
INTRODUCTION
Polycyclic aromatic hydrocarbons (PAHs), like 3-methylcholanthrene (MCA), are procarcinogenic components of air pollutants, tobacco smoke, or overcooked food [1]. According to the National Cancer Institute up to 30% of human cancers in the US are attributed to tobacco smoke and inhaled pollutants (www.cancer.gov). In mouse models, carcinogens like PAHs induce tumors dose-dependently. During early phases of PAH-induced tumor development immune reactions are compromised [2]. Carcinogenic activation of PAHs involves their biotransformation by cytochrome P450 (CYPs) to more reactive diol-epoxides, which bind DNA and thereby induce DNA damage [3]. Among CYP-isoforms, only CYP1A1, CYP1A2, and CYP1B1 activate PAH and its PAH-dihydrodiol. The expression of CYP1A2 is restricted to the liver, while CYP1A1 and CYP1B1 are also expressed in extra hepatic tissue such as lung and mammary glands [4]. PAHs activate the arylhydrocarbon receptor (AhR) and provoke dimerization with the arylhydrocarbon receptor nuclear translocator (ARNT), which causes transcriptional induction of CYP enzymes [3]. So far, the metabolic activation of PAHs has mainly been analyzed in hepatocytes [3, 5-7], while little is known on the involvement of macrophages in this process [8]. However, a recent publication elucidated the importance of Langerhans cells, the skin tissue-resident macrophages, for metabolic activation of PAHs [9]. Besides the metabolism of environmental carcinogens, AhR is also activated by endogenously formed ligands, referring to the development of immune tolerance and increased survival in cancer [10, 11]. In contrast to the application of chemical carcinogens, other mouse models have been established to study tumor formation based on genetic alterations. A common model for breast cancer are transgenic mice expressing the polyoma virus middle T oncoprotein (PyMT) within breast epithelial cells. In this model tumor formation is spontaneous and occurs in all transformed mice [12].
Immune cells like macrophages significantly contribute to all phases of cancer development, a process known as cancer immunoediting [13, 14]. Chronic inflammation, fostered by immune competent macrophages, drives cancer formation for example following infections with human papillomaviruses or Helicobacter pylori [14-16]. If tumor elimination fails, tumors start to expand but still engage interactions with immune cells to perpetuate tumor-associated inflammation [17]. Macrophages are fundamental in supporting cancer immunoediting at the initial phase of tumorigenesis [18]. Moreover, signals in the tumor microenvironment polarize macrophages to a pro-tumor or alternatively-activated phenotype, to promote malignant conversion and immune escape [18]. These tumor-associated macrophages (TAMs) frequently accumulate in hypoxic areas of the tumor [19].
Hypoxia develops due to inadequate blood supply or enhanced oxygen consumption of proliferating cells. As a consequence, hypoxia-inducible factor (HIF) is activated in tumors. HIF is a heterodimer composed of an oxygen-sensitive HIF-α (HIF-1α, HIF-2α, or HIF-3α) subunit and constitutively expressed HIF-1β, which is also known as ARNT [19, 20]. Under normoxic conditions, the HIF-α subunit is hydroxylated by prolyl hydroxylases (PHDs). Following hydroxylation, the α-subunits are recognized by the von Hippel-Lindau (VHL) protein, causing their poly-ubiquitination and proteasomal degradation. Under hypoxia PHDs are inhibited, hydroxylation of HIF-α is reduced, thus allowing its accumulation and dimerization with ARNT in the nucleus [19]. Interestingly, HIF-α shares its dimerization partner HIF-1β/ARNT with AhR. So far, the interaction between the HIF- and the AhR-pathway has been discussed controversially [5, 21, 22].
DNA binding of HIF upregulates a variety of target genes promoting angiogenesis, metastasis, and proliferation [23]. High levels of HIF in tumors correlate with a poor clinical patient outcome [23, 24]. HIF-inhibitors are under consideration as anticancer agents, although the influence of HIF in stromal cells on tumor progression is poorly understood [24-27]. An improvement of conventional chemotherapy for example was shown by targeting PHD2, and thereby activating HIF in endothelial cells, enhancing tumor perfusion and drug delivery [28]. Only few studies addressed the impact of HIF in macrophages on tumor progression, indicating tumor-promoting properties of both HIF-isoforms, although mechanisms largely remain uncertain [19, 29, 30].
To elucidate the role of HIF in macrophages during carcinogenesis, we analyzed tumor formation in wild type (wt) mice or mice carrying a myeloid-specific knockout of Hif-1α, Hif-2α, and Hif-1α/2α in the transgenic PyMT breast cancer model as well as chemical carcinogenesis, induced by subcutaneous injection of MCA. Most strikingly, mice lacking Hif-1α in macrophages did not develop tumors in the MCA model. Mechanistically, a knockout of Hif-1α decreased CYP-mediated metabolic MCA-activation in macrophages and a DNA damage response in fibroblasts.
RESULTS
Myeloid-specific deletion of HIF-1α attenuates tumor formation in MCA-induced carcinogenesis
To explore the role of HIF-α isoforms in carcinogenesis, wt and myeloid conditional knockout mice lacking Hif-1α and/or Hif-2α were used in two different tumor models. Hif-1α expression in peritoneal macrophages (PMs) was reduced by 87% in Hif-1α LysM-/- macrophages compared to wt mice (Supplementary Figure 1A). Hif-2α was expressed in wt macrophages and reduced by 97% in Hif-2α LysM-/- cells (Supplementary Figure 1B). To validate functional consequences of the knockout, we analyzed HIF target gene expression under hypoxia. Adrenomedullin (Adm), a classical HIF-1 target gene, was reduced in macrophages isolated from Hif-1α LysM-/- mice or double knockout macrophages, but not in macrophages from Hif-2α LysM-/- mice (Supplementary Figure 1C). In contrast, Arginase1 (Arg1), a preferred HIF-2 target gene, was downregulated in Hif-2α LysM-/- and double knockout macrophages (Supplementary Figure 1D). To analyze tumor formation, we used two different tumor models in C57BL/6J wt, Hif-1α LysM-/-, Hif-2α LysM-/-, and Hif-1α/2α LysM-/- mice. First, MCA was injected subcutaneously into the right flank of mice to induce fibrosarcomas at the site of injection. Compared to the chemical-induced cancer, tumor formation was also followed in the transgenic PyMT breast cancer model by crossing C57BL/6J conditional Hif-1α LysM-/-, Hif-2α LysM-/-, and Hif-1α/2α LysM-/- mice with mice expressing the PyMT oncoprotein. In the MCA-model 63% of wt mice displayed a tumor after 20 weeks (Figure 1A). Hif-2α LysM-/- mice showed a more rapid tumor formation but the overall rate of tumor development was not significantly different from wt mice. In contrast, tumor frequency in Hif-1α LysM-/- as well as in Hif-1α/2α LysM-/- mice was significantly diminished, basically being absent. Depicted in Figure 1C, the tumor burden in mice lacking Hif-1α (mean tumor burden: 0.303% ± 0.303) and Hif-1α/2α (0.438% ± 0.347) was smaller than in wt mice (1.496% ± 0.379), whereas the mean tumor burden in Hif-2α LysM-/- mice (3.185% ± 0.833) was not significantly enhanced compared to wt mice. Tumors of Hif-2α LysM-/- and wt mice were analyzed for immune cell infiltration and vascularization, but no differences comparing wt with Hif-2α LysM-/- mice became detectable (Supplementary Figure 2A-2D). In the PyMT breast cancer model, no statistical differences in tumor development occurred (Figure 1B) and tumor burden was marginally but not significantly affected (Figure 1D). These data identify HIF-1α as an important factor in tumor formation in chemical PAH-induced carcinogenesis.
