < 0.01), and the significant increase continued throughout the experiment (Figure 1). Body weights of MCP-1-/- mice were lower than those of wild-type mice during the first 3 weeks of experimental feeding (p < 0.05); there were no differences in body weights between MCP-1-/- and wild-type mice receiving the same dietary treatment thereafter (Figure 1).
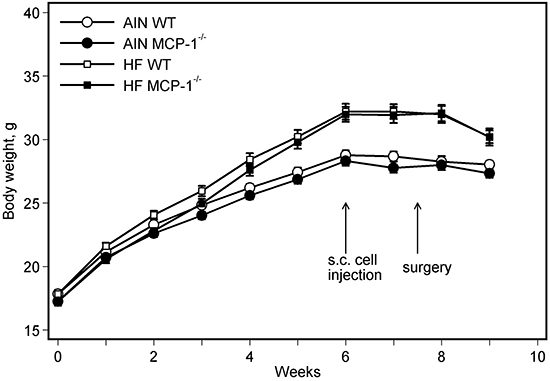
Figure 1: Body weight. Two-way ANOVA and Tukey contrasts were performed to test for differences among the groups. Mice fed the high-fat diet were heavier than those fed the AIN93G diet; the difference was significant starting 3 weeks after initiation of experimental feeding (p < 0.01). Body weights of MCP-1-/- mice were lower than those of wild-type mice during the first 3 weeks of experimental feeding (p < 0.05); there were no differences in body weights between MCP-1-/- and wild-type mice receiving the same dietary treatment thereafter. Values are means ± SEM (n = 21 per group for MCP-1-/- mice, n = 28 per group for wild-type mice). AIN: AIN93G diet; WT: wild-type mice; MCP-1-/-: MCP-1 deficient mice; HF: high-fat diet.
The high-fat diet increased percent body fat mass by 44% (p < 0.01) compared to the AIN93G diet; MCP-1 deficiency increased body fat mass by 27% (p < 0.01) compared to wild-type mice (Figure 2a). Concomitantly, the high-fat diet reduced percent body lean mass by 10% (p < 0.01) and MCP-1 deficiency reduced it by 7% (p < 0.01) compared to their respective controls (Figure 2b). There was no difference in absolute lean mass weight between groups fed the high-fat and the AIN93G diets (Figure 2c); lean mass weights of MCP-1-/- mice were 8% lower than those of wild-type mice (p < 0.01, Figure 2c). Consumption of the high-fat diet increased caloric intake by 7% (p = 0.01) compared to the AIN93G diet; there was no difference in caloric intake between MCP-1-/- and wild-type mice (Figure 2d).
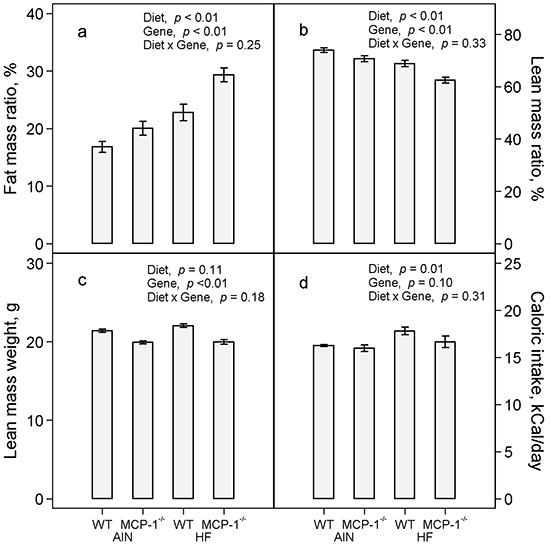
Figure 2: Fat mass:body mass ratio a. lean mass:body mass ratio b. lean mass weight c. and caloric intake d. Two-way ANOVA was performed to test for differences among the groups. Values are means ± SEM (n = 21 per group for MCP-1-/- mice, n = 28 per group for wild-type mice (a,b,c) or n = 6 per group (d)). AIN: AIN93G diet; WT: wild-type mice; MCP-1-/-: MCP-1 deficient mice; HF: high-fat diet.
