
Introduction
Azadirachtaindica, commonly known as neem, is a tree belonging to the Meliceae family, which is native to India and the Indian Subcontinent. Today, neem is well distributed across the world in at least 30 countries including Asia, Africa, and America [1]. Natural compounds that have been identified from various parts of A.indica include azadirachtin, salannin, nimbolide, nimbin, and nimbic acid [2]. Among them, nimbolide is a major tertranortriterpenoid, isolated from the leaves of A.indica. Hence, nimbolideis accessible and affordable, owing to the abundance of neem trees in tropical regions. More importantly, nimbolide is well known for its many uses, such as anti-malaria [3], antibacterial activity against S.aureus and S.coagulase [4], anti-feedant [5], and insecticidal activity [6]. In addition, nimbolidehas been found to possess antioxidant effect and free radical scavenging activities. In comparison toazadirachtin and ascorbic acid (vitamin C), nimbolide was shown to be a more potent antioxidant [7]. Furthermore, nimbolidehas been identified as one of the active ingredients of neem extract, a widely available herbal product used in traditional Indian Ayurvedic medicine to treat acne, wound, gastric ulcer, and infections [8, 9]. In a study by Cohen and co-workers, nimbolide was reported to be the most potent cytotoxic substance among 6 limonoids examined [10]. Through investigation of nimblolide’s action in multiple cancer cell lines, various molecular targets were thus identified. Hence, the mechanistic interpretation of its pharmacodynamic actions can be derived. The major signaling pathways by which nimbolide exerts its effects are represented in Figure 1.
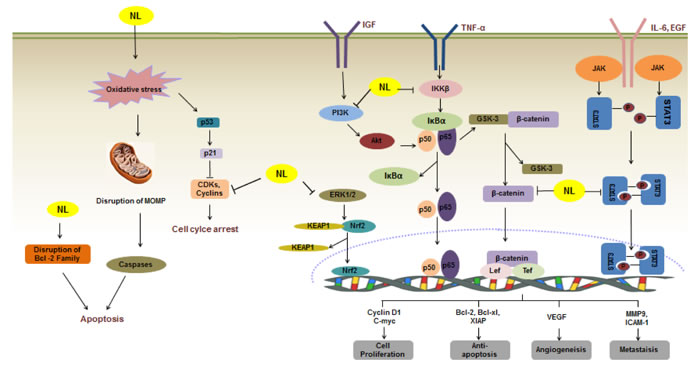
Figure 1: A schematic illustration of the molecular mechanisms of the anticancer action of nimbolide [91]. Nimbolide induces apoptosis through diverse molecular mechanism(s). Nimbolide disrupts MOMP, which promotes caspases activation leading to apoptosis. Nimbolide also reduces the level of CDKs and cyclins, causing cell cycle arrest. Nimbolide also abrogates various signaling cascades including MAPK (ERK1/2), JAK2/STAT3 and PI3K/Akt, leading to suppression of proliferation of a wide variety of human cancer cells. Nimbolide disrupts the Nrf2-KEAP1 complex and promotes the release of Nrf2, thus increasing levels of antioxidant and detoxification enzymes. Nimbolide suppresses IκBα degradation and prevents nuclear translocation of p65, therefore repressing the expression of various genes involved in cell proliferation, anti-apoptosis, angiogenesis and metastasis. Inhibition of the NF-κB pathway also reduces the dissociation of GSK-3β from β-catenin, hence restraining the Wnt/β-catenin signaling pathway. →: activate/induce; Long Left Tack: suppress/inhibit.
In addition, the anticancer and cancer preventive effects of nimbolide were observed in several animal studies [11-13], thereby providing strong evidence to support its further development as a therapeutic agent against cancer. In accordance with the drug development pipeline [14], nimbolide is now at the stage of preclinical pharmacological investigation, where pharmacodynamics (PD), toxicology, and pharmacokinetic (PK) studies should be performed before the drug can proceed to the clinical testing phase. Although the development of various separation techniques [15-18] has facilitated a highly efficient extraction of nimbolide for use in many in vitro experiments to elucidate its potential therapeutic actions, the absence of information on its absorption, distribution, metabolism, and elimination (ADME), as well as its toxicity, in turn hampers further clinical studies on this natural compound. Therefore, research on its PK characteristics and toxicological profile should be carried out to facilitate and accelerate its development as an anticancer drug candidate for clinical trials. The objectives of the present review are to summarize the current status of nimbolideas a chemopreventive and chemotherapeutic agent in specific cancer types based on pre-clinical studies, as well as to highlight the importance of performing toxicological and PK studies that will lay the foundation for its early-phase clinical investigation.
Methodology
Web of Science and Google scholar database searches were conducted during August -September 2015 to identify and retrieve articles related to nimbolide and its derivatives. Search terms included “nimbolide”, ”neem”, “Azadirachtaindica “and” nimbolide derivatives”. The Boolean operator ‘OR’ was placed between each of the search words. The reference list of each article was analysed in detail to identify additional relevant articles. The reviewer read each article in full text (n=70), evaluated its relevance and recorded the findings in a table. The table included a summary of each article’s content and its relevance on a scale rating (not relevant, relevant, very relevant). The “relevant” and “very relevant” articles (n=55) were further reviewed to identify key pharmacodynamic properties of nimbolide.
Chemical structure and properties of nimbolide
The molecular formula of nimbolide [systematic name: (4alpha, 5alpha, 6alpha, 7alpha, 15beta, 17alpha)-7,1521, 23-diepoxy-6-hydroxy-4,8-dimethyl-1-oxo-18,24-dinor-11,12-secochola-2,13,20,22-tetraene-4,11-dicarboxylic acid gamma-lactone methyl ester], is C27H30O7 [19]. Nimbolide has a decalinskeleton [20] and belongs to a group of tetranortriterpenoids called C-secoMeliacins [2]. The 2D chemical structure of nimbolide is illustrated in Figure 2.
Nimbolide was first extracted from fresh leaves of a Nigerian sample of A.indicaby using petroleum spirit. Nimbolide has molecular mass 466, melting point 245-2470C and optical rotation of [a]D+2060. Its infrared radiation (IR) spectrum shows carbonyl absorptions at 1665 (cyclohexenone), 1720 (CO2CH3) and 1770cm-1 (γ-lactone) [17]. Using this method, nimbolide was obtained as colorless plates following crystallization from acetone [15]. Another method of nimbolide isolation by column chromatography from crude extract of A.indicah as also been reported [16].
The significant cell cytotoxicity and anticancer effect of nimbolide was most likely due to the presence of anα, β-unsaturated ketone structural element. In addition, a γ-lactone moiety was also reported to be responsible for its cytotoxicity [15]. Furthermore, structural modifications of nimbolide to various amide derivatives have conferred improved cytotoxicity. In 2006, Sastry et al. reported that two of the nimbolide derivatives possessed stronger inhibitory activities than nimbolide against six cancer cell lines, namely, HT-29 (colon), SW-620 (colon), HOP-62 (lung), A-549 (lung), PC-3 (prostate), and OVCAR-5 (ovary) [21].