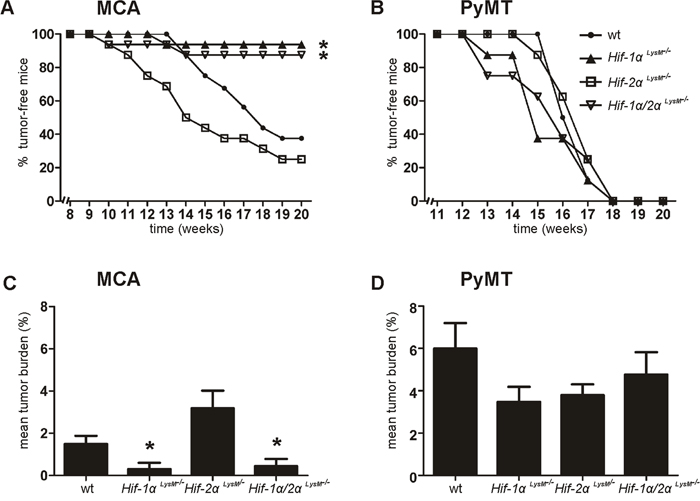
Figure 1: Role of HIF in tumorigenesis. A. Development of 3-methylcholanthrene (MCA)-induced fibrosarcomas (n = 16 per genotype) and B. development of polyoma virus middle T oncoprotein (PyMT) breast cancer (n = 8 per genotype) in wt C57BL/6J, myeloid Hif-1α LysM-/-, Hif-2α LysM-/- and Hif-1α/2α LysM-/- mice was assessed by palpation twice weekly. C. Mean tumor burden [(tumor weight/body weight)/number of mice)] in MCA-injected and D. PyMT mice was calculated at the end of the experiments. Bar graphs show mean values ± SEM. * P < 0.05 compared to wt mice.
Loss of HIF-1α in macrophages impairs metabolic activation of MCA in vivo
The reduced/absent tumor incidence in Hif-1α LysM-/- mice suggests differences in tumor initiation as a likely mechanism for reduced tumor outgrowth. To characterize the role of HIF-1α in myeloid cells at early stages of carcinogenesis, tissue samples from the site of MCA-as well as corn oil (control)-injections were obtained after 5 days. This time point was chosen to allow immune cell infiltration [31] and provided similar results to the analysis performed at day 10 (data not shown). As MCA was dissolved in corn oil, lipid droplets with encapsulated MCA formed in the skin in close proximity to injection sites. The formation and occurrence of these droplets were similar in skin tissue of wt and Hif-1α LysM-/- mice (data not shown). Five days after MCA-injection mainly neutrophils and fewer macrophages invaded the tissue surrounding lipid droplets. Skin of wt and Hif-1α LysM-/- mice showed a similar pattern of neutrophil and macrophage accumulation after the application of MCA, whereas minor neutrophil infiltration was detected following control injections, i.e. corn oil (Supplementary Figure 3A). Furthermore, the production of inflammatory cytokines such as interleukin (IL)-1ß or tumor necrosis factor α (TNFα), reported to be involved in MCA-induced tumor formation [31, 32], revealed no differences comparing wt and Hif-1α LysM-/- mice (Supplementary Figure 3B, 3C). Thus, differences in inflammation at sites of tumor initiation appear highly unlikely to explain a reduced tumor outcome in the absence of myeloid HIF-1. Therefore, metabolic activation of the MCA pre-carcinogen was analyzed 5 days after MCA-injection. In general, the MCA-metabolizing enzyme CYP1A1 was expressed in macrophages in the skin as shown by immunohistochemistry (Supplementary Figure 4A). The expression of Cyp1a1 mRNA in Hif-1α LysM-/- mice was diminished by approximately 75% under control conditions at sites of corn oil-injection compared with wt mice (Figure 2A). Reduced Cyp1a1 expression became more pronounced following MCA injection, whereas the expression of Cyp1b1 was similar in both genotypes in skin tissue (Figure 2B). These findings were confirmed by immunohistochemistry, detecting CYP1A1 and CYP1B1 in skin around lipid droplets. Consistent with mRNA expression, CYP1A1 signals were diminished around lipid droplets in skin of Hif-1α LysM-/- mice compared with wt mice (Figure 2C), while CYP1B1 expression around the injection site remained unchanged (Figure 2C). We then used a functional read-out of metabolic activation and analyzed DNA damage by immunohistochemistry staining for the phosphorylated form of the histone H2AX (γH2AX). Interestingly, DNA damage was detected in tissue close to the MCA-containing lipid droplets of wt mice, but was largely reduced in skin of Hif-1α LysM-/- mice (Figure 2C). We could not detect differences in DNA damage in the skin of HIF-2α LysM-/- compared to wt mice (Supplementary Figure 4B). These results suggest that the loss of Hif-1α, but not of Hif-2α, diminishes CYP1A1-mediated metabolic activation of MCA in skin in vivo, thereby reducing DNA damage.
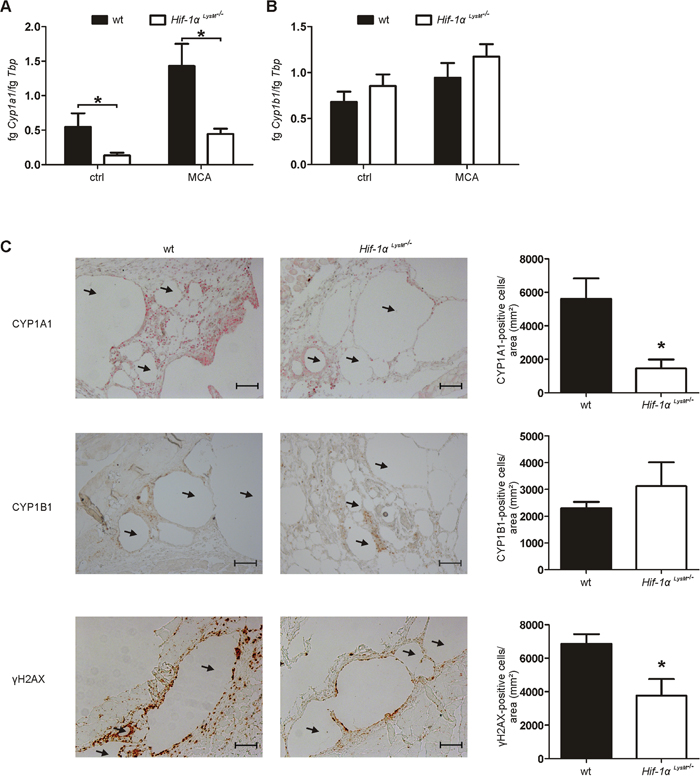
Figure 2: Cyp expression and DNA damage in Hif-1α LysM-/- mice. A. Cyp1a1 and B. Cyp1b1 mRNA expression in whole skin tissue in close proximity to MCA-injection sites 5 days after application was assessed by qPCR. mRNA levels were normalized to Tbp. Values give the mean ± SEM of n = 7. * P < 0.05. C. Representative images and quantification of skin sections 5 days after MCA-injection, stained for CYP1A1 (red), CYP1B1 (brown) and γH2AX (brown) by immunohistochemistry. Arrows indicate lipid droplets. Scale bars: 100 μm. Values are the mean ± SEM of n = 3 (HIF-1α LysM-/-)/ 4 (wt), * P < 0.05 compared to wt.