Subcutaneous injection of LLC cells resulted in a primary tumor at the injection site and pulmonary metastasis. The high-fat diet increased the number of lung metastases in wild-type mice by 64% compared to their AIN93G-fed counterparts (p < 0.05, Figure 3a). Deficiency in MCP-1 reduced the number of metastases in high-fat diet-fed mice by 27% (p < 0.05), but it did not affect the number of metastases in AIN93G-fed mice (Figure 3a). Compared to the AIN93G diet, the high-fat diet increased the cross-sectional area and volume of pulmonary metastases by 46% (p < 0.01, Figure 3b) and 69% (p < 0.01, Figure 3c), respectively. Compared to wild-type mice, deficiency in MCP-1 reduced the cross-sectional area and volume of metastases by 17% (p < 0.05, Figure 3b) and 21% (p < 0.05, Figure 3c), respectively.
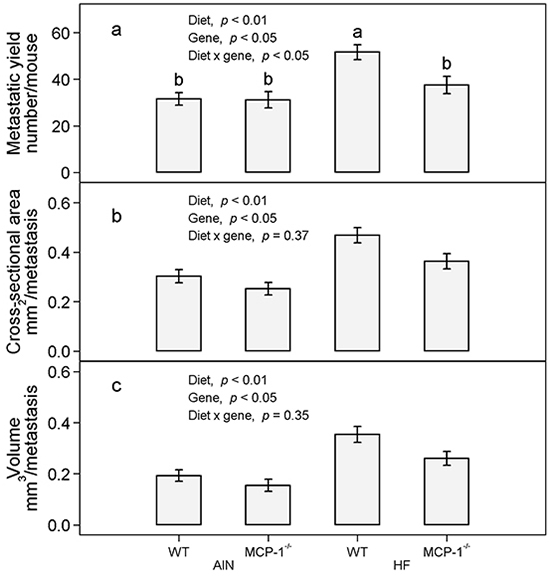
Figure 3: The number a. cross-sectional area b. and volume c. of pulmonary metastases in MCP-1-/- and wild-type mice fed the AIN93G or the high-fat diet. Two-way ANOVA and Tukey contrasts were performed to test for differences among the groups. Values (means ± SEM) with different superscripts are significantly different at p < 0.05 (n = 21 per group for MCP-1-/- mice, n = 28 per group for wild-type mice). AIN: AIN93G diet; WT: wild-type mice; MCP-1-/-: MCP-1 deficient mice; HF: high-fat diet.
In wild-type mice fed the AIN93G diet, LLC increased concentrations of MCP-1 in adipose tissue by 52% (p < 0.01, Figure 4a) and plasma by 89% (p < 0.01, Figure 4b) compared to non-tumor-bearing controls. The high-fat diet further increased concentrations of MCP-1 in adipose tissue by 37% (p < 0.01, Figure 4a) and in plasma by 45% (p < 0.05, Figure 4b) in LLC-bearing mice compared to the AIN93G diet. Monocyte chemotactic protein-1 was not detectable in adipose tissue and plasma of MCP-1-/- mice regardless of diet (Figure 4a and 4b). The concentration of MCP-1 in 24-hour-old serum-free conditioned medium from LLC cells was 2.0 ± 0.1 ng/mg protein (n = 10). Concentrations of MCP-1 in primary tumors of MCP-1-/- and wild-type mice were 235.0 ± 10.1 and 217.0 ± 10.1 pg/mg protein (p = 0.21, n = 20), respectively.
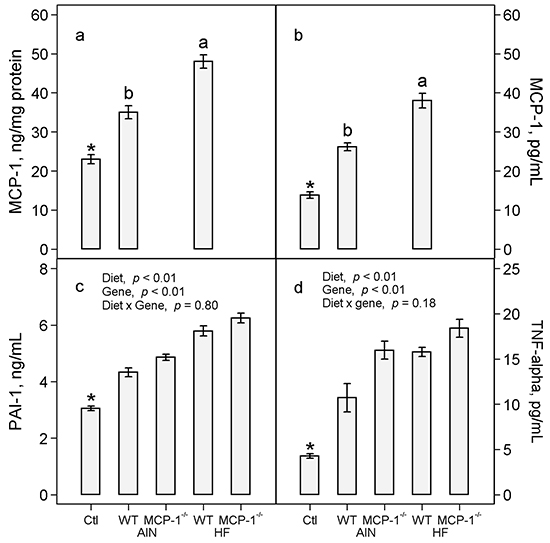
Figure 4: Concentrations of MCP-1 in adipose tissue a. and plasma b. and concentrations of PAI-1 c. and TNF-α d. in plasma. Two-way ANOVA and Tukey contrasts were performed to compare differences among the groups of LLC-bearing mice; a priori contrasts were performed to compare differences in mice fed the AIN93G diet with or without LLC. Values (means ± SEM) with different superscripts are significantly different at p ≤ 0.05 for LLC-bearing groups (n = 10 per group). *p < 0.01 compared to AIN WT. Ctl: non-tumor-bearing wild-type mice fed the AIN93G diet; AIN: AIN93G diet; WT: wild-type mice; MCP-1-/-: MCP-1 deficient mice; HF: high-fat diet.