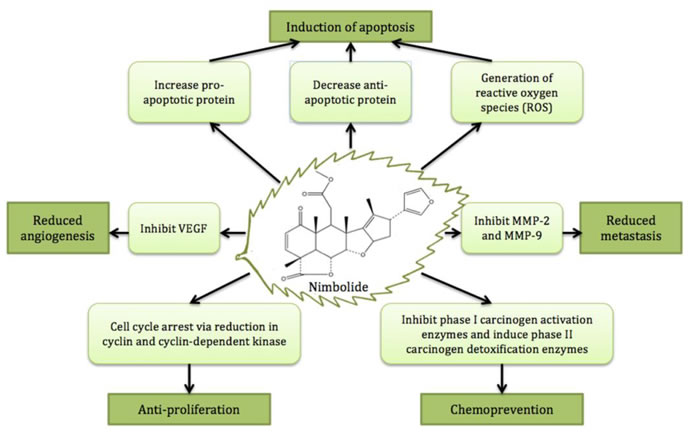
Figure 2: The major anticancer activities and cancer preventive effect of nimbolide.
Anti-cancer activities
Various PD studies of nimbolide on different cancer types have been conducted. There is strong evidence to support its activity against a wide range of malignancies from in vitro studies and in vivo animal models.
In vitro studies
Nimbolide has been shown to exhibit anticancer effects on a variety of cancer cell lines through disturbing the cell cycle of cancer cells. For instance, in human colon cancer cells, nimbolide induced cell cycle arrest and abrogated cell growth [22]. Nimbolide also exerted a strong inhibitory effect on the growth of colon cancer (HT-29) cellsdue to cell cycle arrest. In another study [23], flowcytometric analysis of U937 cells (leukemic cell lines) showed that nimbolide treatment (1–2.5 μM) resulted in cell cycle disruption by decreasing the number of cells in G0/G1 phase. Besides these, the anti-proliferation effect on cancer cells was also evident by the reduction of Ki-67 protein [24] in Waldenstrom macroglobulinemia (a type of B lymphocyte cancer) [11] and glioblastoma multiforme [12].
More importantly, nimbolide showed a strong apoptotic effect on cancer cells. It also has an inhibitory role on metastasis and angiogenesis of cancer cells. For example, nimbolide treatment resulted in 50% inhibition of the prostate cancer PC-3 cell line at a concentration of 2µM. This was accompanied by the induction of cellular apoptosis [25]. Similarly, nimbolide was shown to induce apoptosis in human osteocarcinoma [26], Waldenstrom macroglobulinemia (WM) [11], human cervical cancer [27], and hepatocarcinoma cells [28]. Additionally, an in vitro study further indicated that nimbolide was able to combat metastasis and angiogenesis of cancer cells [29]. For instance, nimbolide have been shown to effectively decrease breast cancer cell invasion and migration via the transwell invasion and wound healing assays [30].
In order to comprehensively analyze the anticancer activity of nimbolide, the half inhibitory concentrations (IC50) of nimbolide in different cancer types were compared, ranked, and presented in Table 1. The IC50values for various cancer types tested range between 0.2 to 15 μM. WM emerged as the best target for nimbolide with the lowest IC50value, which was in the nanomolar range. Since nimbolide showed remarkable potency against WM, the development and research of nimbolide as potential drug candidate for WM should be prioritized. In addition, nimbolide has also been found to be quite effective against leukemia, choriocarcinoma and colon cancer, with IC50values slightly above 1 μM [22, 23, 31]. Further investigation on these 3 cancer types is also warranted.
Table 1: Summary of in vitro IC50 of nimbolide in different cancer types
Rank |
Cancer Type |
In Vitro Concentration (IC50) |
Reference |
1 |
Waldenstrom Macroglobulinemia: cancer of B lymphocytes |
0.20 μM |
[11] |
2 |
Leukemia |
1.12 μM |
[23] |
3 |
Choriocarcinoma: trophoblastic cancer |
1.19 μM |
[31] |
4 |
Colon cancer |
1.25 μM |
[22] |
5 |
Melanoma: skin cancer |
1.74 μM |
[23] |
6 |
Prostate cancer |
2.00 μM |
[25] |
7 |
Glioblastoma multiforme: primary brain tumour |
3.00 μM |
[12] |
8 |
Breast Cancer |
4.00 μM |
[92] |
9 |
Osteocarcinoma: bone cancer |
4.30 μM |
[10] |
10 |
Cervical cancer |
5.00 μM |
[27] |
10 |
Liver cancer |
5.00 μM |
[28] |
12 |
Neuroblastoma |
5.20 μM |
[10] |
13 |
Lung cancer |
15.6 μM |
[21] |
In vivo studies
Beside in vitro investigations, in vivo studies have been performed on the preclinical effect of nimbolide against colorectal, lymphoma, andbrain cancers. Firstly, nimbolide was shown to possess a dose-dependent anticancer effect on colorectal cancer. When nimbolide was injected intraperitoneally (i.p.) for 10 days at a dose of 5mg/kg, a 67% reduction in the volume of colorectal cancer xenografts in mice was observed. At a higher dose of 20 mg/kg, an even more substantial and remarkable 90% reduction in tumour volume was achieved [13]. This was further accompanied by inhibition of angiogenesis and tumour metastasis, there by highlighting the promising activity of nimbolide against this cancer.
However, incongruency between the anticancer potency of nimbolide between in vivo doses and in vitro IC50 has been reported. In a WM tumour xenografted mouse study [11], Chitta et al. reported that nimbolide was only effective against WM tumours at very high doses of 100-200 mg/kg, i.p. after 26days of treatment. These doses arehowever very close to the previously reported median lethal dose (LD50) of nimbolide(280 mg/kg) in adult female mice [32]. Hence, we can conclude that nimbolide has very weak anticancer activity against WM tumours in vivo despite its high sensitivity (IC50 = 0.2 μM) against WM cellsi n vitro. In contrast to the high doses used in WM study, significantly lower doses (5-20 mg/kg) were administered via the same i.p.route in a colorectal cancer study. Here, a good anticancer effect was achieved although nimbolide was less potent in colorectal cancer cells (IC50 = 1.25 μM). Likewise, in another study on glioblastoma multiforme, nimbolide was very potent on brain tumour in vivo at a dose of 0.01 mg/kg via intravenous (i.v.) injection for 7 days, while glioblastoma cells has been found to be much less sensitive to nimbolide, with a relatively high in vitro IC50 (3 μM) [12]. Hence, systematic PK studies need to be carried out to establish the relationship between the in vivo doses and the maximum achievable concentrations in blood and tumour site, which can help to elucidate the gap between the in vitro and in vivo findings.
