
INTRODUCTION
TOPK (T-LAK cell-originated protein kinase) is initially identified as a MAPKK-like protein kinase from lymphokine-activated killer T cell and it is also known as PBK (PDZ-binding kinase) linking with the PDZ2domain of tumor suppressor protein hDlg [1, 2]. TOPK is highly expressed in a variety of tumors including breast cancer, colorectal cancer (CRC) and melanoma, and involves in regulating several cell functions such as tumor cell cycle progression [3–6], transformation [7, 8], proliferation [9] and apoptosis [10, 11]. More and more published reports have suggested that elevated levels of TOPK may be associated with the tumorigenesis, metastasis and poor prognosis of cancers [12–17].
Colon cancer is one of the most prevalent cancers worldwide, as well as one of the leading causes of cancer death. American Cancer Society reported that 1 658 370 new cases of cancer would be diagnosed in the United States in 2015 and about 8% cases would be CRC [18]. In previous study, we found that there existed a positive feedback loop between TOPK and ERK2 (extracellular signal-regulated kinase 2), and therefore promoted tumorigenesis in colon cancer in vitro and in vivo [7]. Moreover, TOPK is a valuable prognostic marker in patients with sporadic CRC, and 30-40% of CRC patients may benefit from the inhibition of TOPK [17]. Deschoolmeester et al also reported that TOPK may be a biomarker in prognosis and a therapeutic target in CRC [19]. TOPK is very important in colon cancer. However, only a few kinases including hDlg [2], Cdk1 [2, 3], ERK2 and p38 [7] are known to phosphorylate TOPK at Thr9, and the mechanism of phosphorylation of TOPK at Thr9 have not yet been identified. Therefore, to find out new upstream kinases and other phosphorylation sites would make the progress of clinical application in TOPK field.
Src is the first transforming protein with tyrosine kinase activity discovered and isolated [20]. It can activate multiple signaling pathways, including the PI3K/Akt, MAPK, Stat3, IL-8, VEGF, and cytoskeletal-formation pathways to regulate cellular functions [21]. Src activity increases in 80% of colon cancer patients [22], and the activation of Src can stimulate Ras-Raf-MEK-ERK1/2 pathway, and in turn, promotes carcinogenesis [23, 24]. After being screened from 45 patients with colorectal carcinoma, Src activity is considered as an independent indicator of poor clinical prognosis in all stages of human colon carcinoma [25-27].
Both Src and TOPK are very important in colon cancer, and furthermore, there exist the Src consensus substrate motif, pY[A/G/S/T/E/D] in TOPK. Therefore, we hypothesize whether Src could phosphorylate TOPK directly in colon cancer. In this study, we found that Src phosphorylated TOPK directly in vitro and in vivo, increased the stability and activity of TOPK. The interaction between Src and TOPK promoted carcinogenesis in colon cancer. Thus, these findings may help to predict prognosis or develop new therapeutic strategies to colon cancer.
RESULTS
Src phosphorylates TOPK at Y74 and Y272 in vitro
Both TOPK and Src play an important role in carcinogenesis and clinical prognosis in CRC, and there is a consensus phosphorylation site (Y272) of Src in TOPK. Therefore, we hypothesized that Src could phosphorylate TOPK. To test this idea, an in vitro kinase assay was performed in the presence of [γ-32P] ATP with Src as an active kinase and TOPK as a substrate. The data indicated that Src could phosphorylate TOPK in vitro (Figure 1A). The potential tyrosine phosphorylation sites of TOPK were predicted by NetPhos 2.0 (Figure 1B) [28]. Five high-score peptides were then designed and synthesized commercially (Y1-Y5) (PEPTIDE 2.0, Houston, TX, USA). The peptides were individually incubated with active Src in the presence of [γ-32P] ATP in an in vitro kinase assay. The results showed that both Y74 and Y272 were phosphorylated by Src, and the phosphorylation signal was even stronger at the site of Y74 (Figure 1C: Y1, Y4). To further confirm the results from peptide mapping, the antibodies recognizing phosphorylatedTOPK (phospho-TOPK (Y74) (p-TOPK (Y74)) or phospho-TOPK (Y272) (p-TOPK (Y272)) were prepared as described in Materials and Methods, but the p-TOPK (Y272) antibody failed to detect phospho-TOPK. The wild type (His-TOPK) (WT), Y74F TOPK (74F), Y272F TOPK (272F) and Y74Y272FF TOPK (His-TOPK) (FF) were purified from E coli respectively, and used as substrates for active Src in an in vitro kinase assay. The results of Western Blot using prepared p-TOPK (Y74) antibody showed that Src could phosphorylate TOPK at Y74, and the signal disappeared in the single mutant TOPK-74F and the double mutant TOPK-FF (Figure 1D). These data suggested that Src did phosphorylate TOPK at Y74 in vitro.
