
INTRODUCTION
Pheochromocytoma/paraganglioma (PCC/PGL) is defined by the presence of metastases at non-chromaffin sites distant from the primary tumor, such as lung, liver, bone and lymph nodes, rather than local invasion [1]. Although PCC/PGL malignancy rate varies with tumor site and the particular inherited mutation in familial diseases, it is difficult to predict malignancy in primary tumors [2, 3]. Therefore, biomarkers that can indicate subsets of tumors that may metastasize must be identified.
Establishment of histopathological malignancy criteria is challenging due to the rarity, heterogeneity and lengthy tumor development time of PCL/PGL. In 2002, Thompson developed the Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) system, consisting of 12 parameters weighted according to relative frequency in benign and malignant adrenal PCC/PGLs [4]. Tumors with a PASS ≥ 4 were identified as potentially aggressive. Although this scoring system has been validated independently, there is debate as to its utility and reproducibility [5, 6]. Consequently, there are no consistently reliable histological features to distinguish malignant from benign tumors or to predict malignancy using resected primary tumor tissue.
Several attempts have been made to establish useful molecular markers for PCC/PGL malignancy. Mutations of the succinate dehydrogenase subunit B gene (SDHB) were associated with high rates of malignancy and extra-adrenal tumor metastasis [7, 8]. Non-diploid tumors were also found to be associated with malignancy [9]. Comparative genomic hybridization (CGH) studies in sporadic PCC/PGL revealed loss of chromosomes 1p, 3q, and 6q and gain of 9q, 16p, 17q, 19p and 19q [10, 11]. One study reported that progression to malignancy was strongly associated with deletions on chromosomes 6q and 17p [10]. Another revealed that alterations of chromosome 11, especially loss of 11q22–23, were more frequent in malignant than benign tumors [11]. In a gene expression profile study, over 80% of differentially expressed genes were downregulated in malignant tumors [17]. In contrast, HSP90 [18], human telomerase reverse transcriptase (hTERT) [19], tenascin [20], N-cadherin [21] and COX-2 [20] were overexpressed in malignant tumors.
Here, we performed array CGH (aCGH) and paired gene expression profiling of fresh, frozen PCC/PGL samples (n = 12), including three malignant tumors, to identify genes that distinguish benign from malignant tumors. Integrated analysis of the data identified phenylethanolamine-N-methyltransferase (PNMT) as a candidate marker. This was validated using real-time quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) and immunohistochemistry (IHC) in an independent set of 62 PCC/PGL samples.
RESULTS
Patient clinicopathological characteristics
Fresh, frozen PCC/PGL specimens were obtained from nine patients with benign tumors and three patients with malignant tumors, classified based on the absence or presence of metastatic lesions. Detailed clinicopathological characteristics of these 12 PCC/PGL patients are presented in Table 1. Additionally, 62 formalin-fixed paraffin-embedded (FFPE) PCC/PGL samples were analyzed, including 19 malignant and 31 benign specimens. Benign and malignant PCC/PGL patient clinicopathological characteristics were compared (Table 2). All patients presented non-syndromic PCC/PGL without a family history of the disease. Of the 62 patients, 40 (64.5%) had benign tumors, while 22 (35.5%) had malignant PCC/PGL, indicated by either lymph node or distant metastasis. Among the 40 patients with benign PCC/PGL, two (5.0%) were < 20 years old. The malignant PCC/PGL group consisted of 12 (54.5%) patients with PCC and 10 (45.5%) with PGL. The median PASS of the malignant PCC/PGL patients was > 4, significantly higher than that of benign PCC/PGL patients (P < 0.001). Malignant PCC/PGL tumors were larger than benign ones (P = 0.039). In addition, recurrence occurred in only 1/40 patient with benign PCC/PGL, with no deaths. Recurrence and death was observed in 14/22 (63.6%) and 4/22 malignant PCC/PGL patients (18.2%), respectively. Statistical analyses revealed no significant differences between benign and malignant PCC/PGL patients with regard to sex (P = 0.822), age (P = 0.535), disease pathology (P = 0.596) or follow-up duration (P = 0.125).
Table 1: Clinicopathologic characteristics of 12 PCC/PGL patients
No. |
Sex |
Age (year) |
Type |
Primary site |
Metastasis site |
Diagnosis |
Size (cm) |
Death |
Recur |
|---|---|---|---|---|---|---|---|---|---|
1 |
M |
51 |
B |
retroperitoneum |
|
PGL |
7 |
No |
No |
2 |
F |
56 |
M |
intraabdominal |
LN |
PGL |
8 |
No |
No |
3 |
M |
54 |
B |
adrenal gland |
|
PCC |
11 |
No |
No |
4 |
M |
51 |
B |
retroperitoneum |
|
PGL |
6 |
No |
No |
5 |
M |
52 |
M |
retroperitoneum |
bone |
PGL |
5.5 |
Yes |
No |
6 |
M |
67 |
B |
abdomen |
|
PGL |
7 |
No |
No |
7 |
F |
41 |
B |
retrocaval |
|
PGL |
5 |
No |
Yes |
8 |
M |
58 |
M |
bladder |
bone, rectum |
PGL |
6.5 |
No |
Yes |
9 |
F |
53 |
B |
adrenal gland |
|
PCC |
9.5 |
No |
No |
10 |
M |
70 |
B |
retroperitoneum |
|
PGL |
7 |
No |
No |
11 |
F |
40 |
B |
retroperitoneum |
|
PGL |
7.5 |
No |
No |
12 |
M |
47 |
M |
retroperitoneum |
LN |
PGL |
6 |
No |
No |
M, Male; F, Female; B, Benign; M, Malignant; PGL, paraganglioma; PCC, pheochromocytoma.