Chemopreventive effect:
In addition to its anticancer effect, nimbolide showed remarkable chemopreventive property. A chemopreventive agent perturbs various steps in cancer initiation, promotion, and progression. For instance, chemopreventive agents hinder tumour initiation by preventing interaction between carcinogens or reactive free radicals and DNA, hence lowering the chance of DNA damage and mutations [33]. Nimbolide acts as an effective chemopreventive agent since it modulates the biotransformation of carcinogens. Carcinogens upon entering the body are either activated or deactivated by xenobiotic-metabolizing enzymes [34-36], which will be elaborated in the next section. The chemo preventive effects of nimbolide were reported in an in vivo study using 7,12-dimethylbenz[a]anthracene (DMBA)-induced hamster buccal pouch (HBP) carcino genesis as a model [7]. In this study, upon oral administration of 0.01 mg/kg of nimbolide for 14 weeks, nimbolide reduced the incidence of tumour and pre-neoplastic lesions, there by highlighting its potent chemopreventive activity and potential as a candidate for cancer prevention.
Mechanism of actions
The major anticancer activities and cancer preventive effect of nimbolideare depicted in Figure 2.
Inhibition of cell growth and proliferation
Cell growth and proliferation are mediated by progression through cell cycle, which is tightly regulated by a group of proteins called cyclin-dependent kinases (CDKs). CDKs bind to a regulatory protein called cyclin to form an activated complex, which drives the transition of cells from one phase to another [37].
Nimbolide caused cell cycle arrest at G1/S phase. Evidently, nimbolide was found to reduce cyclin A level, which is required for colon cancer cells to proceed through S phase, hence inducing cell cycle arrest and resulting ininhibition of cell growth [22]. Similarly, nimbolide also reduced the expression of CDK2/cyclin E complex and increased the level of cyclin D2, thereby preventing colon cancer cells from entering the S phase of the cell cycle [37-39]. In addition, nimbolide directly inhibited CDK4/CDK6 kinase activity, leading to hypophosphorylation of the retinoblastoma protein (Rb), which in turn downregulated the expression of cyclin E, resulting in cell cycle arrest at G1/S and suppressionof glioblastoma growth [12].
Apart from G1/S phasearrest,nimbolidealso caused cell cycle arrest at both the G0/G1 and G2/M phases. For instance, nimbolide significantly suppressed the viability of HeLa cells by inducing cell cycle arrest at G0/G1 phase accompanied by p53-dependent p21 accumulation and downregulation of cyclin B as well as cyclin D1 [27].In nimbolide-treated HT-29 colorectal cancer cells, a substantial downregulationG2/M cell cycle checkpoint proteins, CHK2 and Rad17 were noted [38]. Taken together, nimbolide exhibited inhibitory activity on several CDK/cyclin molecules, there by resulting in cell cycle arrest.
Induction of apoptosis
Nimbolide can induce apoptosis in cancer cells by modulation of apoptotic proteins via both intrinsic and extrinsic pathways of apoptosis [40].The intrinsic pathway is a mitochondria-mediated pathway while the extrinsic pathway is mediated by death receptors (DR).
In the intrinsic pathway, the death signal triggers mitochondria to activate Bax, which forms holes in the outer membrane, facilitating the release of cytochrome c [41]. Cytochrome c interacts with Apaf-1 to form the apoptosome, which activates initiator caspase-9. Caspase-9 in turn activates effector caspase-3, which causes fragmentation of the nucleus, disruption of the cytoskeleton, membrane blebbing, and cellfragmentation [42]. Antiapoptotic proteins and proapoptotic proteins regulate the level of activation of caspase-3. Nimbolide treatmentdecreased the expression of antiapoptotic proteins(Bcl-xL, Bcl-2, survivin, caspase inhibitor molecules) and increased the expression of proapoptotic proteins (cytochrome c, Bax, Bad, Bid, cleaved caspases) in prostate cancer cells [25].
Moreover, a variety of signaling pathways regulate apoptosis by controlling the expression or activity of proapoptotic members of the Bcl-2 family [43]. Activation of the tumor suppressor p53, for example,can induce programmed cell death. Cytosolic p53 can in turn inhibit anti-apoptotic Bcl-xL and promote cytochrome c release.Using this mechanism, nimbolide acted by up-regulation of p53 levelin HeLa cells, thereby priming these cells towards apoptosis by destabilizing the mitochondria [44, 45].
In the extrinsic pathway, extracellular death signals such as death ligand, Fas ligand, tumour necrosis factor (TNF), and TNF-related apoptosis-inducing ligand(TRAIL) bind to death receptors (DRs), which activates caspase-8 [46-48]. Caspase-8 activates caspase-3, leading to apoptosis. Nimbolide up-regulated the expression of both DR5 and DR4 in chronic myeloid leukemia (KBM-5), multiple myeloma (U266), embryonic kidney carcinoma (A293), pancreatic adenocarcinoma (AsPC-1), and breast adenocarcinoma (MDA-MB-231) cells, resulting in activation of the extrinsic apoptotic pathway [49].
Moreover, for DRs up-regulation by nimbolide, activation of ERK and p38 MAPK were required. In nimbolide-treated colorectal cancer cells, apoptosis proceed by activation of caspase3/9 and ERK1/2 in the MAPK signaling pathway [49]. Furthermore, nimbolide resulted in generation of reactive oxygen species (ROS), which are required for ERK activation, which in turn up-regulated DRs and triggered apoptosis via the extrinsic pathway [27, 31]. ROS also induced lipid peroxidation of cellular membranes, generating toxic metabolites such as malondialdehyde (MDA) that can react with DNA to form adducts to induce apoptosis.
Nimbolidecan potentiate apoptosis induced by TNF-α in human leukemia cells [50]. Tumour necrosis factor (TNF-α) is a multifunctional inflammatory cytokine that also induces apoptosis of cancer cells via the extrinsic pathway [51]. Apart from its own antitumour effects, nimbolde can sensitize cancer cells to the effects of other anti-cancer agents such as TNF-related apoptosis-inducing ligand (TRAIL) [49]. In MCF-7 breast cancer cells, nimbolide and TRAIL were minimally effective in inducing apoptosis as single agents. However, when used in combination, significant apoptosis was observed in these cells, thereby extending the therapeutic efficacy of both agents. More importantly, this combination showed selectivity for cancer cells, as it was unable to evoke similar apoptosis in normal breast cells.
Another signaling cascade that nimbolide targets to decrease proliferation and enhance apoptosis of cancer cells is the Insulin-like Growth Factor I (IGF-1) pathway. IGF-I receptor is a receptor tyrosine kinase that can activate two different downstream effector cascades. Under the first scenario, Ras, Raf and mitogen-activated protein kinase (MAPKs) are activated, resulting in the transcription of genes that promote sustained proliferation [52]. In the second case, phosphoinositide 3-kinase (PI3K)/Aktis constitutively activated, which can regulate cellular survival and antiapoptotic signaling events [53]. Nimbolide was found to significantly inhibit IGF-1-mediated PI3K/Akt and MAPK signaling in human MCF-7 breast cancer cells, thereby inducing apoptosis and inhibiting proliferation of these cells [54].