HIF-1α-deficient macrophages reveal an altered expression of xenobiotic metabolizing enzymes and limit DNA damage ex vivo
To verify molecular mechanisms of reduced metabolic MCA-activation, biotransformation was analyzed ex vivo. As it was recently published that tissue-resident macrophages, i.e. Langerhans cells, are the key player for metabolic activation of PAHs in the skin, we used macrophages in our ex vivo studies to explore the role of HIF-1α in metabolic activation of MCA and subsequent DNA damage [9]. In vivo, transformation of fibroblasts induces fibrosarcomas after MCA-application [33]. Therefore, we established a coculture model of PMs and NIH3T3 fibroblasts or mouse embryonic fibroblasts (MEF) to measure DNA damage ex vivo. Although stimulation with MCA caused some DNA damage in NIH3T3 cells in the absence of macrophages, DNA damage was substantially increased in NIH3T3 cells cocultured with wt macrophages without affecting viability or proliferation within the 24 h incubation period (Figure 3A, Supplementary Figure 5A, 5F-5I). PMs isolated from Hif-1α LysM-/- and wt mice were then analyzed for mRNA expression of the MCA-metabolizing enzymes Cyp1a1 and Cyp1b1. Stimulation with MCA induced the expression of Cyp1a1 (Figure 3B), while the increase in Cyp1b1 was marginal (Figure 3C). Similar to the results in skin, the expression of Cyp1a1 was drastically reduced by about 97% in PMs of Hif-1α LysM-/- mice compared to wt PMs under control conditions (Figure 3B). Also, a massive reduction of Cyp1a1 mRNA was detected after MCA-application in macrophages lacking Hif-1α. In addition, PMs of Hif-1α LysM-/- mice also reduced Cyp1b1 expression by roughly 47% under control conditions as well as after MCA-stimulation (Figure 3C). To assess functional consequences of reduced Cyp1a1 and Cyp1b1 mRNA expression, we cocultured NIH3T3 fibroblasts or MEFs with wt PMs or with PMs isolated from Hif-1α LysM-/- mice. Consistent with our observations in vivo, H2AX phosphorylation, a marker of DNA damage, was lower in NIH3T3 cells and MEFs that were cocultured with PMs of Hif-1α LysM-/- mice, but not with PMs of Hif-2α LysM-/- mice, compared to NIH3T3 cells or MEFs cocultured with wt PMs (Figure 3D, 3E, Supplementary Figure 5B-5E). Apparently, macrophages are important to induce DNA damage in fibroblasts after MCA-application requiring CYP expression and subsequent metabolic PAH-activation in myeloid cells.
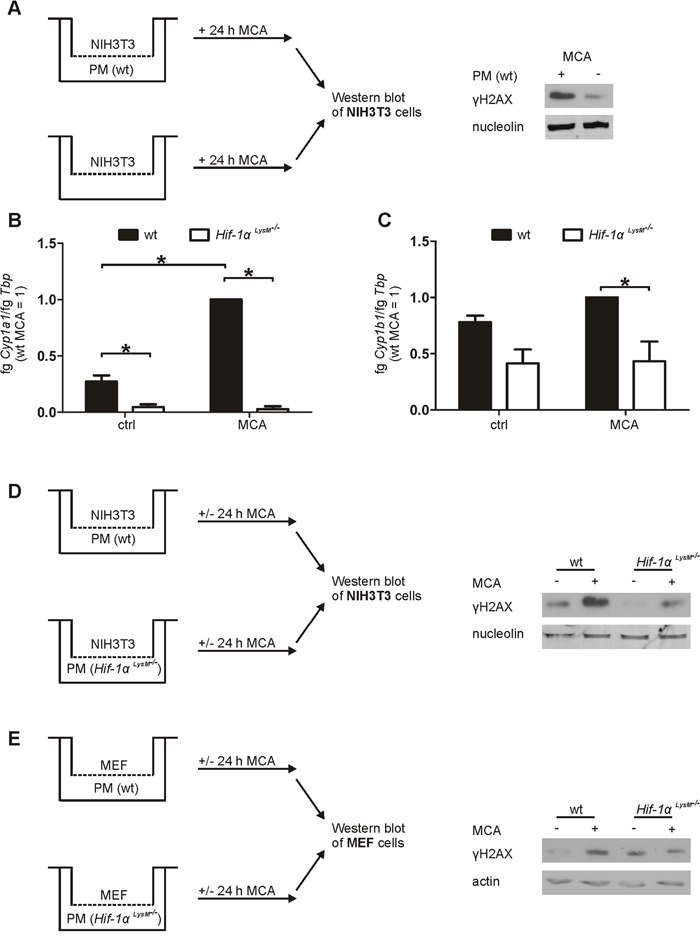
Figure 3: Impaired Cyp1a1 and Cyp1b1 expression in Hif-1α LysM-/- macrophages and fibroblast DNA damage in an in vitro coculture. A. Schematic illustration of the NIH3T3/peritoneal macrophages (PMs) coculture set up, stimulated with MCA for 24 h. A representative Western blot of phosphorylated γH2AX in NIH3T3 cells cocultured with or without PMs is shown. Nucleolin served as a loading control. B. Cyp1a1 and C. Cyp1b1 mRNA expression of PMs that were isolated from wt and Hif-1α LysM-/- mice and stimulated with MCA or dimethylsulfoxide (DMSO: ctrl) for 8 h assessed by qPCR. mRNA levels were normalized to Tbp. The ratio of Cyp1a1 or Cyp1b1 and Tbp in wt mice after stimulation with MCA was set to 1. Mean values ± SEM of n = 4 are presented (each n contains 3 mice/genotype). * P < 0.05. D. Illustration of the cocultures of NIH3T3 cells with PMs isolated from wt or Hif-1α LysM-/- mice E. and the cocultures of MEF cells with PMs isolated from wt or Hif-1α LysM-/- mice, stimulated with MCA or DMSO for 24 h. Representative Western analysis of phosphorylated H2AX in NIH3T3 is shown. Nucleolin or actin served as loading controls.
CYPs account for metabolic activation of MCA
To determine the potential of PMs isolated from Hif-1α LysM-/- mice, showing a reduced Cyp1a1 and Cyp1b1 expression, to activate MCA, we quantified the metabolite MCA-trans-9,10-dihydrodiol (MCA-Diol) by LC-MS/MS. CYP1A1 and CYP1B1 hydroxylate PAHs like MCA to a MCA-9,10-epoxide (PAH-epoxide; Figure 4A), which subsequently is converted by epoxide hydroxylase-1 (Ephx-1) to MCA-9,10-transdihydrodiol that is again hydroxylated by CYP1A1 or CYP1B1 to a more reactive MCA-9,10-trans-diol-epoxide. Therefore, PMs of wt and Hif-1α LysM-/- mice were stimulated with MCA for 8 hours and supernatants were collected for LC-MS/MS measurements. MCA-Diol formation was significantly reduced in supernatants of Hif-1α LysM-/- PMs compared to wt PMs (Figure 4B). In cells that were stimulated with DMSO only (ctrl) no metabolites were detected in the supernatants (data not shown) and the expression of Ephx-1, which catalyzes the formation of MCA-dihydrodiol from MCA-epoxide, did not differ between wt and Hif-1α knockout-cells (Supplementary Figure 6A).