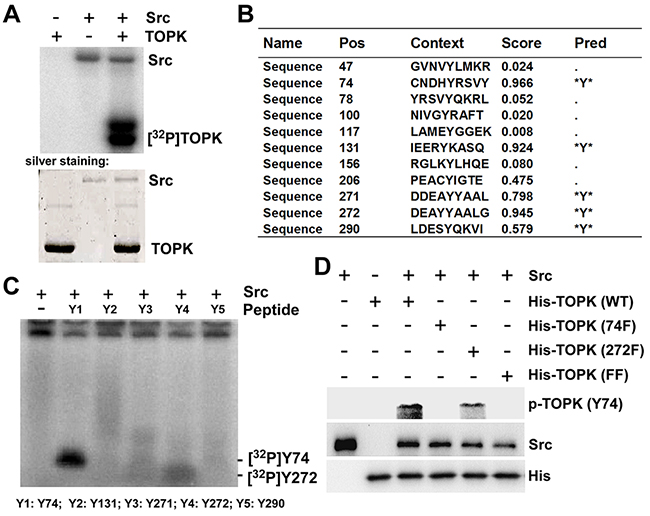
Figure 1: Src phosphorylates TOPK at Y74 and Y272 in vitro. A. Active Src phosphorylated inactive TOPK in vitro in the presence of [γ-32P] ATP as visualized by autoradiograph. B. Potential phosphorylated tyrosine sites of TOPK were predicted by NetPhos 2.0 software program. C. Src phosphorylated TOPK at Y74 or Y272 in peptide mapping. Five synthesized peptides containing potential tyrosine sites were used as substrates in an in vitro kinase assay with active Src in the presence of [γ-32P] ATP and the results were visualized by autoradiography. D. Validation of anti phospho-TOPK (Y74) (p-TOPK (Y74)) in an in vitro kinase assay. Wild type His-TOPK (WT), single mutant His-TOPK (74F), single mutant His-TOPK (272F) or double mutant His-TOPK (FF) as shown was used as substrate for active Src. Reactive products were resolved by SDS-PAGE and visualized by Western blot with p-TOPK (Y74).
Src binds with TOPK and phosphorylates TOPK at Y74 ex vivo
To verify whether Src can phosphorylate TOPK at Y74 ex vivo, we firstly detected the expression of Src and TOPK in four kinds of different colon cancer cell lines. The results showed that the expression of Src was the highest in SW480 cells but the expression of TOPK was lowest (Figure 2A). Then we checked that if Src and TOPK could co-localize in SW480 cells under the confocal microscope. The result showed that Src (red) co-localized with TOPK (green) in both the cytoplasm and nucleus of SW480 cells (Figure 2B). Subsequently, the SW480 cells were harvested and lysed, and Ni-NTA-His-TOPK was used to pull down endogenous Src, and then Src was probed with anti-Src by Western blot. The results indicated that Src could directly bind with TOPK (Figure 2C). Next, we cotransfected pcDNA3-HA-TOPK and pcDNA4-His-Src into HEK293T cells, and the phosphorylation of TOPK at Y74 was analyzed by p-TOPK (Y74). The result indicated that the phosphorylation level of TOPK at Y74 was increased (lane 3) compared to the level of controls (lane 1 and 2) when cotransfected with Src and TOPK, and the level was dramatically increased (lane 4) after being stimulated by EGF (Figure 2D). Moreover, the phosphorylation level of TOPK at Y74 in SW480 cells at 0, 5, 15 or 30 minutes after EGF treatment was tested. The result showed that endogenous phosphorylation of TOPK at Y74 was increased after EGF treatment (Figure 2E). These data indicated that phosphorylation of TOPK at Y74 could be detected ex vivo.
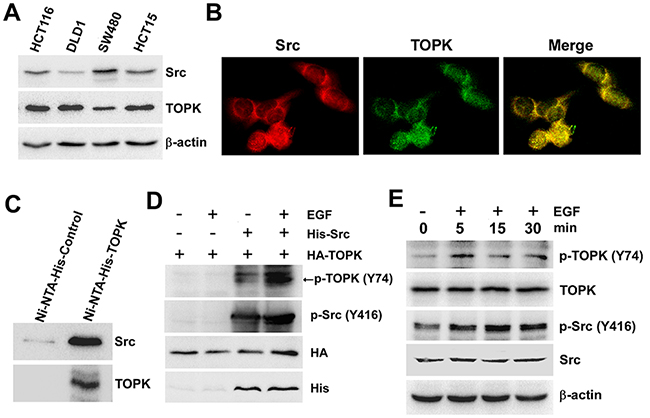
Figure 2: Src binds with TOPK and phosphorylates TOPK at Y74 ex vivo. A. The levels of Src and TOPK in four colon cancer cell lines were shown. B. Colocalization of Src and TOPK was visualized by confocal microscope in SW480 cells. Cytoplasmic and nuclear staining of Src and TOPK was mostly merged together. C. Ni-NTA-His-TOPK bound with endogenous Src of SW480 cells. D. Src promoted phosphorylation of TOPK in 293T cells induced by EGF after co-transfected with pcDNA4-His-Src and pcDNA3-HA-TOPK (EGF 80 ng/ml; 30 min). E. EGF induced a time-dependent phosphorylation of TOPK at Y74 in SW480 cells (EGF 80 ng/ml; 0 min, 5 min, 15 min, 30 min).