Nimbolide can also act on a deregulated nuclear factor kappa-B(NF-κB) oncogenic pathway. NF-κB is a pro-inflammtory transcription factor involved in carcinogenesis, oncogenic progression, and apoptosis evasion [55]. NF-κB is inactivated in the cytoplasm by interacting with inhibitory Iκβ proteins such as IKKα and IKKβ [56]. This interaction results in the inactive NF-κB complex being primarily in the cytoplasm due to a strong nuclear export signal in Iκβ. In many cancer cells, NF-κB is constitutively active and located in the nucleus due to incessant stimulation of the IKK pathway that degrades Iκβ, while in some other cases, the Iκβ gene is mutated and dysfunctional [57-60]. As a result, NF-κB can enter the nucleus and promote the expression of multiple genes involved in tumor initiation, promotion and progression [56, 61]. Nimbolide was found to inhibit IκB degradation and prevent nuclear translocation of NF-κB. This subsequently caused cell cycle arrest by downregulating numerous genes involved in cellular proliferation [62]. Nimbolide can induce apoptosis through inactivation of NF-κB. This led to significant suppression of Bcl-2 with concomitant increase in the expression of Bax, cytochromec, and Smac/DIABLO [28]. Furthermore, nimbolide attenuated the NF-κB mediated Wnt/β-catenin signaling pathway in HepG2 cells. Wnt is a family of glycoproteins that regulates the amount of the transcriptional co-activator β-catenin that controls key developmental gene expression programs during embryonic development and tissue homeostasis. Hence, mutations in the Wnt pathway correlate to human birth defects and cancer [63]. Abrogation of NF-κB and Wnt signaling by nimbolide stimulatedcaspase-mediated apoptosis in HepG2 cells [28]. Unpublished data from our group also indicate that nimbolide can substantially abrogate the activation of another important oncogenic transcription factor, signal transducer and activator of transcription 3 (STAT3) in diverse prostate cancer cells. This effect was found to be mediated via an increased production of reactive oxygen species due to GSH/GSSG imbalance.
Reduction in tumor metastasisandangiogenesis:
Tumour metastasis accounts for approximately 90% of all cancer-related deaths [64]. The cancer cells from the primary site migrate to other organs via either the blood stream (haematogenous spread) or the lymphatic system (lymphatic spread), and form new tumours at the secondary sites [65].The role of matrix metallo proteinases (MMP) and urokinase plasminogen activator (uPA) in matrix degradation and tumour invasion has been well studied [66-68]. MMP and uPA are regulated by tissue inhibitor of matrix metalloproteinases (TIMP) [69].
Nimbolide reducedphorbol 12-myristate 13-acetate(PMA)-induced tumor cell migration and invasion by inhibition of MMP via NF-κB [22]. The downregulation of proinvasive and angiogenic proteins, with up-regulationof their inhibitors by nimbolide that was observed in this study was in line with the anti-invasive and anti-angiogenic potential of neem preparations [70]. In addition, nimbolide reduced mRNA expression of MMP-2, MMP-9, uPA and uPA receptor in breast cancer cells (MCF-7) [30].
Chemokine (C-C motif) ligand(CCL) is a pro-inflammatory chemokine and associates with its receptor, C-C chemokine receptor type (CCR). CCL2 recruits inflammatory leukocytes (such as tumour-associated macrophages) in the tumour microenvironment and facilitates cancer cell progression [71]. CXC ligand (CXCL12) drives tumour invasion by induction of MMP-9 [72]. CXCR4 is responsible for metastatic implantation into other organs [73]. For instance, nimbolide treatment (2, 4 μM in breast cancer MCF-7 cells) was found to significantly reduce CCL2, CXCR4, and CXCL12 mRNA expression compared to control cells [30].Similarly, nimbolide was shown to downregulate the expression of MMP-9, ICAM-1, and CXCR4 in colorectal cancer xenografts in a nude mouse model [13].
Vascular endothelial growth factor (VEGF) is a pro-angiogenic factor that can be activated by hypoxia-inducible factor-1a (HIF-1a) [74, 75]. Phosphorylated epidermal growth factor receptor (pEGFR) isalso associated with increased angiogenesis in breast cancer [76]. Nimbolide down-regulated expression of both pEGFR and VEGF receptors in breast cancer cells.Moreover, CXCL8 or interleukin-8, a pro-angiogenic chemokine, can stimulate both endothelial cell proliferation and migration through CXCR2 as well as enhance VEGF production [73, 77]. Evidently, nimbolide also reduced expression of CXCL8 and CXCR2 in breast cancers, and thus abrogated angiogenesis [30].
Chemoprevention
Nimbolide has potential to prevent pro-carcinogen activation and oxidative DNA damage by inhibiting phase I carcinogen activation enzymes (CYP1A1, CYP1B1) and simultaneously induce phase II carcinogen detoxification enzymes (glutathione-S-transferase (GST) and quinonereductase (QR) [7, 13, 62, 78]. Besides modulation of xenobiotics-metabolizing enzymes, nimbolide also upregulates cellular antioxidant content, which aids in decreasing carcinogen-induced oxidative DNA damage, a potentially critical event in neoplastic transformation. In another in vivo study withnimbolide in the DMBA-painted buccal pouch of hamster, nimbolidetreatment increased the levels of various antioxidant enzymes (glutathione peroxidase, gamma-glutamyltranspeptidase, superoxide dismutase, and catalase) compared to control group [7].
Importance of PK studies
Although accumulating evidence from in vitro and in vivostudes indicated that nimbolide exerts multiple pharmacological effects (antioxidant and anticancer effects), clinical trials to determine the effectiveness of nimbolideas an anticancer drughave not been conducted. The main reason for this is most likely due to the lack of preclinical PK parameters (ADME) of nimbolide. Most natural drug candidates fail to advance in clinical studies due to poor pharmacokinetic properties. Waring et al. (2015) reported that pharmacokinetics and bioavailability remained the third most common cause of attrition, accounting for 16% of failuresof compounds in Phase I trials, among a set of 605 terminated drug candidates developed by 4 major pharmaceutical companies from 2000 to 2010 [79]. For instance, resveratrol, a promising antiaging and anticancer drug candidatederived from grapes, has poor bioavailability despite having favourable in vitro data, thus limiting its use in clinical studies [80].
Oral bioavailability is a key factor to ensure an effective drug concentration is physiologically achievable. PK studies can evaluatethe bioavailability of a drug, which is vital for optimizing the administration route and schedule to achieve therapeutic efficacy [81]. In an in vivo study of nimbolidein colorectal cancer, plasma levels of 222 and 409ng/mL were detected in colorectal cancer xenografted mice 2h after treatment with nimbolide at 5 and 20 mg/kg i.p., respectively. Nimbolide levels of 345 and 868 ng/g of tumor tissue were obtained from the mice treated with nimbolide at 5 and 20 mg/kg i.p., respectively. The concentrations of plasma and tumour tissue samples were analyzed using high performance liquid chromatography (HPLC) [13]. However, this HPLC method for determination of nimbolide in mouse plasma and tissue has not been fully validated according to the US Food Drug Adminsitration (FDA) guidelines [82].