Lewis lung carcinoma increased plasma concentrations of PAI-1 by 42% (p < 0.01, Figure 4c) and TNF-α by 2.5 fold (p < 0.01, Figure 4d) in wild-type mice compared to non-tumor-bearing controls fed the AIN93G diet. The high-fat diet increased plasma PAI-1 by 31% (p < 0.01, Figure 4c) and TNF-α by 28% (p < 0.01, Figure 4d) in LLC-bearing mice compared to the AIN93G diet. Deficiency in MCP-1 increased PAI-1 by 10% (p < 0.01, Figure 4c) and TNF-α by 30% (p < 0.01, Figure 4d) compared to wild-type mice.
Mice with LLC exhibited a 15% increase in plasma concentration of VEGF (p < 0.01, Figure 5a) and a 28% increase in TIMP-1 (p = 0.01, Figure 5b) compared to non-tumor-bearing controls. The high-fat diet increased plasma VEGF by 14% (p < 0.01, Figure 5a) and TIMP-1 by 24% (p < 0.01, Figure 5b) in LLC-bearing mice compared to the AIN93G diet. Deficiency in MCP-1 increased plasma VEGF by 11% (p < 0.01, Figure 5a) and TIMP-1 by 11% (p < 0.05, Figure 5b) compared to wild-type mice.
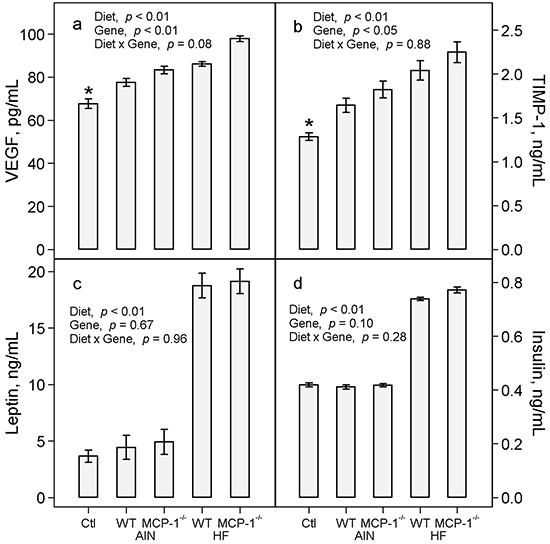
Figure 5: Plasma concentrations of VEGF a. TIMP-1 b. leptin c. and insulin d. Two-way ANOVA was performed to compare differences among the groups of LLC-bearing mice; a priori contrasts were performed to compare differences in mice fed the AIN93G diet with or without LLC. Values are means ± SEM (n = 10 per group). *p ≤ 0.01 compared to AIN WT. Ctl: non-tumor-bearing wild-type mice fed the AIN93G diet; AIN: AIN93G diet; WT: wild-type mice; MCP-1-/-: MCP-1 deficient mice; HF: high-fat diet.
There were no differences in plasma concentrations of leptin and insulin between LLC-bearing and non-tumor-bearing wild-type mice (Figure 5c and 5d). Consumption of the high-fat diet resulted in a 4-fold increase in plasma leptin (p < 0.01, Figure 5c) and an 82% increase in plasma insulin (p < 0.01, Figure 5d) compared to the AIN93G diet. Deficiency in MCP-1 did not affect plasma concentrations of either compared to wild-type mice (Figure 5c and 5d).