The phosphorylation of TOPK at Y74 was inhibited in colon cancer cells expressing low levels of Src
Since Src could phosphorylate TOPK at Y74 in vitro (Figure 1) and ex vivo (Figure 2), what we explored next was if the phosphorylation of TOPK at Y74 was inhibited in colon cancer cells expressing low levels of Src. At first, the Src inhibitor, Dasatinib, was used to test this idea further. Dasatinib is known as a targeted therapeutic small-molecule Src inhibitor [29, 30]. We analyzed the endogenous phosphorylation level of TOPK at Y74 in three different colon cancer cell lines treated with Dasatinib. The results suggested that endogenous phosphorylation level of TOPK at Y74 was gradually decreased in a dose-dependent manner, and p-Src (Y416) was lowered after dasatinib treatment (Figure 3A, 3B and 3C). We then confirmed this idea in shMock- and shSrc-expressing cells. The results suggested that endogenous phosphorylation level of TOPK at Y74 was decreased in shSrc-expressing cells (Figure 3D and 3E).
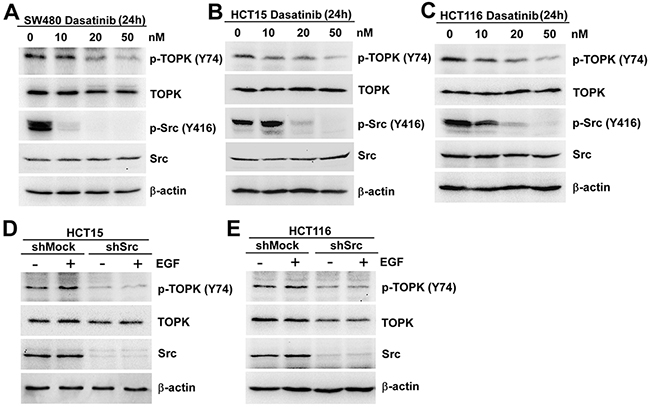
Figure 3: The phosphorylation of TOPK at Y74 was inhibited in colon cancer cells expressing low levels of Src. SW480 A. HCT15 B. and HCT116 C. cells were treated with Dasatinib for 24 h in a dose-dependent manner and the samples were resolved by SDS-PAGE and analyzed by Western blot respectively. The stable shMock and shSrc in HCT15 D. or HCT116 E. cells were treated with EGF (80 ng/ml; 30 min), and analyzed by Western blot, respectively. Data are representatives of results from triplicate experiments.
All the first three figures supported the idea that TOPK could be phosphorylated by Src at Y74 in vitro and ex vivo, which may suggest a novel activation mechanism of TOPK.
The phosphorylation of TOPK at Y74 and Y272 by Src promotes carcinogenesis ex vivo
Next, the function of phosphorylation of TOPK at Y74, Y272, or Y74Y272 by Src was tested respectively. As reported before, TOPK could promote EGF-induced cell transformation and colon cancer development [7]. Thus we constructed TOPK mutants Y74F, Y272F or Y74Y272FF and set up stable cell lines in JB6 and SW480 cell lines in which the expression of endogenous TOPK was low. Growth curves of JB6/ Mock and different JB6/ TOPK cell lines were compared and the results showed that the growth of JB6/ WT cells was remarkably faster than that of JB6/ Mock cells. The growth of JB6/ 74F, 272F, or FF cells were slower than that of JB6/ WT cells.
(Figure 4A). The similar results were observed in SW480 stable cell lines (Figure 4B). Next, the anchorage-independent colony formation ability of stable cell lines was tested. Compared with JB6/ WT group, the colonies formed by JB6/ 74F, JB6/ 272F and JB6/ FF cells were significantly fewer and smaller (Figure 4C). Consistent with the data in the previous study, [7] the colonies formed by JB6/ WT cells were significantly more in number and larger in size than that formed by JB6/ Mock cells (Figure 4C). Similarly, the colonies formed by SW480/ 74F, SW480/ 272F and SW480/ FF cells were also relatively fewer and smaller than those formed by SW480/ WT cells (Figure 4D). All above data showed that mutated TOPK at Y74F, Y272F, or Y74Y272FF could block the anchorage-independent growth ability ex vivo, suggesting that the phosphorylation of TOPK by Src at Y74 and Y272 could promote carcinogenesis ex vivo. TOPK double mutant Y74Y272FF was used in the following experiment since either Y74F or Y272F had function.