Distribution of nimbolide within solid tumours and normal tissues is also an important parameter that requires investigation. During the in vivo study of nimbolide in glioblastomamultiforme, where brain tumour cells were xenografted either by injection at the flank or into the cerebral cortex, the tumour volumes of nimbolide treatment group were about 40% smaller than those of DMSO control group [12]. The favourable PD results indicated that nimbolide might be able to cross blood-brain barrier (BBB) and the concentration could be sufficient for nimbolide to suppress intracranial tumour cell proliferation. Therefore, in vivo PK studies focused on BBB permeability should be carried out to assess blood-brain cerebrospinal fluid and blood-brain extracellular fluid drug concentration relationships, in relation to variation in drug doses and plasma drug levels [83].
More crucially, metabolite evaluation is essential for nimbolide because many natural products are pro-drugs that must undergo metabolic conversion either by the intestinal microflora or mammalian phase I and/or II metabolism before becoming an active metabolite [84]. For instance, Romidepsin (Istodax), isolated fromthe fermentation extract of the rod-shaped bacterium C. violaceum, is a prodrug that is converted in cells to its active form by the reduction of the disulfide bond by glutathione. FDA has approved Romidepsin in 2009 to treat cutaneous T-cell lymphoma [85]. It was reported that nimbolide is a CYP1A1 and CYP1A2 inhibitor as well as a phase II enzymes inducer [7], but more studies need to be carried out to determine whether nimbolide has any active metabolite.
To sum up, the bioavailability evaluation of nimbolidebecomesmore critical ifnimbolideis chosen to be developedas a commercial drug for cancer prevention, in which oral administration is required.PK studies performed with robust and sensitive bio-analytical methods allow us to identify ADME constraints and better understand the bioavailability and other important PK parameters of nimbolide, such as the Cmax (maximum concentration), Tmax (time to reach maximum concentration), t1/2 (elimination half-life), clearance, volume of distribution, and dose linearity/proportionality [86]. So far, there are no systematic studies that have been done to investigate these PK properties of nimbolide. Therefore, the following aspects are urgently needed in getting nimbolide into the clinic. These include i) developing and validating a highly sensitive LC-MS/MS method for determination of nimbolid econcentrations in plasma and tissue samples; ii) performing preclinical PK studies in mice (rodent model), and monkeys (non-rodent model) to define optimized dosage, formulation, and treatment schedule. Based on these preclinical PK parameters, a well-designed phase I clinical trial can be conducted to determine the maximum tolerated dose of nimbolide and response in cancer patients.
Current status on toxicity studies
While PK is a vital part of drug development, preclinical safety/toxicity testing is very important for the development of new drugs;especially in anticancer drugs,since efficacy does not guarantee non-toxicity. During the period from 2000 to 2010, out of 808 drug candidates developed by 4 major pharmaceutical companies, 356 compounds (44%) failed to progress into clinical studies due to toxicity. Preclinical toxicity was the highest cause of attrition accounting for 59% of the failure at the preclinical phase [79]. Hence, the high toxicity-based attrition further highlights the importance of toxicological studies in drug development. In this section, acute toxicity, mutagenicity, and spermicidal effects of nimbolide will be discussed.
The toxicity of nimbolide in human has not been studied. However, nimbolide exhibited acute toxicity in mice, hamster and rats. Nimbolide was more toxic to mice when given i.p.and i.v. at same doses, compared to rats and hamsters. Moreover, in adult male mice, the median lethal dose (LD50) of i.p. route was 225mg/kg, while LD50of i.v. was 24mg/kg body weight, 10 times more toxic than i.p.route. The higher toxicity for i.v.mightbe due to higher peak concentration(Cmax) compared to i.p., which has an absorption phase. The toxicity of nimbolide in mice is markedly reduced when administered intragastrically/orally (i.g.), subcutaneously (s.c) and intramuscularly (i.m.), with LD50 exceeds 600mg/kg body weight [17]. All the above-mentioned toxicological results need to be interpreted based on PK properties of nimbolide,which are currently absent. Hence, a systematic PK study is warranted to determine PK parameters of nimbolide and its metabolite profile.
Furthermore, the spermicidal effect of nimbolide in animals is still inconclusive. In an in vitro study, nimbolide had no significant effect on the viability of rabbit sperm [10]. Upon exposure to nimbolide at 50 μM for 60 min, there was only 20-30% reduction in mobility and viability of the rabbit sperm. However, in another in vivo study, nimbolide, when administered subcutaneously at dose 0.5-1.5 mg/kg to Wistar strain male albino rats, was shown to reduce sperm functional parameters and increase abnormal sperm countin a dose-dependent manner [87]. This result was supported by the in vitro study carried out by the same research group. In this study, 0.5-2 μM of nimbolide was able to deplete the antioxidant defense system in sperm of Wistar strain male albino rats by decreasing activity of antioxidant enzymes (SOD, catalase, GR, GPx); hence increasing the level of production of ROS and inducing oxidative stress in epididymal sperm of rats [88]. With respect to mutagenicity, nimbolide possessed no mutagenic effectusing six tester strains of Salmonella typhimurium [4, 89].
Although toxicity was significantly reduced when nimbolide was administered i.g., s.c. and i.m., the systemic exposure with repeated doses of nimbolide (repeated-dose toxicity studies) should be thoroughly investigated to identify the no observed adverse effect level (NOAEL) in the most appropriate animal species. According to FDA, both a rodent (rat or mouse) and a non-rodent (dog or monkey) general toxicity study of at least 14 days should be conducted [90]. The preclinical data including dose-response relationship, pharmacokinetics and toxicological profile are important for determination of the starting dose in first-in-human studies [90].
Conclusion and future prospects
Nimbolide has been used in traditional Indian medicine to treat various diseases such as infections, gastric ulcers, and cancer. Accumulating evidence from in vitro and in vivo studies supports the anticancer and chemopreventive properties of nimbolide in many cancer types including WM, colon, and brain cancer. Multiple mechanisms of action for nimbolide’s anticancer and chemopreventive effects have been proposed based on its molecular targets identified in several types of cancer. The main anticancer mechanisms of action ofnimbolide include anti-proliferation, induction of apoptosis, inhibition of angiogenesis and migration, as well as suppression of tumourigenesis via modulation of carcinogen metabolizing enzymes. Compared to the great number of in vitro studies that have been conducted, more efforts should be spent on performing in vivo studies to verify the in vitro findings.In addition, there is a contradiction between in vitro concentration and in vivo dose in certain types of cancer. Therefore systematic PK evaluationsare urgently required to interpret the inconsistency between in vitro and in vivo results.To the best of our knowledge, a sensitive bio-analytical method to determine the effective plasma and intracellular concentrations of nimbolide is currently lacking, which is the main obstacle to understanding nimbolide’s biologically relevant concentrations in animal models. Furthermore, comprehensive toxicological studies should be conducted to support the safety of nimbolide in animal models including the non-rodent species.Taken together, nimbolide has demonstrated anticancer and chemopreventive effects on many different types of cancer in preclinical studies. Well-designed PK studies and long-term safety/toxicity investigations can greatly accelerate its development as a potential anticancer drug candidate in early phase clinical trials.