DISCUSSION
Results from the present study are consistent with previous reports that feeding mice an obesogenic, high-fat diet elevates plasma concentration of MCP-1 [18, 19] and enhances pulmonary metastasis [20, 22]. The key finding is that deficiency in MCP-1 reduces high-fat diet-enhanced metastasis, reflected by reductions in both number and size of metastases formed in lungs (Figure 3). It indicates that MCP-1 plays an active role in promoting metastatic progression and that adipose-produced MCP-1 may contribute, at least partly, to high-fat diet-enhanced metastasis. These findings are supported by previous studies showing that treatment of mice with a neutralizing antibody to MCP-1 reduces the growth and metastasis of non-small-cell lung carcinoma [6].
Deficiency in MCP-1 resulted in significant elevations of plasma concentrations of pro-inflammatory cytokines (PAI-1, TNF-α) and angiogenic factors (VEGF, TIMP-1) in LLC-bearing mice (Figures 4 and 5). These elevations apparently compensate for the absence of MCP-1. Plasminogen activator inhibitor-1 promotes metastasis [22, 23] and malignant growth [22, 24]. Tumor necrosis factor-α [25, 26], VEGF [27, 28] and TIMP-1 [29] are potent tumorigenic and angiogenic factors that contribute to cancer progression. Thus, the observed elevations of pro-inflammatory cytokines and angiogenic factors may offset the absence of MCP-1, thereby supporting the metastatic progression of LLC in MCP-1-/- mice.
The mechanisms by which MCP-1 deficiency increases plasma concentrations of pro-inflammatory cytokines and angiogenic factor remain to be elucidated. Available studies show that TNF-α induces the expression of MCP-1 in cancer-associated fibroblasts and mesenchymal stem cells by up-regulating the nuclear factor-κB (NF- κB) pathway [30]. The elevation in plasma concentration of TNF-α in the absence of MCP-1 may be due to a feedback response via the NF-κB/TNF-α pathway.
Concentrations of the adipokine MCP-1 in adipose tissue were elevated in LLC-bearing wild-type mice fed the AIN93G diet (Figure 4). Elevated expression of adipokines during interaction between adipocytes and cancer cells contributes to cancer progression. For example, concentration of MCP-1 is greater in cancer-associated adipocytes from human mastectomy samples than normal adipocytes [31], and MCP-1 expression is higher in peritumoral adipose tissue than control adipose tissue in mice [32]. The LLC cell line used in this study is a variant that metastasizes specifically to lungs [33]. Thus, the observed elevation of MCP-1 in adipose tissue is unlikely a result of interactions between cancer cells and adipocytes but may be through mechanisms related to other aspects of metastasis. This warrants further investigation.
Consistent with our previous reports [18, 22], feeding mice the high-fat diet elevated concentrations of leptin and insulin in plasma (Figure 5). The lack of differences in these measurements between MCP-1-/- and wild-type mice indicates that the observed elevations are independent from MCP-1 status of the mice. Elevations of leptin and insulin contribute to obesity in rodent models [18, 34]. Leptin is angiogenic during tumorigenesis [35] and insulin is involved in type-2 diabetes-mediated mammary tumor progression [36] in mice. Thus, the present results raise the possibility that elevations of leptin and insulin may contribute to the high-fat diet-enhanced metastasis.
The finding that MCP-1-/- mice gained more body fat mass than their wild-type counterparts (Figure 2) was unexpected. However, these results are supported by previous reports that MCP-1-/- mice fed a high-fat diet gain more body fat mass than wild-type mice [37] and that obese MCP-1-/- mice have larger adipocytes [38]. Taken together, these results indicate that MCP-1 deficiency does not attenuate diet-induced adipogenesis but increases body fat mass build-up in this rodent model.
It is generally accepted that it is the fat content of a diet that is responsible for the increases in body adiposity and its associated adipokines. Studies of obesity in mouse models commonly use 45% of energy from fat in diets. We recently reported that restricted feeding of a high-fat diet during the dark cycle (12 hours) does not affect energy intake but reduces body fat mass and plasma concentration of MCP-1 compared to mice fed the same diet ad libitum [39]. It suggests that it is not the fat content of a diet but the disruption of diurnal rhythm of food intake that may be responsible for adipogenesis in mice. This needs to be tested on high-fat diet-enhanced metastasis.