Table 2: Clinicopathologic demographics of patients with benign versus malignant PCC/PGL
Characteristics |
|
Benign |
Malignant |
p-value |
|---|---|---|---|---|
Sex |
Male |
23 (59.0%) |
12 (52.2%) |
0.822 |
|
Female |
17 (42.5%) |
10 (45.5%) |
|
|
|
|
|
|
Age |
≤ 20 |
2 (5.0%) |
0 (0.0%) |
0.535 |
|
> 20 |
38 (95.0%) |
22 (100.0%) |
|
|
|
|
|
|
Pathology |
Pheochromocytoma |
19 (47.5%) |
12 (54.5%) |
0.596 |
|
Paraganglioma |
21 (52.5%) |
10 (45.5%) |
|
|
|
|
|
|
PASS |
median (range) |
2 (0–7) |
5 (0–10) |
< 0.001* |
|
|
|
|
|
Tumor size (cm) |
median (range) |
5.0 (1.6–14.0) |
6.8 (2.0–19.0) |
0.039* |
|
|
|
|
|
F/U duration (month) |
median (range) |
62.5 (0–170) |
55.0 (5–254) |
0.125* |
|
|
|
|
|
Recurrent rate |
|
1/40 (2.5%) |
14/22 (63.6%) |
< 0.01 |
|
|
|
|
|
Death rate |
|
0/40 (0.0%) |
4/22 (18.2%) |
0.013 |
PASS, pheochromocytoma of the adrenal gland scaled score; *Mann-Whitney test.
Genomic copy number alterations in benign and malignant PCC/PGL
We did not observe any noteworthy focal amplifications or deletions via aCGH, and most samples showed few copy number aberrations regardless of malignancy status. Two regions, 1p and 3q, showed relatively frequent heterozygous loss in five and two cases, respectively (log ratio ≈ –0.5) (Figure 1). This indicates that copy number alteration is unlikely to be involved in PCC/PGL carcinogenesis, and other factors such as somatic mutations and gene fusions should be investigated to find relevant driver alterations. Additionally, there was no significant difference in genomic architecture between the malignant and benign samples.
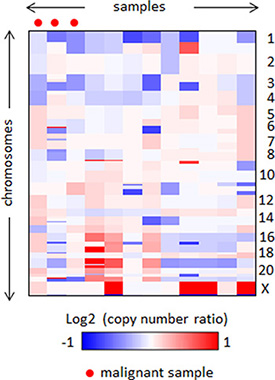
Figure 1: Heatmap of genomic profiles of the segmented copy number data.
PNMT as a candidate marker for malignant PCC/PGL
To identify genes differentially expressed between benign and malignant tumors, we compared the mRNA expression profiles of three malignant and nine benign PCC/PGL specimens. Two hundred genes were overexpressed > 5-fold in malignant tumors. Upregulated genes were involved in either nervous system development (MNX1, NRG1, PITX2, PAX2, FOXA1, POU3F2, TWIST1 and NEUROG2) or synaptic transmission (BDNF, CACNB2, GAD1, NRG1 and DRD4) (Table S1). One hundred ninety genes were downregulated > 5 fold in the same tumors and were not associated with specific biological processes (Table S2). From the 390 differentially expressed genes, we selected phenylethanolamine N-methyltransferase (PNMT), an enzyme involved in the catecholamine biosynthesis pathway, as a candidate malignancy marker. PNMT presented the highest fold difference (benign/malignant fold change of ~160).
Functional analysis of PNMT in PCC/PGL
To better characterize the function of PNMT in PCC/PGL, we analyzed a large (125 samples) public PCC/PGL microarray expression profile dataset (GSE19987) with a variety of mutations in pheochromocytoma susceptibility genes such as RET, VHL, SDHB and SDHD [22]. We performed functional analyses of genes differentially expressed in PNMT-high and -low groups. PNMT-high and -low groups were identified by their distinct normal distributions for PNMT expression in the dataset (Figure 2). About 200 highly upregulated genes [false discovery rate (FDR) < 0.0001] were identified in PNMT-low samples, including TGFBI, VEGFA, CD34, CDH13, EFNA1, EPHB4, MFGE8, NOTCH1 and PGF. Gene Ontology (GO) analysis revealed overrepresentation of gene functions related to blood vessel development, morphogenesis and angiogenesis (Figure 3). We also identified approximately 650 overexpressed genes (FDR < 0.0001) in the PNMT-high group, which were mainly involved in synaptic transmission (GATA3, NTGK1, TFAP2B and PHOX2A), catecholamine metabolism (PNMT, TH, GATA3, HPRT1 and MAOB) and Golgi vesicle transport (VAMP2, CLTC, SCAMP1 and SCFD1)—all functions related to adrenalin hormone synthesis and secretion. These results suggest that low PNMT expression is related to aggressive PCC/PGL tumor development, and is supported by the fact that angiogenesis-related genes are upregulated in low PNMT-expressing tumors.