Abbreviations
A. indica, Azadirachtaindica; ADME, absorption, distribution, metabolism and elimination; Akt, AKT8 virus oncogene cellular homolog; AUC, area under the curve; Bax, Bcl-2-associated X protein;Bcl2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra-large; CDKs, Cyclin dependent kinases; Cmax, maximum concentration;c-Myc, Avian myelocytomatosis virus oncogene cellular;CYP, cytochrome P450; DMBA, 7,12-Dimethylbenzanthracene;EGFR, Epidermal growth factor receptor; ERK, Extracellular-signal-regulated kinase; GSK-3, Glycogen synthase kinase 3; HIF-1a, hypoxia-inducable factor 1a; ICAM, Intercellular adhesion molecule; IGF-1, Insulin-like growth factor 1; IκB, Inhibitor of kappa B; IKK, IκB kinase; IL-6, Interleukin 6; JAK, Janus kinase; JNK, c-jun N-terminal kinase; KEAP, Kelch-like erythroid-cell- derived protein with CNC homology (ECH)-associated protein;LD50, median lethal dose; LEF, Lymphoid enhancer-binding factor; MAPKs, Mitogen-activated protein kinases; MMPs, Matrix metalloproteinases; MOMP, mitochondrial outer membrane potential; NF-κB, Nuclear factor kappa B; NL, Nimbolide; Nrf-2, Nuclear factor (erythroid-derived 2)-like 2;PD, pharmacodynamics; PI3K, Phosphoinositide 3-kinase; PK, pharmacokinetics; PMA, phorbol 12-myristate 13-acetate; ROS, Reactive oxygen species; Smac/DIABLO, second mitochondria-derived activator of caspases; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor; S. aureus, Staphylococcus aureus; S. coagulase, Staphylococcus coagulase; STAT, Signal transducer and activator of transcription; t1/2, elimination half-life;TEF, Transcription enhancer factor; Tmax, time to maximum concentration; TRAIL, TNF-related apoptosis-inducing ligand; uPA, urokinase plasminogen activator; VEGF, Vascular endothelial growth factor;WM, Waldenstrommacroglobulinemia;XIAP, X-linked inhibitor of apoptosis protein.
Acknowledgement
This research is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under Its Research Centres of Excellence Initiative,and Clinician Scientist Award of NMRC for Translational Pipeline: Developing novel therapeutics for cancer treatment, including the role of histone deacetylase inhibitor.
Conflict of interest
None declared.
References
1. Kumar VS and Navaratnam V. Neem (Azadirachta indica): prehistory to contemporary medicinal uses to humankind. Asian Pac J Trop Biomed. 2013; 3(7):505-514.
2. Singh KK. (2008). Neem, a Treatise: I. K. International Pvt Ltd).
3. Rochanakij S, Thebtaranonth Y, Yenjai C and Yuthavong Y. Nimbolide, a constituent of Azadirachta indica, inhibits Plasmodium falciparum in culture. Southeast Asian J Trop Med Public Health. 1985; 16(1):66-72.
4. Rojanapo W, Suwanno S, Somjaree R, Glinsukon T and Thebtaranont Y. Mutagenic and Antibacterial Activity Testing of Nimbolide and Nimbic Acid. Journal of the Science Society of Thailand. 1985; 11(4):177-181.
5. Suresh G, Gopalakrishnan G, Wesley SD, Pradeep Singh ND, Malathi R and Rajan SS. Insect antifeedant activity of tetranortriterpenoids from the Rutales. A perusal of structural relations. J Agric Food Chem. 2002; 50(16):4484-4490.
6. Cohen E, Quistad GB, Jefferies PR and Casida JE. Nimbolide is the principal cytotoxic component of neem-seed insecticide preparations. Pesticide Science. 1996; 48(2):135-140.
7. Priyadarsini RV, Manikandan P, Kumar GH and Nagini S. The neem limonoids azadirachtin and nimbolide inhibit hamster cheek pouch carcinogenesis by modulating xenobiotic-metabolizing enzymes, DNA damage, antioxidants, invasion and angiogenesis. Free Radical Research. 2009; 43(5):492-504.
8. Moghu SN. (2013). Herbal Remedies: 20 Health Benefits Of Neem. India Times.
9. Subapriya R and Nagini S. Medicinal properties of neem leaves: a review. Curr Med Chem Anticancer Agents. 2005; 5(2):149-146.
10. Cohen E, Quistad GB and Casida JE. Cytotoxicity of nimbolide, epoxyazadiradione and other limonoids from neem insecticide. Life Sci. 1996; 58(13):1075-1081.
11. Chitta K, Paulus A, Caulfield TR, Akhtar S, Blake MKK, Ailawadhi S, Knight J, Heckman MG, Pinkerton A and Chanan-Khan A. Nimbolide targets BCL2 and induces apoptosis in preclinical models of Waldenstroms macroglobulinemia. Blood Cancer Journal. 2014; 4.
12. Karkare S, Chhipa RR, Anderson J, Liu XN, Henry H, Gasilina A, Nassar N, Roychoudhury J, Clark JP, Kumar A, Pauletti GM, Ghosh PK and Dasgupta B. Direct Inhibition of Retinoblastoma Phosphorylation by Nimbolide Causes Cell-Cycle Arrest and Suppresses Glioblastoma Growth. Clinical Cancer Research. 2014; 20(1):199-212.
13. Gupta SC, Prasad S, Sethumadhavan DR, Nair MS, Mo YY and Aggarwal BB. Nimbolide, a limonoid triterpene, inhibits growth of human colorectal cancer xenografts by suppressing the proinflammatory microenvironment. Clin Cancer Res. 2013; 19(16):4465-4476.
14. Woodcock J and Woosley R. The FDA Critical Path Initiative and Its Influence on New Drug Development*. Annu Rev Med. 2008; 59:1-12.
15. Kigodi PGK, Blasko G, Thebtaranonth Y, Pezzuto JM and Cordell GA. Spectroscopic and Biological Investigation of Nimbolide and Deoxonimbolide-28 from Azadirachta-Indica. Journal of Natural Products. 1989; 52(6):1246-1251.
16. Nair MS, Gopal S and Issac D. Optimised isolation procedure for biologically active compounds nimbolide and 28-deoxonimbolide from Azadirachta indica leaves. Phytochemistry. 1997; 46(7):1177-1178.
17. Ekong DEU. Chemistry of the meliacins (limonoids). The structure of nimbolide, a new meliacin from Azadirachta indica. Chemical Communications (London). 1967; (16):808a-808a.
18. Ekong DEU and Ibiyemi SA. Biosynthesis of Nimbolide from [2-C-14, (4r)4-H-3(1)]Mevalonic Acid Lactone in the Leaves of Azadirachta-Indica. Phytochemistry. 1985; 24(10):2259-2261.