In summary, results from this study showed that MCP-1 deficiency ameliorates metastasis in high-fat diet-fed mice indicating that adipose-produced MCP-1 contributes, at least partly, to high-fat diet-enhanced metastasis. Furthermore, results that plasma concentrations of pro-inflammatory cytokines and angiogenic factors were elevated in MCP-1-/- mice indicates the existence of compensatory mechanisms that up regulates the production of these cancer promoting factors, which support LLC progression in the absence of MCP-1. These results suggest that inhibition of MCP-1 expression, either by preventive or therapeutic approaches, may attenuate metastatic progression. Elevations in plasma levels of pro-inflammatory cytokines and angiogenic factors in the absence of MCP-1 suggest the existence of compensatory responses that may counteract treatments aimed at MCP-1 inhibition.
MATERIALS AND METHODS
Animals and diets
Four to five-week-old male MCP-1 deficient mice (MCP-1-/-, B6.129S4-Ccl2tm1Rol/J) on a C57BL/6J background and C57BL/6J wild-type mice (The Jackson Laboratory, Bar Harbor, ME) were maintained in a pathogen-free room on a 12:12-hour light/dark cycle with a temperature of 22 ± 1°C. A modified AIN93G formulation [40] providing 16% or 45% (high-fat diet) of energy from corn oil was used in this study (Table 1). Gross energy of each diet (Table 1) was analyzed by oxygen bomb calorimetry (Model 6200, Oxygen Bomb Calorimeter, Parr Instrument, Moline, IL). All diets were powder diets; they were stored at −20°C until being provided to mice. Mice had free access to their diets and deionized water; food intake was recorded for 3 weeks before injection of cancer cells. Mice were weighed weekly; body composition was assessed in conscious, immobilized mice using quantitative magnetic resonance imaging 1 week before the cancer cell injection (Echo whole-body composition analyzer, Model 100, Echo Medical System, Houston, TX).
Table 1: Composition of experimental diets
Ingredient |
Modified AIN93G g/kg |
High-fat g/kg |
|---|---|---|
Corn starch |
390 |
34 |
Casein |
200 |
239 |
Dextrin |
132 |
239 |
Sucrose |
100 |
120 |
Corn oil |
70 |
229 |
Cellulose |
50 |
60 |
AIN93 mineral mix |
35 |
42 |
AIN93 vitamin mix |
10 |
12 |
L-cystine |
4.4 |
5 |
L-methionine |
0.3 |
0.4 |
Choline bitartrate |
2.5 |
3 |
Sodium carbonate |
6.22 |
7 |
t-Butylhydroquinone |
0.01 |
0.05 |
Total |
1000 |
1000 |
Energy |
% |
% |
Protein |
20 |
20 |
Fat |
16 |
45 |
Carbohydrate |
64 |
35 |
Gross energya, kCal/g |
4.4 ± 0.1 |
5.4 ± 0.1 |
an =3 for each diet.
Lewis lung carcinoma cells
Lewis lung carcinoma cells (provided by Dr. Pnina Brodt, McGill University, Montreal, Quebec, Canada) were cultured with RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum and maintained in a humidified atmosphere of 5% CO2 in air at 37°C. Cells used for animal studies were in vivo-selected once [21]; they were monitored for phenotype (by microscopic examination of cell morphology), proliferation properties (by growth curve analysis) and metastatic capability (by injecting cells subcutaneously into mice and examining metastatic formation in lungs). Cells were free of mycoplasma by Hoechst DNA staining and direct culture tests (performed by the American Type Culture Collection, Manassas, VA). Such assessments showed that cell identity and metastatic behavior were similar to those of original stocks from the institution providing the cell line.
Experimental design
This study was approved by the Animal Care and Use Committee of the U.S. Department of Agriculture, Agricultural Research Service, Grand Forks Human Nutrition Research Center. The procedures followed the National Institutes of Health guidelines for the care and use of laboratory animals [41]. Mice (n = 21 per group for MCP-1-/- mice, n = 28 per group for wild-type mice) were fed the AIN93G control or high-fat diet for 7 weeks before they were subcutaneously injected with 2.5 x 105 viable LLC cells per mouse into the lower dorsal region. The resulting primary tumor was removed surgically 11 days later when it was approximately 1 cm in diameter; after that mice were maintained on their respective diets for an additional 10 days. Mice fed the AIN93G diet but not injected with LLC cells were used as non-tumor-bearing controls (n = 18) in comparison of plasma concentrations of pro-inflammatory cytokines, angiogenic factors and insulin to LLC-bearing mice fed the same AIN93G diet. At termination, mice were euthanized with an intraperitoneal injection of a mixture of ketamine and xylazine; their lungs were harvested and fixed with Bouin’s solution. The number of pulmonary metastases was counted and the cross-sectional area and the average diameter of each metastasis were measured using an ImagePro-Plus software- (Media Cybernetics, Silver Spring, MD) and camera-equipped stereomicroscope. The cross-sectional area was defined as the surface area of each lung metastasis. The average diameter was measured at two degree intervals joining two outline points and passing through the centroid. The volume of each metastasis was estimated with the assumption that metastases were spherical and calculated using the average diameter measured [42]. Epididymal adipose tissue and plasma were collected and stored at −80°C for further analyses.