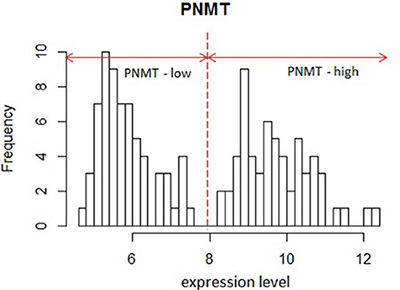
Figure 2: Histogram of PNMT expression in GSE19987. Two distinct distributions are shown.
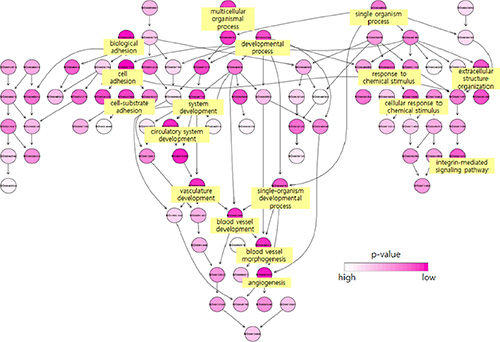
Figure 3: Biological functions of genes overexpressed in the PNMT-low group. Overrepresentation of blood vessel development-related functions was observed upon analysis of the 200 highly upregulated genes in the PNMT-low group. Each circle represents a selected enriched gene ontology (GO) term, and darker color represents increased statistical significance.
Correlation of PNMT with hereditary PCC/PGL
In an analysis of the Pearson correlations between PNMT and all other genes in the microarray platform, the RET proto-oncogene showed the highest correlation with PNMT levels (0.91 and 0.77 in our dataset and GSE19987, respectively; Figure 4). RET is a well-known PCC/PGL susceptibility gene whose germ-line mutations are associated with hereditary disease. However, the correlation between PNMT and RET in this study was independent of RET mutation status (Figure 4A). Hereditary tumors harboring RET mutations overexpressed PNMT, while those harboring either SDH or VHL mutations downregulated PNMT (Figure 4B). This supports a previous study wherein unsupervised hierarchical cluster analysis of gene expression profiles of approximately 200 PCC/PGL samples separated hereditary tumors into two groups: RET/NF1- and SDH/VHL-related, with adrenergic and noradrenergic phenotypes, respectively [23]. The adrenergic/noradrenergic characteristics of PCC/PGL with RET, SDH, or VHL mutations largely reflect their origins from two types of chromaffin cells that can be distinguished based on PNMT expression. This provides a new platform to explore the pathogenic development of hereditary tumors from two different chromaffin cell populations.
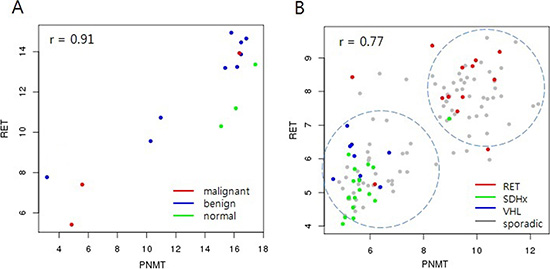
Figure 4: Correlation between PNMT and RET. RET showed the highest correlation (r = 0.91) with PNMT in our data (A) Malignant tumors showed low expression of both PNMT and RET. RET showed the highest correlation (r = 0.77) with PNMT in the GSE19987 dataset, which includes hereditary PCC/PGL (B) Tumors harboring RET germline mutations showed high PNMT and RET expression, while SDHx/VHL germline-mutated tumors showed low expression.
Validation of gene expression by quantitative IHC and qRT-PCR
IHC analyses revealed PNMT staining predominantly within the cytoplasm, and the percentage of PNMT-positive cells was variable. PNMT staining was quantified by calculating the percentage of positively stained tumor cells, and was scored accordingly (Table 3). The median percentage of positive cells was 20% (range, 0–100) in benign PCC/PGL samples compared with 3% (range, 0–80) in malignant samples (P = 0.038; Figure 5A). In benign PCC/PGL, 46.2% of the samples presented > 50% positive cells, while 20.8% of the malignant samples stained > 50% for PNMT (P = 0.031, Figure 6). PNMT mRNA expression was successfully quantified by qRT-PCR in 52 of the 62 FFPE and 4 normal adrenal gland samples. The remaining cases failed to yield reliable qualities and/or quantities of RNA owing to the small size of tumor sections. We observed variable PNMT expression in normal adrenal gland tissues and benign and malignant PCC/PGL, ranging from 9.995–1610.673, 0.005–447.70 and 0.006–396.05, respectively, with median values of 48.3365, 8.55 and 3.44, respectively. PNMT expression was lower in benign than in malignant PCC/PGL, although this was not statistically significant (P = 0.069). PNMT mRNA and protein levels were correlated in PCC/PGL samples (r = 0.444, P = 0.001; Figure 5B) as well as in PCC (P = 0.076) and PGL samples (P = 0.133).