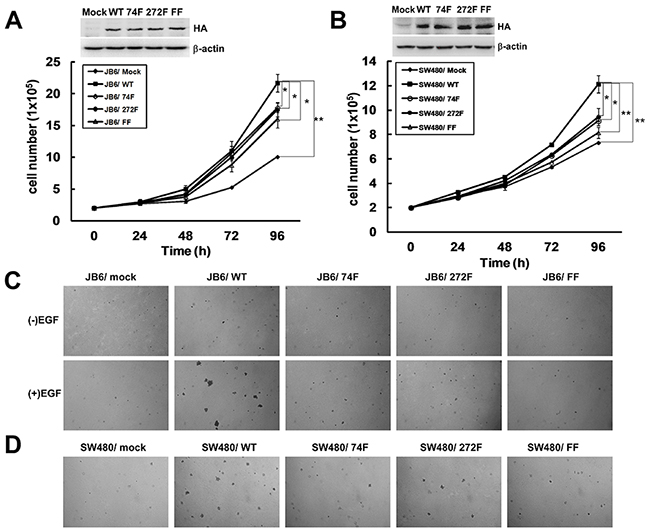
Figure 4: The phosphorylation of TOPK at Y74 and Y272 by Src promotes carcinogenesis ex vivo. A. Growth curves of JB6 cells stably expressing pcDNA3-Mock (JB6/ Mock), pcDNA3-TOPK-WT (JB6/ WT), pcDNA3-TOPK-74F (JB6/ 74F), pcDNA3-TOPK-272F (JB6/ 272F), or double-mutant pcDNA3-TOPK-FF (JB6/ FF). Inset (top) showed verification of the cell lines identified by Western blot. Data are represented as means±SD of triplicate experiments. *, significantly (P < 0.05) decrease in cell number in JB6/ 74F, JB6/ 272F or JB6/ FF cells compared with JB6/ WT cells respectively. **, significantly (P < 0.01) increase in cell number in JB6/ WT cells compared with JB6/ Mock cells. B. Growth curves of SW480 cells stably expressing pcDNA3-mock (SW480/ Mock), pcDNA3-TOPK-WT (SW480/ WT), pcDNA3-TOPK-74F (SW480/ 74F), pcDNA3-TOPK-272F (SW480/ 272F), or double-mutant pcDNA3-TOPK-FF (SW480/ FF). Inset (top) showed verification of the cell lines identified by Western blot. Data are represented as means±SD of triplicate experiments. *, respective (P < 0.05) decrease in cell number in SW480/ 74F or 272F compared with SW480/ WT cells. **, significantly (P < 0.01) decrease in cell number in SW480/ FF cells compared with SW480/ WT cells and significantly (P < 0.01) increase in cell number in SW480/ WT cells compared with SW480/ Mock cells. C. Transfectants of JB6/ Mock, WT, 74F, 272F, or FF were compared for EGF-induced colony formation in soft agar. D. Transfectants of SW480/ Mock, WT, 74F, 272F, or FF were compared for colony formation in soft agar.
The phosphorylation of TOPK at Y74, Y272 by Src enhances the activity of TOPK
Since the phosphorylated TOPK by Src promoted cell transformation, the next question that attracted us was whether the activity of TOPK was enhanced. The phosphorylated Histone H3 at Ser10 is the most widely established and used mitotic marker [31, 32], and is one of TOPK’s substrates confirmed by J. H. Park et al [14]. The results showed that the level of phospho-Histone H3 (p-Histone H3 (S10)) dramatically decreased in JB6/ FF cells (Figure 5A), suggesting the decreasing activity of TOPK with double-mutation. Similar result was observed in Src knockout (Src-/-) cells (Figure 5B). Meanwhile, the levels of p-Histone H3 (S10) also decreased with 50nM dasatinib treatment in the three colon cancer cells (Figure 5C, 5D and 5E). These data implied that the phosphorylation of TOPK at Y74, Y272 by Src enhanced the activity of TOPK.