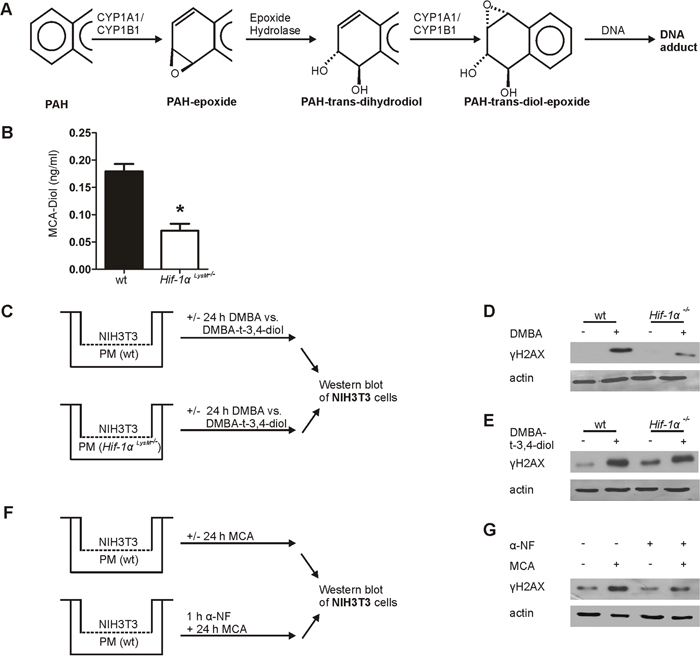
Figure 4: Metabolism of 3-methylcholanthrene (MCA) in peritoneal macrophages. A. Schematic overview of the metabolic activation of polycyclic arylhydrocarbons (PAHs). B. Quantification of MCA-Diol in the supernatants of isolated peritoneal macrophages (PMs) from wt and Hif-1α LysM-/- mice stimulated with MCA for 8 h and quantified by LC-MS/MS. Bars are the mean ± SEM of n = 4 (each n containing 3 mice/genotype). * P < 0.05 compared to wt. C. Schematic illustration of the coculture of NIH3T3 cells with PMs isolated from wt or Hif-1α LysM-/- mice stimulated with 7,12-dimethylbenz[a]anthracene (DMBA), DMBA-trans-3,4-dihydrodiol (DMBA-t-3,4-diol), or DMSO for 24 h. Representative Western blot of phosphorylated γH2AX in NIH3T3 cell stimulated with D. DMBA or E. DMBA-t-3,4-diol from 4 experiments is shown. Actin served as a loading control. F. Schematic illustration of the coculture of NIH3T3 and wt PMs, prestimulated for 1 h with or without 10 nM α-napthoflavone (α-NF), a CYP-inhibitor, following stimulation with MCA or without for 24 h. G. Representative Western blot of phosphorylated γH2AX in NIH3T3 cells cocultured with wt peritoneal macrophages from 5 experiments is shown. Actin served as a loading control.
To prove the relevance of PAH-metabolism in myeloid cells in causing DNA damage we used the PAH 7,12-dimethylbenz[a]anthracene (DMBA) and its commercially available active metabolite DMBA-trans-3,4-dihydrodiol (DMBA-t-3,4-diol) in our coculture system (Figure 4C-4E). Switching to DMBA was necessary because the active MCA-metabolite is not available. Following incubations of DMBA with wt PMs elicited a DNA damage response in fibroblasts, similarly to MCA. The γH2AX signal was largely attenuated when using macrophages from myeloid Hif-1α LysM-/- mice (Figure 4C, 4D, Supplementary Figure 6B). However, the metabolite DMBA-t-3,4-diol was equally potent in causing a DNA damage response when incubated with either wt or Hif-1α LysM-/- PMs (Figure 4C, 4E, Supplementary Figure 6C). This underscores the role of macrophage PAH-metabolism, dependent on HIF-1, for a DNA damage response in fibroblasts. To verify the relevance of CYP1A1 and CYP1B1 in this set up, we employed the CYP inhibitor α-naphtoflavone (α-NF). Co-stimulation of PMs with MCA and α-NF decreased H2AX phosphorylation compared to MCA-stimulation alone (Figure 4F, 4G, Supplementary Figure 6D). α-NF reduced H2AX phosphorylation to a level comparable to that seen in cocultures of NIH3T3 cells with PMs lacking Hif-1α. To summarize, formation of a DNA damaging PAH-metabolite demands HIF-1α in macrophages and is correlated to diminished CYP1A1 and CYP1B1 expression as well as reduced DNA damage.
The loss of HIF-1α decreases ARNT levels in macrophages
Next, we questioned whether HIF-1 directly regulates CYP1A1/CYP1B1. Therefore, Cyp1a1 and Cyp1b1 expression was analyzed in isolated PMs under hypoxic conditions or following their exposure to the HIF-stabilizer dimethyloxaloylglycine (DMOG). Macrophages failed to enhance Cyp1a1 or Cyp1b1 expression after 16 hours hypoxia (1% O2) or DMOG-stimulation (Supplementary Figure 7A, 7B). Regulation of CYP1A1 also remained unaffected by the loss of myeloid Hif-2α (Supplementary Figure 7A). Furthermore, treatment of cocultures with the specific PHD inhibitor IOX2 did not alter DNA damage responses in fibroblasts (Supplementary Figure 5D, 5E). As these data exclude a direct regulation of CYPs by HIF-1, we assumed that HIF-1α indirectly regulates CYP enzymes by affecting the AhR/ARNT pathway. Indeed, expression of Arnt mRNA was significantly reduced in macrophages of Hif-1α LysM-/- mice, whereas Ahr mRNA levels remained unchanged (Figure 5A, 5B). A decrease in Arnt mRNA expression lowered the amount of the ARNT protein in macrophages with a knockout of Hif-1α (Figure 5C, 5D, Supplementary Figure 7C, 7D). Aldolase 3A1 (Aldh3a1) and NADPH:quinone oxidoreductase 1 (Nqo1) mRNA, known AhR/ARNT target genes, were also significantly downregulated in Hif-1α LysM-/- PMs (Figure 5E, 5F). Similar to the behavior of CYPs, Aldh3a1 expression was not only impaired after MCA-stimulation but also at basal level.
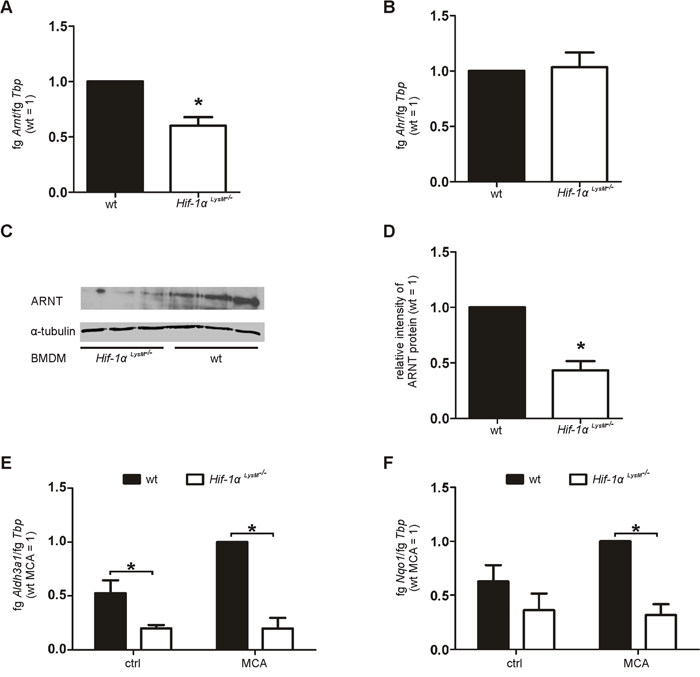
Figure 5: Reduced Arnt expression in Hif-1αLysM-/- macrophages. A. Arnt and B. Ahr mRNA expression in PMs isolated from wt and Hif-1α LysM-/- mice assessed by qPCR. mRNA levels were normalized to Tbp. The ratio of Arnt or Ahr and Tbp in wt mice was set to 1. Values are the mean ± SEM of n = 4 (each n contains 3 mice/genotype). * P < 0.05 compared to wt. C. Representative Western analysis of ARNT protein in BMDMs isolated from wt and Hif-1α LysM-/- mice. Tubulin served as a loading control. D. Quantification of ARNT protein expression. To quantitate the ARNT protein, band intensity of each experiment (3 mice per genotype) was measured. The medium intensities of wt BMDMs were set to 1 and compared to the medium intensities of Hif-1α LysM-/- BMDMs. Experiment including quantification was performed three times. E. Aldh3a1 and F. Nqo1 mRNA expression in PMs isolated from wt and Hif-1α LysM-/- mice, stimulated with MCA or DMSO (ctrl) for 8 h assessed by qPCR. mRNA levels were normalized to Tbp. The ratio of Aldh3a1 and Tbp in wt mice after stimulation with MCA was set to 1. Values are the mean ± SEM of n = 4 (each n contains 3 mice/genotype), * P < 0.05.