Table 3: PNMT immunohistochemical analysis in benign and malignant PCC/PGL samples were scored based on percentage of positive-staining cells
|
Benign (n = 39a) |
Malignant (n = 22) |
|
|---|---|---|---|
PNMT |
|
|
P-value |
0 |
12 (30.8%) |
9 (40.9%) |
0.031b |
1+ |
2 (5.1%) |
7 (31.8%) |
|
2+ |
7 (17.9%) |
2 (9.1%) |
|
3+ |
18 (46.2%) |
4 (18.2%) |
|
a The single remaining benign PCC/PGL case was not stained due to unavailability of a FFPE sample.
b Cochran-Armitage trend test.
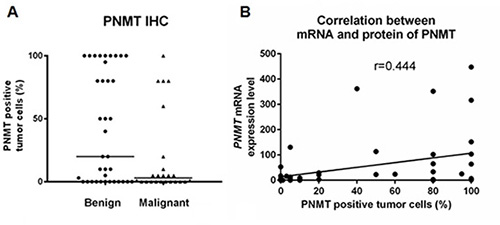
Figure 5: Comparison of benign and malignant PCC/PGL. Comparative quantification of PNMT protein levels by immunohistochemistry (IHC) (A) Correlation between PNMT mRNA and protein levels (B).
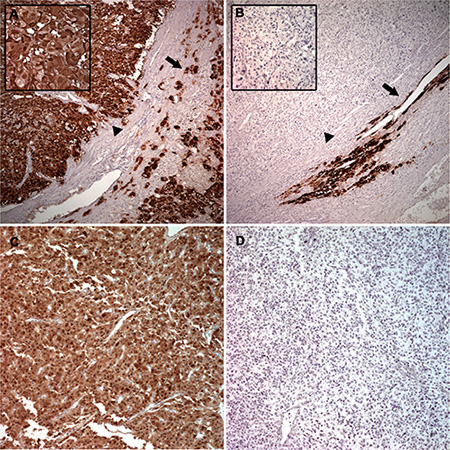
Figure 6: Representative PNMT protein levels in benign and malignant PCC and PGL. Benign PCC (arrowhead) showed the same immunostaining intensity as the adjacent normal adrenal medulla (arrow) (A) Strong cytoplasmic staining was observed (left upper inset). Malignant PCC (arrowhead) exhibited complete absence of PNMT compared with the adjacent normal adrenal medulla (arrow) (B) Left upper inset represents higher magnification. Benign PGL showed diffuse and strong PNMT staining (C) Malignant PGL showed complete absence of PNMT positivity (D).
Correlation between PASS and PNMT immunoexpression
A trend towards an inverse relationship between PASS and PNMT immunoexpression was observed, although this was not statistically significant (r = –0.140, P = 0.282; Figure 7). The case of patient number 7 was particularly interesting. Briefly, a 27-year old woman presented with hypertension and palpitation in March 1997. She had no family history of hypertension or any other known genetic disorder. After workup, she was diagnosed with extra-adrenal PCC in the right para-aortic area, which was treated by excision. In 2011, 14 years after the initial surgical excision, she once again presented with hypertension that lasted for three months. An abdominal and pelvis computerized tomography (CT) scan detected a 4.7 × 3.2 cm well-defined solid mass in the retrocaval space. The mass was excised and pathologically confirmed as recurrent extra-adrenal PCC. The initial PASS for this patient was < 4, suggesting a benign tumor. The PASS was 4 in the recurrent tumor, suggesting a potentially aggressive tumor. Although this patient relapsed, the recurrence site was the retroperitoneum, which did not meet criteria for malignancy. Therefore, this case was classified as benign for gene expression profiling in the current study. However, the gene expression profile of this patient was similar to those of malignant tumors. In addition, IHC revealed complete absence of PNMT protein expression in the primary and recurrent tumors (Figure 8), while qRT-PCR revealed low PNMT mRNA level, similar to malignant PCC/PGL. This finding suggests that the tumor might have a malignant genotype/phenotype and indicates limitations in the clinical and histopathological diagnosis of malignant PCC/PGL.
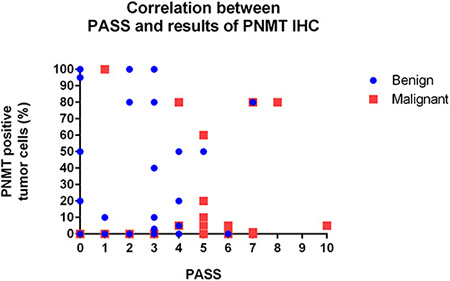
Figure 7: Correlation between PASS and PNMT protein levels.
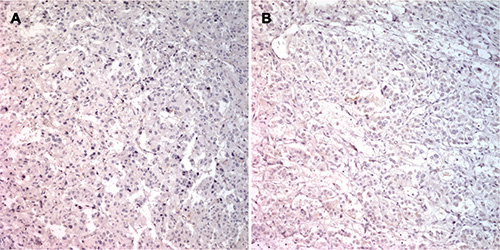
Figure 8: PNMT immunostaining in specimens from patient 7. Primary extra-adrenal pheochromocytoma in the right para-aortic area with complete absence of PNMT positivity (A) Recurrent extra-adrenal pheochromocytoma in the retrocaval space with complete absence of PNMT positivity (B).