19. Anand Solomon K, Malathi R, Rajan SS, Anitha G, Josepha Lourdu Raj J, Narasimhan S, Suresh G and Gopalakrishnan G. The isomeric compounds nimbolide and isonimbolide. Acta Crystallogr C. 2005; 61(Pt 2):o70-72.
20. Gore VK, Desai SR, Mayelvaganan T, Padmakumar R, Hadimani SB and Bhat SV. Convenient Synthesis of Decalin Systems of Bioactive Terpenoids. Tetrahedron. 1993; 49(13):2767-2782.
21. Sastry BS, Babu KS, Babu TH, Chandrasekhar S, SrinivaS PV, Saxena AK and Rao JM. Synthesis and biological activity of amide derivatives of nimbolide. Bioorganic & Medicinal Chemistry Letters. 2006; 16(16):4391-4394.
22. Babykutty S, Priya PS, Nandini RJ, Kumar MAS, Nair MS, Srinivas P and Gopala S. Nimbolide retards tumor cell migration, invasion, and angiogenesis by downregulating MMP-2/9 expression via inhibiting ERK1/2 and reducing DNA-binding activity of NF-kappa B in colon cancer cells. Molecular Carcinogenesis. 2012; 51(6):475-490.
23. Roy MK, Kobori M, Takenaka M, Nakahara K, Shinmoto H, Isobe S and Tsushida T. Antiproliferative effect on human cancer cell lines after treatment with nimbolide extracted from an edible part of the neem tree (Azadirachta indica). Phytotherapy Research. 2007; 21(3):245-250.
24. Scholzen T and Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000; 182(3):311-322.
25. Singh PR, Arunkumar R, Sivakamasundari V, Sharmila G, Elumalai P, Suganthapriya E, Mercy AB, Senthilkumar K and Arunakaran J. Anti-proliferative and apoptosis inducing effect of nimbolide by altering molecules involved in apoptosis and IGF signalling via PI3K/Akt in prostate cancer (PC-3) cell line. Cell Biochemistry and Function. 2014; 32(3):217-228.
26. Liu JF, Hou CH, Lin FL, Tsao YT and Hou SM. Nimbolide Induces ROS-Regulated Apoptosis and Inhibits Cell Migration in Osteosarcoma. Int J Mol Sci. 2015; 16(10):23405-23424.
27. Priyadarsini RV, Murugan RS, Sripriya P, Karunagaran D and Nagini S. The neem limonoids azadirachtin and nimbolide induce cell cycle arrest and mitochondria-mediated apoptosis in human cervical cancer (HeLa) cells. Free Radical Research. 2010; 44(6):624-634.
28. Kavitha K, Priyadarsini RV, Anitha P, Ramalingam K, Sakthivel R, Purushothaman G, Singh AK, Karunagaran D and Nagini S. Nimbolide, a neem limonoid abrogates canonical NF-kappa B and Wnt signaling to induce caspase-dependent apoptosis in human hepatocarcinoma (HepG2) cells. European Journal of Pharmacology. 2012; 681(1-3):6-14.
29. Mahapatra S, Young CYF, Kohli M, Karnes RJ, Klee EW, Holmes MW, Tindall DJ and Donkena KV. Antiangiogenic Effects and Therapeutic Targets of Azadirachta indica Leaf Extract in Endothelial Cells. Evidence-Based Complementary and Alternative Medicine. 2012.
30. Elumalai P, Mercy AB, Arunkamar R, Sharmila G, Bhat FA, Balakrishnan S, Singh PR and Arunakaran J. Nimbolide inhibits invasion and migration, and down-regulates uPAR chemokine gene expression, in two breast cancer cell lines. Cell Proliferation. 2014; 47(6):540-552.
31. Kumar GH, Mohan KVPC, Rao AJ and Nagini S. Nimbolide a limonoid from Azadirachta indica inhibits proliferation and induces apoptosis of human choriocarcinoma (BeWo) cells. Investigational New Drugs. 2009; 27(3):246-252.
32. Glinsukon T, Somjaree R, Piyachaturawat P and Thebtaranonth Y. Acute toxicity of nimbolide and nimbic acid in mice, rats and hamsters. Toxicol Lett. 1986; 30(2):159-166.
33. Steward WP and Brown K. Cancer chemoprevention: a rapidly evolving field. Br J Cancer. 2013; 109(1):1-7.
34. Guengerich FP. Metabolism of chemical carcinogens. Carcinogenesis. 2000; 21(3):345-351.
35. Williams JA and Phillips DH. Mammary expression of xenobiotic metabolizing enzymes and their potential role in breast cancer. Cancer Res. 2000; 60(17):4667-4677.
36. Landi S. Mammalian class theta GST and differential susceptibility to carcinogens: a review. Mutat Res. 2000; 463(3):247-283.
37. Malumbres M and Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005; 30(11):630-641.
38. Roy MK, Kobori M, Takenaka M, Nakahara K, Shinmoto H and Tsushida T. Inhibition of colon cancer (HT-29) cell proliferation by a triterpenoid isolated from Azadirachta indica is accompanied by cell cycle arrest and up-regulation of p21. Planta Medica. 2006; 72(10):917-923.
39. Meyyappan M, Wong H, Hull C and Riabowol KT. Increased expression of cyclin D2 during multiple states of growth arrest in primary and established cells. Mol Cell Biol. 1998; 18(6):3163-3172.
40. Fulda S and Debatin KM. Targeting apoptosis pathways in cancer therapy. Curr Cancer Drug Targets. 2004; 4(7):569-576.
41. Costantini P, Jacotot E, Decaudin D and Kroemer G. Mitochondrion as a novel target of anticancer chemotherapy. J Natl Cancer Inst. 2000; 92(13):1042-1053.
42. Geoffrey M. Cooper REH. (2013). The Cell: A Molecular Approach: Sinauer Associates).
43. Gross A, McDonnell JM and Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes & development. 1999; 13(15):1899-1911.
44. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P and Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003; 11(3):577-590.
45. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M and Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004; 303(5660):1010-1014.
46. Ashkenazi A, Holland P and Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol. 2008; 26(21):3621-3630.
47. Bazzoni F and Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996; 334(26):1717-1725.
48. Roy S and Nicholson DW. Cross-talk in cell death signaling. J Exp Med. 2000; 192(8):F21-25.
49. Gupta SC, Reuter S, Phromnoi K, Park B, Hema PS, Nair M and Aggarwal BB. Nimbolide sensitizes human colon cancer cells to TRAIL through reactive oxygen species- and ERK-dependent up-regulation of death receptors, p53, and Bax. J Biol Chem. 2011; 286(2):1134-1146.
50. Gupta SC, Prasad S, Reuter S, Kannappan R, Yadav VR, Ravindran J, Hema PS, Chaturvedi MM, Nair M and Aggarwal BB. Modification of cysteine 179 of IkappaBalpha kinase by nimbolide leads to down-regulation of NF-kappaB-regulated cell survival and proliferative proteins and sensitization of tumor cells to chemotherapeutic agents. J Biol Chem. 2010; 285(46):35406-35417.