Quantification of cytokines, angiogenic factors and related biomarkers
Concentrations of MCP-1 in epididymal adipose tissue, plasma, 24-hours-old serum-free conditioned medium from LLC cells and primary tumors were quantified by using a sandwich enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN). Primary tumors frozen in liquid nitrogen were pulverized under liquid nitrogen. Total protein from adipose tissue and primary tumors was extracted in RIPA buffer with protease and phosphatase inhibitors [43]. Protein concentrations of the adipose and primary tumor extract and LLC cell lysate were estimated by using the BCA protein assay (Thermo Scientific, Waltham, MA). Concentrations of MCP-1 in adipose tissue, primary tumor and conditioned medium were normalized to protein content. Concentrations of plasminogen activator inhibitor-1 (PAI-1, Molecular Innovations, Inc., Novi, MI), insulin (Mercodia, Inc., Winston Salem, NC), tumor necrosis factor-α (TNF-α), leptin, vascular endothelial growth factor (VEGF) and tissue inhibitor of metalloproteinase-1 (TIMP-1, R&D Systems, Minneapolis, MN) in plasma were quantified by using ELISA kits following manufacturers’ protocols. Samples were read within the linear range of the assay, and the accuracy of the analysis was confirmed with the controls provided in each kit.
Statistical analyses
The effects of diet (AIN93G or high-fat), genotype (MCP-1-/- or wild-type) and their interaction were tested by using two-way analysis of variance (ANOVA). In case of a significant interaction between diet and genotype, Tukey contrasts were performed to compare the 4 dietary groups. To examine the effect of LLC on plasma concentrations of cytokines, angiogenic factors and insulin, a priori contrasts were used to test for differences in wild-type mice fed the AIN93G diet with or without LLC. A mixed model ANOVA with mouse as the random blocking factor and with diet, genotype and their interaction as fixed effects was used to test for differences in cross-sectional area and volume of metastases among the groups. All data are presented as means ± standard error of the mean (SEM). Differences with a p-value of 0.05 or less were considered statistically significant. All analyses were performed by using SAS software (version 9.4, SAS Institute, Cary, NC).
ACKNOWLEDGMENTS
The authors greatly acknowledge the assistance of the following staff of Grand Forks Human Nutrition Research Center: Lana DeMars and Kay Keehr for technical support, LuAnn Johnson for advice in statistical analysis, Dr. Forrest Nielsen for providing input in manuscript revision, James Lindlauf for preparing experimental diets and vivarium staff for providing high quality animal care.
CONFLICTS OF INTEREST
The authors have declared that no competing interests exist.
GRANT SUPPORT
This work was supported by the U.S. Department of Agriculture, Agriculture Research Service, Research Project 3062-51000-050-00D.
Notes
The U.S. Department of Agriculture, Agricultural Research Service, Plains Area, is an equal opportunity/affirmative action employer and all agency services are available without discrimination. Mention of trade names or commercial products in this article is solely for providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
REFERENCES
1. Robinson EA, Yoshimura T, Leonard EJ, Tanaka S, Griffin PR, Shabanowitz J, Hunt DF and Appella E. Complete amino acid sequence of a human monocyte chemoattractant, a putative mediator of cellular immune reactions. Proc Natl Acad Sci U S A. 1989; 86:1850-1854.
2. Carr MW, Roth SJ, Luther E, Rose SS and Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994; 91:3652-3656.
3. Lu Y, Cai Z, Galson DL, Xiao G, Liu Y, George DE, Melhem MF, Yao Z and Zhang J. Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate. 2006; 66:1311-1318.