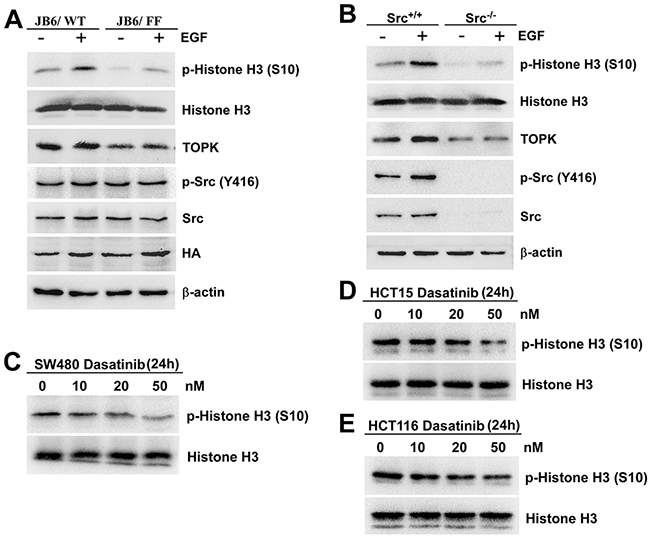
Figure 5: The phosphorylation of TOPK at Y74, Y272 by Src enhances the activity of TOPK. A. JB6/ WT and JB6/ FF cells were treated with EGF (20 ng/ml; 15 min), and the samples were analyzed by Western blot. B. Src+/+ and Src-/- MEFs were treated with EGF (20 ng/ml; 15 min), and analyzed by Western blot. SW480 C. HCT15 D. and HCT116 E. cells were treated with Dasatinib for 24 h in a dose-dependent manner. Then Histones were isolated, resolved by SDS-PAGE and analyzed by Western blot, respectively. Data are representatives of results from triplicate experiments.
The phosphorylation of TOPK by Src enhances the stability of TOPK
As shown in Figure 5B and Figure 3D and 3E, we found that the level of TOPK was obviously decreased in Src-/- cells and shSrc-expressing cells. This attracted us to explore whether the phosphorylation of TOPK by Src could affect the stability of TOPK. First, His-Src, Flag-ubiquitin or HA-TOPK were cotransfected into HEK293T cells. 48 hours later, cell extracts from each group were immunoprecipitated with anti-HA, and Flag-ubiquitin was detected by Western blot. The results indicated that the overexpression of Src dramatically reduced the ubiquitination level of TOPK compared with the control (Figure 6A). Next, to further illuminate these result, we examined the half-life of TOPK. pcDNA3-HA-TOPK wild type (TOPK-WT) and pcDNA3-HA-TOPK double mutant (TOPK-FF) were transiently transfected into HEK293T cells. Then the cells were treated with EGF for 30 min followed by treatment with CHX to assess the stability of TOPK at various time points. The result showed that TOPK-FF had shorter half-life than that of TOPK-WT (Figure 6B). Subsequently, the half-life of endogenous TOPK in Src+/+ cells was also compared with Src-/- cells. The results indicated that the half-life of TOPK in Src+/+ cells was much longer than that in Src-/- cells (Figure 6C). In summary, these results elucidate that phosphorylation of TOPK by Src enhances the stability of TOPK.
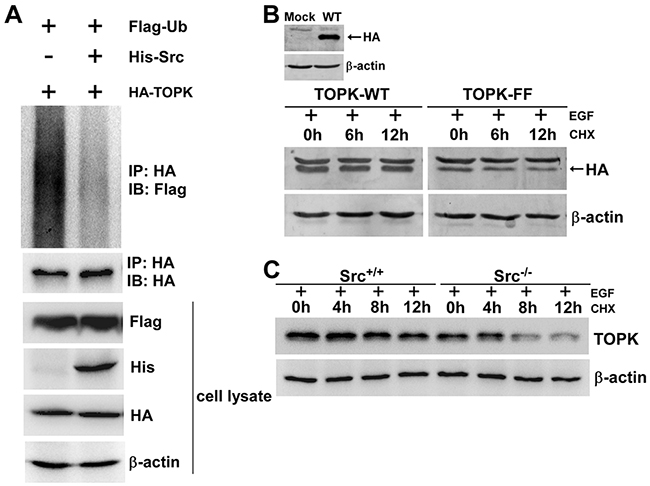
Figure 6: The phosphorylation of TOPK by Src enhances the stability of TOPK. A. Phosphorylation of TOPK by Src prevented TOPK ubiquitination. HEK293T cells were transfected with pCMV-Flag-ubiquitin (Flag-Ub), pcDNA3-HA-TOPK (HA-TOPK), and pcDNA4-His-Src (His-Src) as indicated. The cells were harvested 48 h after transfection. Then the samples were immunoprecipitated (IP) with anti-HA and detected with anti-Flag by Western blot. The transfection efficiency and equal protein loading were verified by Western blot using the whole cell lysate. B. HEK293T cells were transfected with pcDNA3-HA-TOPK wild type (TOPK-WT) and pcDNA3-HA-TOPK double mutant (TOPK-FF), and then were treated with EGF (80 ng/ml; 30 min) followed by addition of CHX (100 μg/ml) to prevent new protein synthesis. The time-dependent stability of TOPK was detected with anti-HA by Western blot. C. Src+/+ and Src-/- MEFs were treated with EGF (20 ng/ml; 15 min) followed by addition of CHX (100 μg/ml) to prevent new protein synthesis. Lysates were collected at the indicated time points. The protein level of TOPK was detected by Western blot.