To test a direct regulation of ARNT by HIF-1, we analyzed the expression of Arnt following HIF-activation with hypoxia and DMOG. Arnt levels were not significantly affected by HIF activation (Supplementary Figure 7E) thus, excluding a direct regulation of ARNT by HIF-1. Further, we tested if the knockout of Hif-1α alters Arnt mRNA stability. Therefore, primary macrophages were time-dependently treated with actinomycin D (actD). A loss of Hif-1α did not alter Arnt mRNA stability (Supplementary Figure 7F). These findings suggest that metabolic activation of MCA is attenuated in Hif-1α LysM-/- macrophages, likely as a consequence of reduced ARNT transcription.
Metabolic MCA-activation is attenuated by a knockdown of ARNT
To verify the crucial role of ARNT in MCA-metabolism, we used an siRNA approach to knock down Arnt. Murine macrophages were isolated from wt mice and transiently transfected with siRNA targeting Arnt (siArnt) compared to a non-targeting siRNA control (siCtrl). As it was not possible to transfect PMs, we used bone marrow-derived macrophages (BMDMs) for the transient knockdown of Arnt. Silencing of Arnt was confirmed at the mRNA level, showing a reduction of 83% compared to siCtrl BMDMs (Figure 6A). Following treatments with MCA, cells harboring a knockdown of Arnt showed a reduced expression of the metabolizing enzymes Cyp1a1, Cyp1b1, and Aldh3a1 (Figure 6B, 6C, 6D) compared to siCtrl-treated BMDMs. The impact of the Arnt knockdown on DNA damage was analyzed with our coculture assay. Similar to coculture experiments with PMs of Hif-1α LysM-/- mice, phosphorylation of γH2AX was reduced in NIH3T3 cells following their coculture with siArnt BMDMs in the presence of MCA (Figure 6E). These data link the loss of Hif-1α, decreased ARNT expression and attenuated CYP1A1 and CYP1B1 induction. Reduced biotransformation of MCA and tumor development in myeloid Hif-1α LysM-/- mice is thus explained by diminished ARNT expression and the subsequently reduced metabolic activation of PAHs by CYP1A1 and CYP1B1.
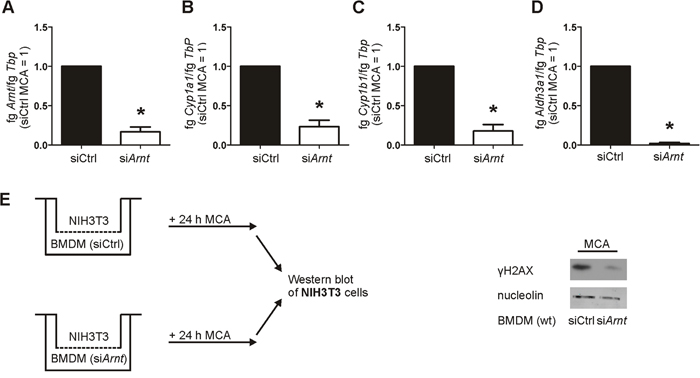
Figure 6: Reduced Arnt expression impairs AhR-mediated gene expression and DNA damage. A. Arnt, B. Cyp1a1, C. Cyp1b1, and D. Aldh3a1 mRNA expression in bone marrow-derived macrophages (BMDMs) transiently transfected with siRNA against Arnt (siArnt) or control siRNA (siCtrl), followed by stimulation with MCA for 8 h, assessed by qPCR. mRNA levels were normalized to Tbp. The ratio of Arnt, Cyp1a1, Cyp1b1, or Aldh3a1 and Tbp in siCtrl BMDMs after MCA-stimulation was set to 1. Values are the mean ± SEM of n = 4 (each n containing 3 mice/genotype), * P < 0.05 compared to siCtrl MCA. E. Illustration of the coculture of NIH3T3 cells with wt BMDMs that were transfected with siRNA against Arnt (siArnt) and control siRNA (siCtrl) and incubated with MCA for 24 h. Representative Western analysis of phosphorylated γH2AX in NIH3T3 cells from 4 experiments. Nucleolin served as a loading control.
DISCUSSION
Although the role of myeloid HIF-1α and HIF-2α during inflammation is well described their impact on carcinogenesis is less well understood [30, 34]. In the oncogene-driven PyMT model, tumor latency was unaffected, while tumor burden was non-significantly reduced in both knockout strains. This corroborates earlier data in PyMT mice with a myeloid-specific Hif-1α LysM-/- knockout, demonstrating no influence on tumor initiation but a reduced tumor mass [29]. Mechanistically, a loss of Hif-1α in macrophages attenuated their ability to suppress T-cell proliferation and activation in vitro thus, favoring cell death at early stages of PyMT tumor progression, assumingly lowering tumor mass in vivo. In this model the role of myeloid HIF-2α is unexplored. We noticed a slightly decreased tumor burden at the end of the experiment (week 20), pointing to a tumor-promoting ability of HIF-2α in the PyMT tumor model. In a murine melanoma model, activation of macrophage HIF-2α potentiates tumor-suppressive effects of GM-CSF and enhances survival of mice, while effects of HIF-activation in the absence of GM-CSF during tumor development remain unclear [35]. In a xenograft model of Lewis Lung carcinoma (LLC) and B16BL6 melanoma cells activation of both HIF-isoforms, by ablating PHD2 in macrophages and T-cells, attenuated tumor development due to altered cell death and proliferation of tumor cells [36]. These findings suggest an organ- and context-specific effect of HIF on spontaneous tumor development.
In contrast to minor effects in the oncogene-driven cancer model the loss of Hif-1α in myeloid cells almost completely attenuated tumor development in the chemical MCA-carcinogen driven model. The model is characterized for its inflammatory component during early tumor formation and thus, interfering with inflammation lowers the tumor incidence [31, 37]. Consistent with these data, we noticed the accumulation of neutrophils and macrophages in close proximity to lipid droplets, formed at the injection sites, but no dependency on HIF-1 towards these signs of inflammation. These observations are supported by the notion that inflammatory cytokines are equally produced in wt and Hif-1α LysM-/- mice, suggesting that early inflammatory markers do not account for differences in tumor progression.
PAHs are prevalent carcinogens that require biotransformation and thus, metabolic activation by CYP1A1 and CYP1B1 [6, 38-40]. These CYPs are not only abundant in the liver but are also expressed in extra hepatic tissue and are crucial for the DNA damage response of PAHs in these tissues [39]. Our data suggest that myeloid cells in the skin contribute to metabolic PAH activation and subsequent DNA damage in fibroblasts, thereby initiating tumorigenesis. Consistent, targeting or depleting myeloid cells in vivo revealed the importance of these cells for tumor induction in different cancer types before [41, 42]. Our data confirm and extend a recent publication identifying Langerhans cells as an essential component for CYP-mediated PAH-metabolism in the 7,12-dimethylbenz[a]anthracene (DMBA) two-stage cutaneous chemical carcinogenesis model [9]. These observations and our data link biotransformation and activation of PAH carcinogens in the skin to cells of the myeloid compartment. Depleting myeloid cells via clodronate to prove this assumption in vivo is technically not possible in long-term experiments such as the MCA-model. As Langerhans cells also express Lysozyme M (LysM) [43] and thus, are depleted of HIF-1 in our conditional knockout mouse as well, it can be speculated that these tissue-associated cells also relay on HIF-1 to enhance chemical carcinogenesis via PAH-metabolism. Besides pointing to myeloid cells in the skin as a necessary component for PAH-metabolisms, our data provide evidence for a role of HIF-1 but not HIF-2 in this process and furthermore emphasize that downregulation of CYPs in myeloid cells is sufficient to attenuate tumor outgrowth in the skin. Replacing the PAH DMBA by its DNA-damaging metabolite DMBA-trans-3,4-dihydrodiol (DMBA-t-3,4-diol), we could restore reduced DNA damage in NIH3T3 cells cocultured with HIF-1α LysM-/- PMs, thereby confirming limited metabolic activation as the crucial parameter for attenuated DNA damage. Upregulation of CYP1A1 and CYP1B1 by PAH requires dimerization of AhR with ARNT [3]. We noticed reduced Arnt expression in Hif-1α knockout macrophages as a likely explanation for the attenuated CYP induction. Previous studies already suggested interactions between the AhR/ARNT and HIF-α/ARNT pathways. Studying the competition between HIF-1α and AhR for ARNT revealed attenuated AhR/ARNT target gene expression under hypoxic conditions [5, 22, 44]. This goes along with studies confirming a higher affinity of HIF-1α compared to AhR for the constitutively expressed ARNT [21]. Moreover, upregulation of ARNT by hypoxia or HIF-stabilizers has been described, although molecular mechanisms remain obscure [45-50]. Interestingly, reduced Arnt mRNA levels under normoxic conditions in vitro in Min6 cells after a knockdown of HIF-1α as well as ex vivo in islets from Hif-1α null-mice were shown, without characterizing potential consequences [51]. To date, the complex interaction of HIF-α isoforms, ARNT, and AhR is not fully understood. So far we excluded an altered mRNA stability and direct regulation of ARNT by HIF-1, which may point to transcriptional regulation of ARNT in Hif-1α LysM-/- mice. To mechanistically explain how a loss of Hif-1α in macrophages downregulates ARNT future investigations are required.