DISCUSSION
In this study, we performed aCGH and gene expression profiling to identify candidate marker genes that might distinguish benign from malignant PCC/PGL. Microarray analysis revealed differential expression of 390 genes. We focused on PNMT for further investigation since it exhibited the highest fold change between benign and malignant samples. The PNMT enzyme plays a key role in adrenal medulla function, and its expression was downregulated in malignant PCC/PGL compared to benign tumors. We also performed functional analysis of genes that were differentially expressed in PNMT-high and -low expressing tumors. Approximately 200 highly upregulated genes (FDR < 0.0001) were identified in the PNMT-low samples. GO analysis revealed that these genes were commonly involved in angiogenesis.
The human PNMT gene is located on chromosome 17q21-q22 [24]. PNMT catalyzes the final step in the catecholamine biosynthesis pathway, in which norepinephrine is methylated to epinephrine [25, 26]. Although it is predominantly found in the chromaffin cells of the adrenal medulla, where a large proportion of the body’s adrenaline is synthesized, PNMT activity has also been reported in various organs such as the heart [27, 28], kidney [29], lung, thymus and different parts of the brain [30]. PNMT gene expression was detected in the majority of these tissues [31–35].
There are few published studies on PNMT expression in PCC/PGL. Isobe, et al. [36] reported higher PNMT mRNA levels in normal adrenal medulla than in PCCs, which might explain why sporadic PCCs predominantly produce noradrenaline. The PNMT promoter contains glucocorticoid responsive elements (GREs) and PNMT transcription is modulated by glucocorticoids [37]. Therefore, elevated PNMT mRNA levels in normal adrenal medulla may be induced by stimulated glucocorticoid release from the adrenal cortex [38]. Low PNMT mRNA levels in PCCs might reflect an insufficient concentration of plasma cortisol in the artery supplying the tumor [36]. While it is generally believed that PNMT is absent in PGL, some investigators observed positive immunohistochemical PNMT staining in these tumors [39–41]. In our study, four cases of PGL in the head and neck showed loss of PNMT immunoexpression and extremely low PNMT mRNA levels. However, we did not observe any difference in PNMT protein and mRNA levels between PCC and PGL in our validation studies.
Eisenhofer, et al. reported that PCCs associated with multiple endocrine neoplasia type 2 (MEN 2) are adrenergic, whereas those associated with von Hippel-Lindau syndrome (VHL) are noradrenergic due to lower PNMT expression [42]. PNMT protein levels are reported to be significantly different between spontaneously occurring benign and locally invasive “malignant” PCC in mice [43]. Loss of PNMT regulatory mechanisms is characteristic of aggressive PCCs and is associated with a less mature phenotype, because PNMT is expressed after other catecholamine-synthesizing enzymes during embryogenesis [43]. This is consistent with our finding that over 80% of differentially expressed genes were downregulated in malignant tumors, and supports the hypothesis that malignant potential is related to a less differentiated expression pattern [43].
Although the association between PNMT and malignant PCC/PGL in humans was not previously reported, efforts have been made to find biochemical markers to predict malignancy [44]. Van der Hast, et al. reported that high dopamine, high norepinephrine and a high ratio of epinephrine to total catecholamine are associated with decreased metastasis-free survival [45]. Eisenhofer, et al. found that plasma methoxytyramine, the O-methylated metabolite of dopamine, was correlated with malignancy [46]. In our study, PNMT was downregulated in malignant PCC/PGL and PNMT protein expression differed between benign and malignant PCC/PGL. Although we did not evaluate plasma epinephrine or norepinephrine, our findings suggest that serum epinephrine is likely less abundant in patients with malignant PCC/PGL. Norepinephrine-producing tumors lacking PNMT are less differentiated than epinephrine-producing tumors. Accordingly, Kimura, et al. recently proposed a grading system for adrenal PCC/PGL (GAPP) that considers a tumor’s biochemical phenotype and type of catecholamine produced when assessing metastasis risk [47].
Recently, PCC/PGL was divided into two main clusters based on tumor transcription profiles [48]. Cluster 1 was associated with hypoxia-related signals; cluster 2 with increased kinase signaling. Typically, patients with cluster 1 tumors present dopaminergic, noradrenergic, or a mix of dopaminergic and noradrenergic biochemical profiles. Those with cluster 2 tumors present either adrenergic or a mix of noradrenergic and adrenergic biochemical phenotypes. Tumors with noradrenergic profiles have an immature secretory phenotype, whereas those with either adrenergic or mixed noradrenergic/adrenergic profiles have a mature secretory phenotype. Metastasis risk is inversely correlated with maturity profile [48]. Thus, low PNMT expression and lack of epinephrine production might be associated with malignancy.
Use of PNMT as a diagnostic marker for malignancy could aid in the clinical management of PCC/PGL. IHC analysis of protein expression in FFPE samples is an effective and routine clinical diagnostic tool. Patients with clinically and histopathologically benign tumors at higher risk of malignancy based on low PNMT expression would benefit from frequent follow-ups, along with regular biochemical testing and imaging, to detect recurrent or persistent tumors. [48]. In light of the current practices, low PNMT expression with relation to catecholamine type might be associated with malignant behavior.