Src phosphorylates TOPK to promote tumorigenesis of colon cancer in vivo
Based on the ex vivo data, we then verified the above results in vivo. We compared the ability of SW480/ WT cells to form tumors in athymic nude mice with that of SW480/ FF cells. SW480/ WT or SW480/ FF cells (3×106) were injected subcutaneously into the right flank of athymic Balb/c mice. 5 out of the 8 mice injected with both experimental cells developed tumors. The tumors from mice injected with SW480/ WT cells grew from 0 mm3 to nearly 1000 mm3 within 5 weeks. And the tumors from SW480/ FF cells inoculated mice were much smaller (Figure 7A). Tumor growth curves indicated that the increasing growth rate in the mice injected with SW480/ FF cells was significantly lower than that in the mice inoculated with SW480/ WT cells (Figure 7B). After the tumors of mice injected with SW480/ WT cells grew to 1000 mm3, the mice were euthanized and the tumors were dissected and then sent for hematoxylin & eosin (H&E) staining and immunohistochemical (IHC) analysis. The results showed a solid arrangement of tumor cells with high nuclear/cytoplasmic ratio, marked nuclei pleomorphism, visible mitosis, and numerous blood vessels in some areas (Figure 7C left panel). It suggested both of them were poorly differentiated adenocarcinoma cells. IHC analysis of the tumor samples from two groups of mice showed that p-Histone H3 (S10) was more highly expressed in SW480/ WT cells than that in SW480/ FF cells (Figure 7C right panel). Therefore, the tumorigenic properties of SW480/ FF cells significantly reduced, suggesting that Y74 and Y272 were important sites for TOPK function in vivo.
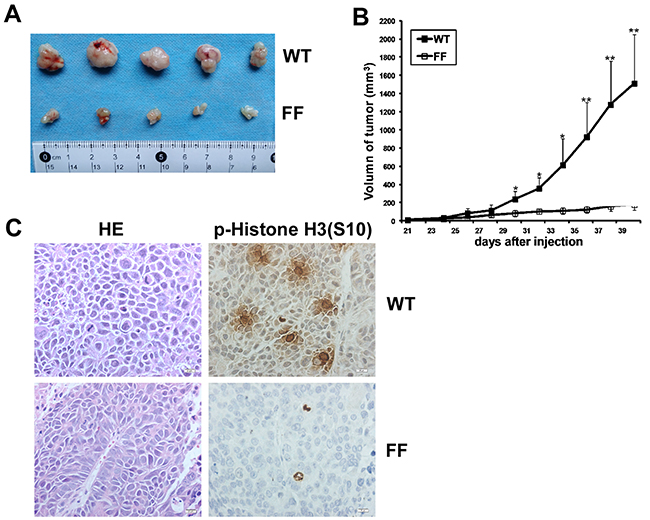
Figure 7: Src phosphorylates TOPK to promote the tumorigenesis of colon cancer in vivo. A. Tumors dissected from SW480/ WT or SW480/ FF group were shown. B. Tumor growth curve from mice injected with SW480/ WT or SW480/ FF cells. Data are expressed as means±SE of 5 mice in each group. The asterisk indicated a significant increase in tumor size in SW480/ WT injected mice compared with SW480/ FF injected mice. *P < 0.05, **P < 0.01. C. Left, H&E stained tumor sections from mice injected with either SW480/ WT or SW480/ FF cells. Poorly differentiated adenocarcinoma cell clusters were found in the two groups. Right, IHC analysis of p-Histone H3 (S10) expression in tumor sections from mice injected with either SW480/ WT or SW480/ FF cells. Nuclear expression of p-Histone H3 (S10) was detected in the tumor sections from SW480/ WT cells, while very weak staining of p-Histone H3 (S10) was detected from SW480/ FF cells. Magnification, 400×.
DISCUSSION
Several groups of scientists have reported that TOPK can regulate cellular proliferation and promote tumorigenesis. Furthermore, TOPK is considered to be an unfavorable prognostic indicator in colon cancer patients and this provides the opportunity to predict prognosis in these patients using anti-TOPK [19]. However, non-phospho-TOPK antibody can not fully represent the activity of TOPK, and the phosphorylation TOPK at T9 can not promote tumorigenesis (Supplementary Figure S1). There must exist other activation mechanisms of TOPK related to tumorigenesis. So far it is only reported that hDlg [2], P53 [33], E2F-CREB/ATF [34], c-Myc-E2F1 [35], Cdk1 [2, 3, 36], p38 and ERK2 [7] could interact with TOPK and regulate TOPK at the transcriptional or posttranscriptional level. Among them, ERK2, p38 or Cdk1 can phosphorylate TOPK at T9 and the positive feedback loop between TOPK and ERK2 seemed to contribute to transformation in colon cancer [7]. In this study, Src was identified to be a novel upstream kinase of TOPK in vitro and ex vivo. The phosphorylation of TOPK at Y74, Y272 by Src can promote tumorigenesis of colon cancer cells ex vivo and in vivo and activate TOPK–Histone H3 pathway.