The loss of Hif-2α left tumor initiation unaffected, but accelerated tumor development and enhanced tumor burden in the MCA model. This may point to a tumor-repressing ability of HIF-2α. The impact of HIF-2α on tumorigenesis was already analyzed in a model of murine chemically-induced colitis-associated cancer (CAC) and hepatocellular carcinoma (HCC). Impaired tumor cell proliferation and progression in the CAC model after the myeloid loss of Hif-2α was attributed to reduced TAM infiltration with no effect on HCC progression [30]. We neither observed reduced immune cell infiltration with a knockout of Hif-2α, nor differences in vascularization, which implied tumor- and/or organ-specific effects of myeloid HIF-2α.
Collectively, the loss of Hif-1α in myeloid cells attenuated CYP-mediated metabolic activation of PAHs, subsequent in vivo and ex vivo DNA damage, and reduced tumor development. PAHs are part of tobacco smoke and air pollutants. [52-55]. An increasing potential of carcinogens as part of air pollution causes most cancer deaths, besides triggering cardiovascular diseases [1]. Others have already revealed the need for nonepithelial stromal cells to activate PAH mutagens [9, 56]. The marked resistance of myeloid cells with a knockout of Hif-1α to chemical carcinogenesis establishes the capacity of HIF-1 to substantially enhance toxicity of environmental agents. Our data are consistent with a cooperative carcinogenicity scenario, where CYP1A1/CYP1B1 metabolize MCA to a DNA damaging agent, which is delivered to adjacent fibroblasts to initiate DNA damage and start tumorigenesis. These studies open the possibility that HIF-1 in resident macrophages is a prerequisite for PAH-induced mutations and tumor development within other epithelial tissues, contributing to the risk of colon, lung, or genitourinary carcinomas. Further studies need to prove whether HIF-1 specific inhibitors can be used to prevent chemically-induced skin or lung carcinoma.
MATERIALS AND METHODS
Myeloid-specific knockout of HIF-1α and HIF-2α
Hif-1αflox/flox/lysozyme M Cre (LysMCre) mice (Hif-1α LysM-/-) were generated and kindly provided by Prof. R.S. Johnson. Hif-2αflox/flox mice were obtained from Prof. M.C. Simon and crossed to LysMCre mice to obtain myeloid-specific Hif-2α knockout mice (Hif-2α LysM-/- ). Mice were established as 129 Sv x C57BL/6 mice containing floxed sites flanking exon 2 of the Hif-1α or Hif-2α gene and were backcrossed into the C57BL/6 background. Hif-1α/2αflox/flox/LysMCre (Hif-1α/2α LysM-/- ) mice were bred by crossing Hif-1αflox/flox/LysMCre mice containing loxP sites around exon 2 of Hif-2α. Mice having flanked loxP sites but were negative for the Cre recombinase served as controls (wt mice).
PyMT breast cancer model
For details see Supplementary Methods.
MCA induced fibrosarcomas
Male mice were subcutaneously (s.c.) injected in the right thigh with 100 μg MCA dissolved in 100 μl corn oil (Sigma-Aldrich, Steinheim, Germany). Tumor progression in mice as well as mice weight was evaluated twice weekly by palpation [29, 31]. Tumors developed within week 8-20 and mice were sacrificed after 21 weeks. Tumor tissue was extracted and weighed. Part of the tumor tissue was fixed in 4% paraformaldehyde (PFA) or zinc fixation buffer for immunohistochemistry (IHC), while remaining tissue was prepared for flow cytometry analysis. For studies at early time points, mice were injected with the indicated MCA concentration in one thigh and with 100 μl corn oil as a control in the other thigh. Skin tissue around the injection site with a diameter of approximately 1.5 cm was isolated 5 days after injection and either fixed in 4% PFA or frozen in liquid nitrogen for RNA isolation.
Flow cytometry
For details see Supplementary Methods.
Isolation of primary mouse macrophages
Peritoneal macrophages (PMs) and bone marrow-derived macrophages (BMDMs) were isolated as described previously [57].
Cell culture and incubation procedures
NIH3T3, bone-marrow-derived macrophages (BMDMs), and peritoneal macrophages (PMs) were cultured in Dulbecco’s Modified Eagle’s Medium DMEM (high glucose) supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin (all from PAA). Cells were kept at 37°C in a humidified atmosphere with 5% CO2. BMDMs were differentiated for 7 days using 20 ng/ml macrophage colony-stimulating factor (M-CSF, Peprotech, Hamburg, Germany). PMs were cultured for 2 days before treatment. For hypoxic exposure cells were incubated either with 1% O2 in a hypoxia workstation InVivo400 (Ruskinn Technology, Leeds, UK) or 1 mM dimethyloxalylglycine (DMOG; Alexis Biochemicals, Lörrach, Germany) for 16 h. For RNA analysis, primary macrophages were incubated with MCA (5 μg/ml) or dimethylsulfoxide (DMSO) as a control for 8 h. To analyze mRNA stability, differentiated BMDMs were treated with 2.5 μg/ml actinomycin D (Sigma-Aldrich) for times indicated.
siRNA transfection
For transfection, isolated BMDMs were differentiated with 20 ng/ml M-CSF for 1 week and transfected afterwards with 150 nM SMARTpool ON-TARGET plus siArnt or ON-TARGET plus Non-targeting pool siCtrl (both Thermo Scientific, Karlsruhe, Germany) using Hiperfect transfection reagent (Quiagen, Hilden, Germany). Therefore, BMDMs were incubated in medium without FCS containing siRNA and Hiperfect. After 6 h, medium containing M-CSF (final concentration of 20 ng/ml) was added and after 24 h cells were stimulated with MCA (5 μg/ml) or dimethylsulfoxide (DMSO) as a control for 8 h.
Fibroblast-macrophage cocultures
PMs or BMDMs were isolated as described and plated into the lower compartment of transwell inserts (6-well plates), while NIH3T3 fibroblasts were seeded in the upper well of the inserts (0.1 μm pore size). The following day, cells were stimulated with MCA (5 μg/ml), 7,12-dimethylbenz[a]anthracene (DMBA: 2 μg/ml), DMBA-trans-3,4-dihydrodiol (DMBA-t-3,4-diol: 2 μg/ml) or DMSO (0.1%) for 24 h. Afterwards, NIH3T3 cells were harvested for Western analysis. Alternatively, cells were prestimulated for 1 h with 10 nM α-naphtoflavone (α-NF) (Sigma-Aldrich), followed by the addition of MCA for 24 h.
RNA isolation
For RNA preparation from skin, tissue was obtained as described above, frozen and dissected by an IKA®T25 digital UltraTurrax® (IKA®-Werke GmbH & Co. KG, Staufen, Germany) in peqGold (Peqlab, Erlangen, Germany). RNA from skin and cells was isolated and qPCR was performed as described before [58].