In conclusion, by combining aCGH with expression array analysis and validation studies, we identified PNMT as a candidate gene to distinguish malignant from benign PCC/PGL. This is the first demonstration of the association between PNMT downregulation and PCC/PGL malignancy in humans, and our findings identify PNMT as one of the most differentially expressed genes between malignant and benign tumors.
MATERIALS AND METHODS
Sample collection and study design
Fresh, frozen samples were obtained from 12 PCC/PGL patients according to a protocol approved by the Institutional Review Board between 2007 and 2013. Two pathologists studied hematoxylin/eosin-stained slides prepared from all frozen samples and confirmed that all samples contained tumor areas > 80%. All 12 samples were included in aCGH and expression microarray analyses. Normal adrenal gland medullas from three patients who underwent surgery for renal cell carcinoma were used as normal controls for expression microarray analysis.
To validate the results of altered gene expression and copy numbers analyses, 62 additional PCC/PGL FFPE samples were analyzed, including 19 malignant and 31 benign tumors. Eligibility criteria for including patients in the study were as follows: histological confirmation of PCC/PGL, availability of sufficient tissue for biomarker tests, and complete clinical and outcome information.
Microarray analysis
Molecular profiling of the tumors was done using aCGH and expression microarray (Agilent Technologies, 60 K) following the manufacturer’s protocol. Following segmentation using the GLAD algorithm, significantly amplified or deleted regions were identified by GISTIC (parameters: -ta 0.5 –td -0.5 –qvt 0.01). Genes differentially expressed in benign and malignant samples were selected based on their fold changes. Unsupervised hierarchical clustering was performed with a correlation metric to analyze similarity in expression among the 15 samples (12 pheochromocytoma, 3 normal adrenal medulla). The 390 genes showing the highest variance [interquartile range (IQR) > 4] across the 15 samples were selected for hierarchical clustering. To characterize PNMT, we analyzed a large public dataset (GSE19987; http://www.ncbi.nlm.nih.gov/gds/) that consisted of 125 pheochromocytomas with RET, VHL, and SDHx germline mutation data. PNMT-high and -low groups were defined according to the distribution of PNMT levels in the dataset. We used SAM (Significance Analysis of Microarrays) to identify genes that were differentially expressed between PNMT-high and -low groups. Biological functions related to PNMT were investigated by GO analysis using the R package GOstats (http://bioconductor.org/).
RT-PCR analysis of PNMT
RNA was isolated from FFPE tumor samples using the RNeasy FFPE RNA Isolation Kit (Qiagen, Valencia, CA), and cDNA synthesis was performed using the SuperScript III first-strand kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturers’ instructions. The PCR primers and probes are described in Table S3. HPRT1 and GUSB were used as endogenous control genes. RT-PCR analysis was performed with an ABI 7900HT Fast Real-time PCR system (Applied Biosystems, Foster City, CA, USA) using SYBR Green. Briefly, 1 μL of cDNA product was used as template in a 10 μL PCR reaction containing 5 μL of Power SYBR Green PCR master mix (Applied Biosystems) and 200 nM of each primer. All reactions were performed in triplicates. The amplification protocol was as follows: 95°C for 10 min; 40 cycles of 95°C for 10 s and 60°C for 60 s; and one cycle for melting curve analysis. The relative expression of PNMT compared with that of the reference genes was calculated using ΔCt (cycle threshold).
IHC for PNMT
Tissues were fixed in 10% formalin solution, dehydrated through a graded ethanol series, cleared in xylene, and processed for embedding in paraffin wax according to routine protocol. The sections were placed in a 0.3% H2O2 solution for 15 min to inhibit endogenous peroxidase activity. They were then incubated with anti-PNMT primary antibody (1:400; ab119784, Abcam, Cambridge, UK) for 1 h at room temperature. The EnVision+ detection system for mouse antibodies (K4001, DAKO, Glostrup, Denmark) was used according to the manufacturer’s instructions. Slides were then stained with liquid diaminobenzidine tetrahydrochloride (DAB+) using a high-sensitivity substrate-chromogen system (K3468, DAKO), and counterstained with Meyer’s hematoxylin. Images were visualized with an Olympus BX40 light microscope (Olympus, Tokyo, Japan). Cytoplasmic staining was considered PNMT-positive, expressed as the percentage of positively stained tumor cells. The following criteria were used to score samples based on percent positive cells: 0, < 1%; 1, 1%–9%; 2, 10%–49%; and 3, > 50%.
Statistical analyses
Either the χ2 or Fisher’s exact test was used to examine associations between benign and malignant PCC/PGLs and their clinicopathological parameters. Differences in median PASS score, tumor size and follow-up duration between benign and malignant PCC/PGL were evaluated by the Mann-Whitney non-parametric test. PNMT mRNA level differences among normal adrenal tissue, benign PCC/PGL and malignant PCC/PGL was evaluated by the Kruskal-Wallis non-parametric test. Correlations between the percentage of PNMT protein immunopositive cells and PNMT mRNA level were investigated using the Spearman’s rank correlation test. Correlations were evaluated using the Spearman’s rank correlation coefficient. We used the Cochran-Armitage test to evaluate the PNMT immunoreactive score trend in benign and malignant PCC/PGL.