Colon cancer patients could benefit both from the detection of the disease at early stages and from the advance of targeted therapy [37, 38]. In clinic, it is reported previously that Src activity gradually increases with the progression from benign polyps to primary colonic lesions to liver metastatic cancer [24, 27]. TOPK is highly expressed in CRC compared with the matched normal colorectal tissue [7]. In this study, the activity of TOPK was first time reported dramatically increased after phosphorylated by Src at Y74 and Y272. The phosphorylation of TOPK by Src at Y74 and Y272 were closely related to tumorigenic. We generated p-TOPK (Y74) and p-TOPK (Y272) antibodies in this study, and only p-TOPK (Y74) could be used in vitro and ex vivo. Both antibodies failed to detect in IHC study. More specific phosphorylated TOPK (Y74) and (Y272) antibodies are needed urgently to be used in clinicopathological study in colon cancer, because accurate understanding TOPK activity in colon cancer patients will provide more valuable information.
Our study showed TOPK could be degraded through ubiquitination pathway, the phosphorylation of TOPK by Src inhibited this process, and the half-life of TOPK dramatically decreased in Src-/- MEFs compared with Src+/+ MEFs. It has been reported that FBW7 can bind with ERK [39] or c-Jun [40], and TOPK can bind with ERK [7] or c-Jun [41] as well. Therefore, FBW7 may be related to the ubiquitination of TOPK.
In addition to the above, our results may bring benefits to the option of potential double-target for novel colon cancer targeted therapy. Although some single-agent Src inhibitors have been applied in clinical trials, preclinical studies hinted that a combined therapy might greatly increase the efficiency of Src inhibitors in colon cancer [42]. At the same time, TOPK gradually becomes an attractive molecular target for the treatment of a wide range of human cancers. At present, Caffeic acid (CaA) [43], HI-032 [44] and OTS964 [45] are the reported inhibitors for TOPK, and OTS964 is considered to be the best one for its high bioavailability. Coincidentally, the kinase profile analysis in that research indicated that OTS964 inhibited not only 79.7% of TOPK activity, but also 87.6% of Src activity [45]. In our study, the activity and stability of TOPK were increased by Src, and therefore the inhibition of both Src and TOPK, like OTS964, will be very efficient to cure colon cancer. It is promising to develop more drugs targeting both TOPK and Src for anticancer therapy.
In conclusion, our study elucidates that Src is a novel upstream kinase of TOPK, the phosphorylation of TOPK at Y74, Y272 by Src increases TOPK stability and promotes tumorigenesis of colon cancer. The specific phosphorylated TOPK antibodies would be made to predict prognosis for colon cancer patients instead of non-phospho-TOPK antibody. And it may develop new therapeutic strategies to colon cancer.
MATERIALS AND METHODS
Antibodies and reagents
TOPK and β-actin were purchased from Santa Cruz Technology, Inc (Santa Cruz, CA, USA). Phospho-Src (Tyr416) (D49G4) Rabbit mAb, Src (32G6) Rabbit mAb, Histone H3 (D1H2) XP Rabbit mAb, phospho-Histone H3 (Ser10) (D2C8) XP Rabbit mAb, and HA-Tag Rabbit polyclonal antibody were purchased from Cell Signaling Technology, Inc (Boston, MA, USA). Ubiquitin Rabbit polyclonal antibody was purchased from Proteintech Group, Inc (Chicago, USA). His-Tag Mouse monoclonal antibody (4E6) and HA-Tag Mouse monoclonal antibody (1B10) were obtained from Pregene, Inc (Beijing, China). HRP-labeled Goat anti Mouse IgG (H+L) and Goat anti Rabbit IgG (H+L) were purchased from EarthOx, LLC (San Francisco, CA, USA). Donkey anti-Rabbit IgG (H+L) Alexa Fluor® 546 Red and Donkey anti-Mouse IgG (H+L) Alexa Fluor® 488 Green were purchased from Invitrogen, Inc (Carlsbad, CA, USA). Phospho-TOPK at Y74 or Y272 antibodies were prepared by Abgent, Inc (Suzhou, China). All antibodies were used following the instructions of the respective manufacturers. The small hairpin RNA constructs against Src (5′-GACAGACCTGTCCTTCAAGAA-3′) used in this study was from the BioMedical Genomics Center at the University of Minnesota. Dasatinib was purchased from Selleckchem, Inc (Houston, TX, USA).