Quantitative real time PCR (qPCR)
For details see Supplementary Methods.
Western analysis
As PMs yield not enough protein for Western analysis, the protein level of ARNT was examined in BMDMs instead of PMs. Cells were washed in PBS, lysed in cell lysis buffer (6,65 M urea, 10% glycerol, 1% SDS, 10 mM Tris/HCl, pH 6.8; pH adjusted to 7.4) and sonicated following centrifugation (16000 g, 10 min). After quantification of the protein amount using a protein assay kit (Bio-Rad) 60 μg protein was loaded on 15% SDS-polyacrylamide gels and blotted onto a Immobilon-FL polyvinylidene difluoride (PVDF)-membrane (Millipore). Western analysis of ARNT was performed with 100 μg protein on a 10% SDS-polyacrylamide gel. Membranes were incubated using antibodies against phospho-Histone H2AX (γH2AX: Millipore), ARNT (Abcam), α-tubulin (Sigma-Aldrich), actin (Sigma-Aldrich), and nucleolin (SantaCruz, Heidelberg, Germany) overnight at 4°C. Blots were incubated with the appropriate secondary antibodies and detected using either Enhanced Chemiluminescence (ECL) or the Odyssey infrared imaging system (Li-COR Biosciences, Bad Homburg, Germany). ARNT blots were quantified by ImageJ 1.3 (NIH).
Immunohistochemistry
Tissue samples were dehydrated in alcohol (Sigma-Aldrich) and embedded in paraffin. Sections of 4 μm were deparaffinized in xylene (Sigma-Aldrich) and rehydrated in alcohol. For antigen retrieval, sections were boiled for 12 min in TRS buffer (DAKO, Hamburg, Germany). Endogenous Avidin and Biotin were blocked by using a biotin-blocking system (DAKO). For staining, a Catalyzed Signal Amplification (CSA) kit (DAKO) was used. Primary antibodies were incubated at 4°C overnight. Primary antibodies used: anti-mouse F4/80 (eBioscience, Frankfurt, Germany), anti-CD31 (Chemicon International, Temecula, CA, USA), anti-mouse Ym1 rabbit polyclonal antibody (StemCell Technologies, Grenoble, France), anti-phospho-Histone H2AX (Millipore, Darmstadt, Germany; Abcam, Cambridge, UK), CYP1A1 (Abcam, Cambridge, UK), and Ly6B.2 allogenic antibody (AbD Serotec, Puchheim, Germany). Afterwards, corresponding biotinylated secondary antibodies (biotinylated anti-rat from DAKO and biotinylated anti-rabbit from Axxora, Loerrach, Germany) were applied. For color development Diaminobenzidin (DAB) (contained in the CSA kit) was added as a substrate to the peroxidase-coupled secondary antibody. Sections were analyzed using the microscope Axioskop 40 (Carl Zeiss AG, Göttingen, Germany). CD31 positive signals were quantified using HistoQuest 2.X (Tissue Gnostics, Wien, Austria).
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS)
For LC-MS/MS detection, PMs were incubated with MCA or DMSO for 8 h, followed by collection of cell supernatants. To determine analyte retention times and mass fragmentation, the standard 3-methylcholanthrene-cis-11,12-dihydrodiol (11,12-diOH-3-MCA) was obtained from MRIGlobal, Kansas City, MO, USA. For sample extraction, 600 μl of each culture media was mixed with 50 μl standard/methanol and 20 μl internal standard solution (10 μg/ml of 1-pyrenol in methanol (Sigma-Aldrich)) and extracted twice with ethyl acetate (1 ml and 600 μl). The organic supernatant was evaporated under a gentle stream of nitrogen at 45°C following reconstitution in 50 μl methanol. Samples were injected into the liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS). The system consisted of a hybrid triple quadrupole-ion trap mass spectrometer QTrap 4000 (AB Sciex, Darmstadt, Germany) equipped with a Turbo-V-source, an Agilent 1100 chromatographic system (Agilent, Waldbronn, Germany), and an HTC Pal autosampler (CTC Pal, Zwingen, Switzerland). The system was operated in positive ionization mode at 5500 V and 400°C in MS3 scan modus.
For chromatographic separation a Max-RP column (150 x 2 mm, Phenomenex, Aschaffenburg, Germany) was used with a linear gradient at a flow rate of 300 μl/min. Mobile phase A was water/ammonia (100:0.01, v/v) and mobile phase B was methanol/ammonia (100:0.01, v/v). Total run times were 40 min and sample volumes for injection were 20 μl. Calibration curves ranged from 0.1 to 10 ng/mL for 11,12-diOH-3-MCA. Samples were quantified using the internal standard method and the software Analyst 1.4 (AB Sciex, Darmstadt, Germany).
Statistical analysis
All experiments were performed at least 3 times and results are expressed as means ± SEM. Graphs showing tumor development were generated with Kaplan-Meier analysis and compared using the log-rank test. Student’s t-test was performed to evaluate differences between wt and Hif-1α LysM-/- samples and multiple-group comparisons were calculated after analysis of variance (ANOVA) and Bonferroni’s test. Differences at p < 0.05 were considered statistically significant.
Study approval
Animal experiments were approved by local authorities (approval number F144/05) and performed following the guidelines of Hessian animal care and use committee.
ACKNOWLEDGMENTS
We are grateful to Prof. R.S. Johnson (Department of Physiology, Development and Neuroscience, University of Cambridge, UK) and Prof. M.C. Simon (Department of Cancer Biology, University of Pennsylvania School of Medicine, Philadelphia, PA) for providing myeloid Hif-knockout mice. Special thanks go to Tanja Keppler, Bettina Wenzel, and Sandra Labocha for their excellent technical support.
FINANCIAL SUPPORT
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 815, project A8 (BB) and A14 (GG) and SFB 1039 (GG) project Z01) and Deutsche Krebshilfe (BB) (109599).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Kessler R. Prevention: Air of danger. Nature. 2014; 509:S62-63.
2. Schreiber TH, Podack ER. A critical analysis of the tumour immunosurveillance controversy for 3-MCA-induced sarcomas. Br J Cancer. 2009; 101:381-386.
3. Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science. 1992; 256:1193-1195.
4. Omiecinski CJ, Remmel RP, Hosagrahara VP. Concise review of the cytochrome P450s and their roles in toxicology. Toxicol Sci. 1999; 48:151-156.
5. Allen JW, Johnson RS, Bhatia SN. Hypoxic inhibition of 3-methylcholanthrene-induced CYP1A1 expression is independent of HIF-1alpha. Toxicol Lett. 2005; 155:151-159.
6. Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004; 95:1-6.
7. Kondraganti SR, Fernandez-Salguero P, Gonzalez FJ, Ramos KS, Jiang W, Moorthy B. Polycyclic aromatic hydrocarbon-inducible DNA adducts: evidence by 32P-postlabeling and use of knockout mice for Ah receptor-independent mechanisms of metabolic activation in vivo. Int J Cancer. 2003; 103:5-11.
8. Monteiro P, Gilot D, Le Ferrec E, Lecureur V, N'Diaye M, Le Vee M, Podechard N, Pouponnot C, Fardel O. AhR- and c-maf-dependent induction of beta7-integrin expression in human macrophages in response to environmental polycyclic aromatic hydrocarbons. Biochem Biophys Res Commun. 2007; 358:442-448.
9. Modi BG, Neustadter J, Binda E, Lewis J, Filler RB, Roberts SJ, Kwong BY, Reddy S, Overton JD, Galan A, Tigelaar R, Cai L, Fu P, Shlomchik M, Kaplan DH, Hayday A, et al. Langerhans cells facilitate epithelial DNA damage and squamous cell carcinoma. Science. 2012; 335:104-108.
10. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EM, Macchiarulo A, Vacca C, Iannitti R, Tissi L, Volpi C, Belladonna ML, Orabona C, Bianchi R, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014; 511:184-190.
11. Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X, Anastasov N, Lehmann I, Hofer T. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget. 2014; 5:1038-1051. doi: 10.18632/oncotarget.1637.
12. Fantozzi A, Christofori G. Mouse models of breast cancer metastasis. Breast Cancer Res. 2006; 8:212.
13. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
14. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr Opin Immunol. 2014; 27C:16-25.
15. Lee K, Hwang H, Nam KT. Immune Response and the Tumor Microenvironment: How They Communicate to Regulate Gastric Cancer. Gut Liver. 2014; 8:131-139.
16. Cuninghame S, Jackson R, Zehbe I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology. 2014; 456-457C:370-383.
17. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883-899.
18. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010; 11:889-896.
19. Mamlouk S, Wielockx B. Hypoxia-inducible factors as key regulators of tumor inflammation. Int J Cancer. 2013; 132:2721-2729.
20. Mandl M, Depping R. Hypoxia-Inducible Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT) (HIF-1beta): Is It a Rare Exception? Mol Med. 2014; 20:215-220.
21. Gradin K, McGuire J, Wenger RH, Kvietikova I, fhitelaw ML, Toftgard R, Tora L, Gassmann M, Poellinger L. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the Arnt transcription factor. Mol Cell Biol. 1996; 16:5221-5231.
22. Nie M, Blankenship AL, Giesy JP. Interactions between aryl hydrocarbon receptor (AhR) and hypoxia signaling pathways. Environ Toxicol Pharmacol. 2001; 10:17-27.
23. Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008; 15:678-685.
24. Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000; 157:411-421.
25. Song IS, Wang AG, Yoon SY, Kim JM, Kim JH, Lee DS, Kim NS. Regulation of glucose metabolism-related genes and VEGF by HIF-1alpha and HIF-1beta, but not HIF-2alpha, in gastric cancer. Exp Mol Med. 2009; 41:51-58.
26. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999; 59:5830-5835.
27. Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009; 13:2780-2786.
28. Leite de Oliveira R, Deschoemaeker S, Henze AT, Debackere K, Finisguerra V, Takeda Y, Roncal C, Dettori D, Tack E, Jonsson Y, Veschini L, Peeters A, Anisimov A, Hofmann M, Alitalo K, Baes M, et al. Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell. 2012; 22:263-277.
29. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010; 70:7465-7475.
30. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, Hammond R, Gimotty PA, Keith B, Simon MC. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010; 120:2699-2714.
31. Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA, Apte RN. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007; 67:1062-1071.
32. Ziolkowska M, Nowak JJ, Janiak M, Ryzewska A. Recombinant tumor necrosis factor alpha inhibits growth of methylcholanthrene-induced sarcoma and enhances natural killer activity of tumor-infiltrating lymphocytes in aging rats. Arch Immunol Ther Exp (Warsz). 1993; 41:357-364.
33. Zhang J, Chen L, Liu X, Kammertoens T, Blankenstein T, Qin Z. Fibroblast-specific protein 1/S100A4-positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res. 2013; 73:2770-2781.
34. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003; 112:645-657.
35. Roda JM, Wang Y, Sumner LA, Phillips GS, Marsh CB, Eubank TD. Stabilization of HIF-2alpha induces sVEGFR-1 production from tumor-associated macrophages and decreases tumor growth in a murine melanoma model. J Immunol. 2012; 189:3168-3177.
36. Mamlouk S, Kalucka J, Singh RP, Franke K, Muschter A, Langer A, Jakob C, Gassmann M, Baretton GB, Wielockx B. Loss of prolyl hydroxylase-2 in myeloid cells and T-lymphocytes impairs tumor development. Int J Cancer. 2014; 134:849-858.
37. Qin Z, Kim HJ, Hemme J, Blankenstein T. Inhibition of methylcholanthrene-induced carcinogenesis by an interferon gamma receptor-dependent foreign body reaction. J Exp Med. 2002; 195:1479-1490.
38. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011; 331:1565-1570.
39. Shimada T, Yun CH, Yamazaki H, Gautier JC, Beaune PH, Guengerich FP. Characterization of human lung microsomal cytochrome P-450 1A1 and its role in the oxidation of chemical carcinogens. Mol Pharmacol. 1992; 41:856-864.
40. Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 1996; 56:2979-2984.
41. Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther. 2008; 7:788-799.
42. Reusser NM, Dalton HJ, Pradeep S, Gonzalez-Villasana V, Jennings NB, Vasquez HG, Wen Y, Rupaimoole R, Nagaraja AS, Gharpure K, Miyake T, Huang J, Hu W, Lopez-Berestein G, Sood AK. Clodronate inhibits tumor angiogenesis in mouse models of ovarian cancer. Cancer Biol Ther. 2014; 15:1061-1067.
43. Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med. 2008; 205:2839-2850.
44. Vorrink SU, Severson PL, Kulak MV, Futscher BW, Domann FE. Hypoxia perturbs aryl hydrocarbon receptor signaling and CYP1A1 expression induced by PCB 126 in human skin and liver-derived cell lines. Toxicology and applied pharmacology. 2014; 274:408-416.
45. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92:5510-5514.
46. Chilov D, Camenisch G, Kvietikova I, Ziegler U, Gassmann M, Wenger RH. Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1alpha. J Cell Sci. 1999; 112:1203-1212.
47. Zhong H, Hanrahan C, van der Poel H, Simons JW. Hypoxia-inducible factor 1alpha and 1beta proteins share common signaling pathways in human prostate cancer cells. Biochem Biophys Res Commun. 2001; 284:352-356.
48. Vavilala DT, Ponnaluri VK, Vadlapatla RK, Pal D, Mitra AK, Mukherji M. Honokiol inhibits HIF pathway and hypoxia-induced expression of histone lysine demethylases. Biochem Biophys Res Commun. 2012; 422:369-374.
49. Mandl M, Kapeller B, Lieber R, Macfelda K. Hypoxia-inducible factor-1beta (HIF-1beta) is upregulated in a HIF-1alpha-dependent manner in 518A2 human melanoma cells under hypoxic conditions. Biochem Biophys Res Commun. 2013; 434:166-172.
50. Wolff M, Jelkmann W, Dunst J, Depping R. The Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT/HIF-1beta) is influenced by hypoxia and hypoxia-mimetics. Cell Physiol Biochem. 2013; 32:849-858.
51. Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ, O'Connell PJ, Loudovaris T, Kay TW, Kulkarni RN, Okada T, Wang XL, Yim SH, Shah Y, Grey ST, Biankin AV, et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J Clin Invest. 2010; 120:2171-2183.
52. Shields PG, Bowman ED, Harrington AM, Doan VT, Weston A. Polycyclic aromatic hydrocarbon-DNA adducts in human lung and cancer susceptibility genes. Cancer Res. 1993; 53:3486-3492.
53. Hecht SS. Tobacco smoke carcinogens and breast cancer. Environ Mol Mutagen. 2002; 39:119-126.
54. Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002; 21:7435-7451.
55. Kinoshita N, Gelboin HV. The role of aryl hydrocarbon hydroxylase in 7,12-dimethylbenz(a)anthracene skin tumorigenesis: on the mechanism of 7,8-benzoflavone inhibition of tumorigenesis. Cancer Res. 1972; 32:1329-1339.
56. Gould MN, Cathers LE, Moore CJ. Human breast cell-mediated mutagenesis of mammalian cells by polycyclic aromatic hydrocarbons. Cancer Res. 1982; 42:4619-4624.
57. Weichand B, Weis N, Weigert A, Grossmann N, Levkau B, Brune B. Apoptotic cells enhance sphingosine-1-phosphate receptor 1 dependent macrophage migration. Eur J Immunol. 2013; 43:3306-3313.
58. Jennewein C, Kuhn AM, Schmidt MV, Meilladec-Jullig V, von Knethen A, Gonzalez FJ, Brune B. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J Immunol. 2008; 181:5646-5652.