51. van Horssen R, Ten Hagen TL and Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006; 11(4):397-408.
52. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008; 8(12):915-928.
53. Manning BD and Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129(7):1261-1274.
54. Elumalai P, Arunkumar R, Benson CS, Sharmila G and Arunakaran J. Nimbolide inhibits IGF-I-mediated PI3K/Akt and MAPK signalling in human breast cancer cell lines (MCF-7 and MDA-MB-231). Cell Biochemistry and Function. 2014; 32(5):476-484.
55. Hoesel B and Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Molecular Cancer. 2013; 12(1):1-15.
56. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006; 25(51):6680-6684.
57. Chai EZ, Siveen KS, Shanmugam MK, Arfuso F and Sethi G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem J. 2015; 468(1):1-15.
58. Li F, Zhang J, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Kumar AP, Ahn KS and Sethi G. NF-kappaB in cancer therapy. Arch Toxicol. 2015; 89(5):711-731.
59. Li F and Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. 2010; 1805(2):167-180.
60. Sethi G and Tergaonkar V. Potential pharmacological control of the NF-kappaB pathway. Trends Pharmacol Sci. 2009; 30(6):313-321.
61. Romashkova JA and Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999; 401(6748):86-90.
62. Kumar GH, Priyadarsini RV, Vinothini G, Letchoumy PV and Nagini S. The neem limonoids azadirachtin and nimbolide inhibit cell proliferation and induce apoptosis in an animal model of oral oncogenesis. Investigational New Drugs. 2010; 28(4):392-401.
63. MacDonald BT, Tamai K and He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009; 17(1):9-26.
64. Spano D, Heck C, De Antonellis P, Christofori G and Zollo M. Molecular networks that regulate cancer metastasis. Semin Cancer Biol. 2012; 22(3):234-249.
65. Wong SY and Hynes RO. Lymphatic or Hematogenous Dissemination: How Does a Metastatic Tumor Cell Decide? Cell cycle (Georgetown, Tex). 2006; 5(8):812-817.
66. Schmalfeldt B, Prechtel D, Harting K, Spathe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W and Lengyel E. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res. 2001; 7(8):2396-2404.
67. de Bart ACW, Quax PHA, Löwik CWGM and Verheijen JH. Regulation of plasminogen activation, matrix metalloproteinases and urokinase-type plasminogen activator-mediated extracellular matrix degradation in human osteosarcoma cell line MG63 by interleukin-1 alpha. Journal of Bone and Mineral Research. 1995; 10(9):1374-1384.
68. Hofmann UB, Westphal JR, Van Muijen GN and Ruiter DJ. Matrix metalloproteinases in human melanoma. J Invest Dermatol. 2000; 115(3):337-344.
69. Duffy MJ, McGowan PM and Gallagher WM. Cancer invasion and metastasis: changing views. J Pathol. 2008; 214(3):283-293.
70. Manikandan P, Letchoumy PV, Gopalakrishnan M and Nagini S. Evaluation of Azadirachta indica leaf fractions for in vitro antioxidant potential and in vivo modulation of biomarkers of chemoprevention in the hamster buccal pouch carcinogenesis model. Food and Chemical Toxicology. 2008; 46(7):2332-2343.
71. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA and Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011; 475(7355):222-225.
72. Goda S, Inoue H, Umehara H, Miyaji M, Nagano Y, Harakawa N, Imai H, Lee P, Macarthy JB, Ikeo T, Domae N, Shimizu Y and Iida J. Matrix metalloproteinase-1 produced by human CXCL12-stimulated natural killer cells. Am J Pathol. 2006; 169(2):445-458.
73. Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D and Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004; 64(23):8604-8612.
74. Roskoski R, Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007; 62(3):179-213.
75. Jain RK and Xu L. alphaPlGF: a new kid on the antiangiogenesis block. Cell. 2007; 131(3):443-445.
76. Magkou C, Nakopoulou L, Zoubouli C, Karali K, Theohari I, Bakarakos P and Giannopoulou I. Expression of the epidermal growth factor receptor (EGFR) and the phosphorylated EGFR in invasive breast carcinomas. Breast Cancer Res. 2008; 10(3):R49.
77. Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001; 12(4):375-391.
78. Talalay P. Chemoprotection against cancer by induction of Phase 2 enzymes. BioFactors. 2000; 12(1-4):5-11.
79. Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O and Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015; 14(7):475-486.
80. Smoliga JM and Blanchard O. Enhancing the delivery of resveratrol in humans: if low bioavailability is the problem, what is the solution? Molecules. 2014; 19(11):17154-17172.
81. FDA. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. Center for Drug Evaluation and Research (CDER). 2003.
82. FDA. Guidance for Industry: Bioanalytical Method Validation. Center for Drug Evaluation and Research (CDER). 2001.
83. Au-Yeung SC, Rurak DW, Gruber N and Riggs KW. A pharmacokinetic study of diphenhydramine transport across the blood-brain barrier in adult sheep: potential involvement of a carrier-mediated mechanism. Drug Metab Dispos. 2006; 34(6):955-960.
84. Kumar SV, Saravanan D, Kumar B and Jayakumar A. An update on prodrugs from natural products. Asian Pac J Trop Med. 2014; 7s1:S54-59.
85. VanderMolen KM, McCulloch W, Pearce CJ and Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J Antibiot (Tokyo). 2011; 64(8):525-531.
86. Li L, Brunner I, Han AR, Hamburger M, Kinghorn AD, Frye R and Butterweck V. Pharmacokinetics of alpha-mangostin in rats after intravenous and oral application. Mol Nutr Food Res. 2011; 55 Suppl 1:S67-74.
87. Aladakatti R, Sukesh, B., Jadaramkunti, UC and Hiremath, MB. Effect of graded doses of nimbolide (a major component of Azadirachta indica leaves) on biochemical and sperm functional parameters in male albino rats. J Laborat Anim Sci. 2011; 1:24-30.
88. Kumbar SB. In-vitro effect of nimbolide, an isoprenoid of neem leaf, on antioxidant system of rat cauda epididymal spermatozoa: A dose dependent study. Journal of Applied Pharmaceutical Science. 2012.
89. Uwaifo AO. The mutagenicities of seven coumarin derivatives and a furan derivative (nimbolide) isolated from three medicinal plants. Journal of Toxicology and Environmental Health. 1984; 13(4-6):521-530.
90. FDA. Guidance for industry. M3 (R2) nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. Center for Drug Evaluation and Research (CDER). 2010; 10903:20993-20002.
91. Bodduluru LN, Kasala ER, Thota N, Barua CC and Sistla R. Chemopreventive and therapeutic effects of nimbolide in cancer: the underlying mechanisms. Toxicol In Vitro. 2014; 28(5):1026-1035.
92. Elumalai P, Gunadharini DN, Senthilkumar K, Banudevi S, Arunkumar R, Benson CS, Sharmila G and Arunakaran J. Induction of apoptosis in human breast cancer cells by nimbolide through extrinsic and intrinsic pathway. Toxicol Lett. 2012; 215(2):131-142.