GRANT SUPPORT
This study was supported by the R & D Program of the Society of the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2013M3C8A1078501) and a grant (SBRI C-B1–133–1 and SMX1132711) from the Samsung Biomedical Research Institute, Seoul, Korea.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
REFERENCES
1. DeLellis RA, Lloyd RV, U. Heitz P, Eng C. The WHO Classification of Tumours of Endocrine Organs. 69008 Lyon, France: International Agency for Research on Cancer (IARC); 2004.
2. Choi SE, Kim YC, Kim TS, Noh DY, Youn YK, Choe KJ, Oh SK. Clinical Analysis of Malignant Pheochromocytoma. Cancer Res Treat. 1999; 31:1307–1314.
3. Pheochromocytoma. J Pathol Transl Med. 1982; 16:790–795.
4. Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002; 26:551–566.
5. Wu D, Tischler AS, Lloyd RV, DeLellis RA, de Krijger R, van Nederveen F, Nose V. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol. 2009; 33:599–608.
6. Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, Brennan M, Ghossein RA. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery. 2008; 143:759–768.
7. Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001; 69:49–54.
8. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X, Network C. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003; 63:5615–5621.
9. Nativ O, Grant CS, Sheps SG, O’Fallon JR, Farrow GM, van Heerden JA, Lieber MM. The clinical significance of nuclear DNA ploidy pattern in 184 patients with pheochromocytoma. Cancer. 1992; 69:2683–2687.
10. Dannenberg H, Speel EJ, Zhao J, Saremaslani P, van Der Harst E, Roth J, Heitz PU, Bonjer HJ, Dinjens WN, Mooi WJ, Komminoth P, de Krijger RR. Losses of chromosomes 1p and 3q are early genetic events in the development of sporadic pheochromocytomas. Am J Pathol. 2000; 157:353–359.
11. Edstrom E, Mahlamaki E, Nord B, Kjellman M, Karhu R, Hoog A, Goncharov N, Teh BT, Backdahl M, Larsson C. Comparative genomic hybridization reveals frequent losses of chromosomes 1p and 3q in pheochromocytomas and abdominal paragangliomas, suggesting a common genetic etiology. Am J Pathol. 2000; 156:651–659.
12. Brouwers FM, Elkahloun AG, Munson PJ, Eisenhofer G, Barb J, Linehan WM, Lenders JW, De Krijger R, Mannelli M, Udelsman R, Ocal IT, Shulkin BL, Bornstein SR, et al. Gene expression profiling of benign and malignant pheochromocytoma. Ann N Y Acad Sci. 2006; 1073:541–556.
13. Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, Hodin R, Heitritter S, Moore F, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005; 1: 72–80.
14. Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, Giordano TJ, Greene LA, Goldstein DS, Lehnert H, Manger WM, Maris JM, Neumann HP, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer. 2004; 11:423–436.
15. Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM, Munson PJ, Mannelli M, Goldstein DS, Elkahloun AG. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer. 2004; 11:897–911.
16. Thouennon E, Elkahloun AG, Guillemot J, Gimenez-Roqueplo AP, Bertherat J, Pierre A, Ghzili H, Grumolato L, Muresan M, Klein M, Lefebvre H, Ouafik L, Vaudry H, et al. Identification of potential gene markers and insights into the pathophysiology of pheochromocytoma malignancy. J Clin Endocrinol Metab. 2007; 92:4865–4872.
17. Waldmann J, Fendrich V, Holler J, Buchholz M, Heinmoller E, Langer P, Ramaswamy A, Samans B, Walz MK, Rothmund M, Bartsch DK, Slater EP. Microarray analysis reveals differential expression of benign and malignant pheochromocytoma. Endocr Relat Cancer. 2010; 17: 743–756.
18. Boltze C, Lehnert H, Schneider-Stock R, Peters B, Hoang-Vu C, Roessner A. HSP90 is a key for telomerase activation and malignant transition in pheochromocytoma. Endocrine. 2003; 22:193–201.
19. Elder EE, Xu D, Hoog A, Enberg U, Hou M, Pisa P, Gruber A, Larsson C, Backdahl M. KI-67 AND hTERT expression can aid in the distinction between malignant and benign pheochromocytoma and paraganglioma. Mod Pathol. 2003; 16:246–255.
20. Salmenkivi K, Haglund C, Arola J, Heikkila P. Increased expression of tenascin in pheochromocytomas correlates with malignancy. Am J Surg Pathol. 2001; 25:1419–1423.
21. Khorram-Manesh A, Ahlman H, Jansson S, Nilsson O. N-cadherin expression in adrenal tumors: upregulation in malignant pheochromocytoma and downregulation in adrenocortical carcinoma. Endocr Pathol. 2002; 13:99–110.
22. Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010; 42:229–233.
23. Burnichon N, Vescovo L, Amar L, Libe R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, Plouin PF, Jeunemaitre X, Favier J, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011; 20:3974–3985.
24. Hoehe MR, Plaetke R, Otterud B, Stauffer D, Holik J, Byerley WF, Baetge EE, Gershon ES, Lalouel JM, Leppert M. Genetic linkage of the human gene for phenylethanolamine N-methyltransferase (PNMT), the adrenaline-synthesizing enzyme, to DNA markers on chromosome 17q21-q22. Hum Mol Genet. 1992; 1:175–178.