4. Lebrecht A, Grimm C, Lantzsch T, Ludwig E, Hefler L, Ulbrich E and Koelbl H. Monocyte chemoattractant protein-1 serum levels in patients with breast cancer. Tumour Biol. 2004; 25:14-17.
5. Yoshidome H, Kohno H, Shida T, Kimura F, Shimizu H, Ohtsuka M, Nakatani Y and Miyazaki M. Significance of monocyte chemoattractant protein-1 in angiogenesis and survival in colorectal liver metastases. Int J Oncol. 2009; 34:923-930.
6. Fridlender ZG, Kapoor V, Buchlis G, Cheng G, Sun J, Wang LC, Singhal S, Snyder LA and Albelda SM. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am J Respir Cell Mol Biol. 2011; 44:230-237.
7. Nam JS, Kang MJ, Suchar AM, Shimamura T, Kohn EA, Michalowska AM, Jordan VC, Hirohashi S and Wakefield LM. Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of dysadherin in human breast cancer cells. Cancer Res. 2006; 66:7176-7184.
8. Bussard KM, Okita N, Sharkey N, Neuberger T, Webb A and Mastro AM. Localization of osteoblast inflammatory cytokines MCP-1 and VEGF to the matrix of the trabecula of the femur, a target area for metastatic breast cancer cell colonization. Clin Exp Metastasis. 2010; 27:331-340.
9. Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, Zlabinger GJ and Stulnig TM. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab. 2008; 93:3215-3221.
10. Bruun JM, Lihn AS, Pedersen SB and Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. J Clin Endocrinol Metab. 2005; 90:2282-2289.
11. Van Coillie E, Van Damme J and Opdenakker G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. 1999; 10:61-86.
12. Christiansen T, Richelsen B and Bruun JM. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes. 2005; 29:146-150.
13. Daniell HW. Increased lymph node metastases at mastectomy for breast cancer associated with host obesity, cigarette smoking, age, and large tumor size. Cancer. 1988; 62:429-435.
14. Loi S, Milne RL, Friedlander ML, McCredie MR, Giles GG, Hopper JL and Phillips KA. Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol Biomarkers Prev. 2005; 14:1686-1691.
15. Bassett WW, Cooperberg MR, Sadetsky N, Silva S, DuChane J, Pasta DJ, Chan JM, Anast JW, Carroll PR and Kane CJ. Impact of obesity on prostate cancer recurrence after radical prostatectomy: data from CaPSURE. Urology. 2005; 66:1060-1065.
16. Amling CL, Riffenburgh RH, Sun L, Moul JW, Lance RS, Kusuda L, Sexton WJ, Soderdahl DW, Donahue TF, Foley JP, Chung AK and McLeod DG. Pathologic variables and recurrence rates as related to obesity and race in men with prostate cancer undergoing radical prostatectomy. J Clin Oncol. 2004; 22:439-445.
17. Takahashi K, Mizuarai S, Araki H, Mashiko S, Ishihara A, Kanatani A, Itadani H and Kotani H. Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. J Biol Chem. 2003; 278:46654-46660.
18. Yan L, Graef GL, Claycombe KJ and Johnson LK. Effects of voluntary running and soy supplementation on diet-induced metabolic disturbances and inflammation in mice. J Agric Food Chem. 2013; 61:9373-9379.
19. Sundaram S, Bukowski MR, Lie WR, Picklo MJ and Yan L. High-Fat Diets Containing Different Amounts of n3 and n6 Polyunsaturated Fatty Acids Modulate Inflammatory Cytokine Production in Mice. Lipids. 2015; doi: 10.1007/s11745-015-4093-x.
20. Yan L and Combs GFJ. Consumption of a high-fat diet abrogates inhibitory effects of methylseleninic acid on spontaneous metastasis of Lewis lung carcinoma in mice. Carcinogenesis. 2014; 35:2308-2313.
21. Yan L and Demars LC. Effects of dietary fat on spontaneous metastasis of Lewis lung carcinoma in mice. Clin Exp Metastasis. 2010; 27:581-590.
22. Yan L and DeMars LC. Effects of a high-fat diet on spontaneous metastasis of Lewis lung carcinoma in plasminogen activator inhibitor-1 deficient and wild-type mice. PLoS One. 2014; 9:e110869.