25. Axelrod J. Purification and properties of phenylethanolamine-N-methyl transferase. J Biol Chem. 1962; 237:1657–1660.
26. Kirshner N, Goodall M. The formation of adrenaline from noradrenaline. Biochim Biophys Acta. 1957; 24:658–659.
27. Ebert SN, Baden JM, Mathers LH, Siddall BJ, Wong DL. Expression of phenylethanolamine n-methyltransferase in the embryonic rat heart. J Mol Cell Cardiol. 1996; 28: 1653–1658.
28. Kennedy B, Ziegler MG. Ontogeny of epinephrine metabolic pathways in the rat: role of glucocorticoids. Int J Dev Neurosci. 2000; 18:53–59.
29. Kennedy B, Bigby TD, Ziegler MG. Nonadrenal epinephrine-forming enzymes in humans. Characteristics, distribution, regulation, and relationship to epinephrine levels. J Clin Invest. 1995; 95:2896–2902.
30. Pendleton RG, Gessner G, Sawyer J. Studies on the distribution of phenylethanolamine N-methyltransferase and epinephrine in the rat. Res Commun Chem Pathol Pharmacol. 1978; 21:315–325.
31. Andreassi JL, 2nd, Eggleston WB, Stewart JK. Phenylethanolamine N-methyltransferase mRNA in rat spleen and thymus. Neurosci Lett. 1998; 241:75–78.
32. Jelokova J, Rusnak M, Kubovcakova L, Buckendahl P, Krizanova O, Sabban EL, Kvetnansky R. Stress increases gene expression of phenylethanolamine N-methyltransferase in spleen of rats via pituitary-adrenocortical mechanism. Psychoneuroendocrinology. 2002; 27:619–633.
33. Krizanova O, Micutkova L, Jelokova J, Filipenko M, Sabban E, Kvetnansky R. Existence of cardiac PNMT mRNA in adult rats: elevation by stress in a glucocorticoid-dependent manner. Am J Physiol Heart Circ Physiol. 2001; 281: H1372–1379.
34. Kubovcakova L, Micutkova L, Bartosova Z, Sabban EL, Krizanova O, Kvetnansky R. Identification of phenylethanolamine N-methyltransferase gene expression in stellate ganglia and its modulation by stress. J Neurochem. 2006; 97:1419–1430.
35. Warthan MD, Freeman JG, Loesser KE, Lewis CW, Hong M, Conway CM, Stewart JK. Phenylethanolamine N-methyl transferase expression in mouse thymus and spleen. Brain Behav Immun. 2002; 16:493–499.
36. Isobe K, Nakai T, Yukimasa N, Nanmoku T, Takekoshi K, Nomura F. Expression of mRNA coding for four catecholamine-synthesizing enzymes in human adrenal pheochromocytomas. Eur J Endocrinol. 1998; 138:383–387.
37. Sabban EL, Kvetnansky R. Stress-triggered activation of gene expression in catecholaminergic systems: dynamics of transcriptional events. Trends Neurosci. 2001; 24:91–98.
38. Betito K, Diorio J, Meaney MJ, Boksa P. Adrenal phenylethanolamine N-methyltransferase induction in relation to glucocorticoid receptor dynamics: evidence that acute exposure to high cortisol levels is sufficient to induce the enzyme. J Neurochem. 1992; 58:1853–1862.
39. Funahashi H, Imai T, Tanaka Y, Tobinaga J, Wada M, Matsuyama T, Tsukamura K, Yamada F, Takagi H, Narita T, et al. Discrepancy between PNMT presence and relative lack of adrenaline production in extra-adrenal pheochromocytoma. J Surg Oncol. 1994; 57:196–200.
40. Lloyd RV, Sisson JC, Shapiro B, Verhofstad AA. Immunohistochemical localization of epinephrine, norepinephrine, catecholamine-synthesizing enzymes, and chromogranin in neuroendocrine cells and tumors. Am J Pathol. 1986; 125:45–54.
41. Partanen M, Rapoport SI, Reis DJ, Joh TH, Stolk JM, Linnoila I, Teitelman G, Hervonen A. Catecholamine-synthesizing enzymes in paraganglia of aged Fischer-344 rats. Immunohistochemistry and fluorescence microscopy. Cell Tissue Res. 1984; 238:217–220.
42. Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001; 86:1999–2008.
43. Hill GD, Pace V, Persohn E, Bresser C, Haseman JK, Tischler AS, Nyska A. A comparative immunohistochemical study of spontaneous and chemically induced pheochromocytomas in B6C3F1 mice. Endocr Pathol. 2003; 14:81–91.
44. Korevaar TI, Grossman AB. Pheochromocytomas and paragangliomas: assessment of malignant potential. Endocrine. 2011; 40:354–365.
45. van der Harst E, de Herder WW, de Krijger RR, Bruining HA, Bonjer HJ, Lamberts SW, van den Meiracker AH, Stijnen TH, Boomsma F. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur J Endocrinol. 2002; 147:85–94.
46. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, Mannelli M, Linehan WM, Adams K, Timmers HJ, Pacak K. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012; 48:1739–1749.
47. Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, Rakugi H, Ikeda Y, Tanabe A, Nigawara T, Ito S, Kimura I, Naruse M, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2014; 21:405–414.
48. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014; 14:108–119.