23. Tsuchiya H, Sunayama C, Okada G, Matsuda E, Tomita K and Binder BR. Plasminogen activator inhibitor-1 accelerates lung metastasis formation of human fibrosarcoma cells. Anticancer Res. 1997; 17:313-316.
24. McMahon GA, Petitclerc E, Stefansson S, Smith E, Wong MK, Westrick RJ, Ginsburg D, Brooks PC and Lawrence DA. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. J Biol Chem. 2001; 276:33964-33968.
25. Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A and Balkwill F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007; 67:585-592.
26. Waters JP, Pober JS and Bradley JR. Tumour necrosis factor and cancer. J Pathol. 2013; 230:241-248.
27. Volm M, Koomagi R and Mattern J. Prognostic value of vascular endothelial growth factor and its receptor Flt-1 in squamous cell lung cancer. Int J Cancer. 1997; 74:64-68.
28. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS and Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993; 362:841-844.
29. Kopitz C, Gerg M, Bandapalli OR, Ister D, Pennington CJ, Hauser S, Flechsig C, Krell HW, Antolovic D, Brew K, Nagase H, Stangl M, von Weyhern CW, Brucher BL, Brand K, Coussens LM, et al. Tissue inhibitor of metalloproteinases-1 promotes liver metastasis by induction of hepatocyte growth factor signaling. Cancer Res. 2007; 67:8615-8623.
30. Katanov C, Lerrer S, Liubomirski Y, Leider-Trejo L, Meshel T, Bar J, Feniger-Barish R, Kamer I, Soria-Artzi G, Kahani H, Banerjee D and Ben-Baruch A. Regulation of the inflammatory profile of stromal cells in human breast cancer: prominent roles for TNF-alpha and the NF-kappaB pathway. Stem Cell Res Ther. 2015; 6:87.
31. Fujisaki K, Fujimoto H, Sangai T, Nagashima T, Sakakibara M, Shiina N, Kuroda M, Aoyagi Y and Miyazaki M. Cancer-mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res Treat. 2015; 150:255-263.
32. Wagner M, Bjerkvig R, Wiig H, Melero-Martin JM, Lin RZ, Klagsbrun M and Dudley AC. Inflamed tumor-associated adipose tissue is a depot for macrophages that stimulate tumor growth and angiogenesis. Angiogenesis. 2012; 15:481-495.
33. Brodt P. Characterization of two highly metastatic variants of Lewis lung carcinoma with different organ specificities. Cancer Res. 1986; 46:2442-2448.
34. Fenton JI, Nunez NP, Yakar S, Perkins SN, Hord NG and Hursting SD. Diet-induced adiposity alters the serum profile of inflammation in C57BL/6N mice as measured by antibody array. Diabetes Obes Metab. 2009; 11:343-354.
35. Rene Gonzalez R, Watters A, Xu Y, Singh UP, Mann DR, Rueda BR and Penichet ML. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009; 11:R36.
36. Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S and LeRoith D. Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes. 2010; 59:686-693.
37. Inouye KE, Shi H, Howard JK, Daly CH, Lord GM, Rollins BJ and Flier JS. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes. 2007; 56:2242-2250.
38. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K and Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006; 116:1494-1505.
39. Sundaram S and Yan L. Time-restricted feeding reduces adiposity in mice fed a high-fat diet. Nutr Res. 2016; doi: 10.1016/j.nutres.2016.02.005.
40. Reeves PG, Nielsen FH and Fahey GCJ. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993; 123:1939-1951.
41. Institute for Laboratory Animal Research. (2011). Guide for the Care and Use of Laboratory Animals. (Washington, D.C.: National Academies Press).
42. Welch DR, Neri A and Nicolson GL. Comparison of ‘spontaneous’ and ‘experimental’ metastasis using rat 13726 mammary adenocarcinoma metastatic cell clones. Invasion Metastasis. 1983; 3:65-80.
43. Ortega FJ, Moreno-Navarrete JM, Mayas D, Garcia-Santos E, Gomez-Serrano M, Rodriguez-Hermosa JI, Ruiz B, Ricart W, Tinahones FJ, Fruhbeck G, Peral B and Fernandez-Real JM. Breast cancer 1 (BrCa1) may be behind decreased lipogenesis in adipose tissue from obese subjects. PLoS One. 2012; 7:e33233.