Cell culture, plasmids and transfection
JB6 Cl41, HEK293T, SW480, HCT116 and HCT15 cells were purchased from American Type Culture Collection (ATCC). Cells were cultured following the procedures provided by ATCC and were used within 6 months of resuscitation. Src wild-type (Src+/+) and knockout (Src-/-) mouse embryonic fibroblasts (MEFs) were gifts from Imamoto A (University of Chicago, Chicago, IL 60637, USA). The cells were cultured with Dubelco’s minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS). TOPK mutants at Y74, Y272, or Y74Y272 (designated Y74F, Y272F, and FF) were performed with the QuikChange Mutagenesis Kit (Stratagene, Inc., La Jolla, CA, USA). The mutant plasmids were sent to Sangon Biotech, Inc. (Shanghai, China) for DNA sequencing.
The transfect reagent, Simple-Fect, was purchased from Signaling Dawn Biotech (Wuhan, China). G418 (Sigma, St. Louis, USA) was used to set up stable cell lines. The pcDNA3-HA-TOPK, pcDNA4-His-Src and vector plasmids were transfected using Simple-fect into 293T cells at 50- 60% confluence following the manufacturer’s suggested protocol.
Bacterial expression and purification of the His-TOPK
PET-His-TOPK-WT and pET-His-TOPK-mutants were expressed in E. coli BL21 bacteria. Bacteria were grown at 37°C to an absorbance of 0.6–0.8 at 600 nm, induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) at 30°C for 4h. All proteins were purified using nickel-nitrilotriacetic acid agarose (Qiagen, Inc., Valencia, CA, USA) overnight at 4°C and eluted with 200 mM imidazole. After protein quantitation, the samples were separated by 10% SDS polyacrylamide gel for electrophoresis (SDS-PAGE), and visualized by Coomassie Brilliant Blue staining.
In vitro kinase assay
The Src active kinase and 10×kinase buffer were purchased from Millipore Corp. (Billerica, MA, USA). The inactive substrate (2 μg) and the active kinase (0.2 μg in a 30 μl reaction) were incubated at 32°C for 40 min in 1×kinase buffer containing 100 μmol/L unlabeled ATP or 1 μCi [γ-32P] ATP. The samples were added with 5×SDS buffer and then resolved by SDS-PAGE and visualized by autoradiography or Western blot.
Confocal laser scanning fluorescence microscopy
SW480 cells were fixed in methanol (−20°C) and blocked in 5% normal goat serum at room temperature for 1 h. Then the cells were incubated overnight with the primary antibodies to detect TOPK and Src at 4°C. On the second day, the cells were incubated for 1 h at room temperature with the Alexa Fluor 546 (red for Src) or Alexa Fluor 488 (green for TOPK) conjugated secondary antibody while being protected from light. Colocalization of proteins was observed by laser scanning confocal microscopy (NIKON C1si Confocal Spectral Imaging System, NIKON Instruments Co., Japan). 5% normal goat serum instead of the primary antibodies was used as a negative control.
Western blot and immunoprecipitation
Cells (8×105) were cultured in 10 cm diameter dishes to 70-80% confluence, starved for 24 hours and treated with EGF ( R&D lot: HLM481304). After that the cells were harvested and disrupted in 300 μl of RIPA buffer (1X PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1mmol/L Na3VO4, and 1 mmol/L aprotinin and 1 mmol/L phenylmethylsulfonyl fluoride). The samples were sonicated 15 seconds for three times and centrifuged at 13,000 rpm for 10 minutes. Histones were isolated as described previously [10]. Then the quantity of protein was determined by the Bradford method [46]. The samples (30-50 μg protein) with 5×SDS loading buffer were heated at 95°C for 10 minutes, and then cooled on ice. After that, the samples were separated on a 10%-15% SDS-PAGE and subsequently transferred onto a PVDF membrane (Millipore, Billerica, MA, USA). Then antibody-bound proteins were detected by chemiluminescence (BIO-RAD, USA). The untreated cell samples were used as negative controls. The samples for immunoprecipitation were harvested in 1% CHAPS instead of RIPA buffer. Equal amounts of protein (1–2mg) were subjected to immunoprecipitation following the manufacturer’s suggested protocol. (http://www.scbt.com/protocols.html?protocol=immunoprecipitation).
Growth curve analysis
Cells were plated at 2 × 105 cells per 10 cm dish and counted in triplicate at different time points using a hemacytometer for generating a growth curve.
Anchorage-independent cell transformation assay
Different cell lines (8 × 103/well) in a 6-well plate were exposed or not exposed to EGF (20 ng/ml) and cultured in 1ml of 0.33% BME (Eagle basal medium, Sigma-Aldrich Corp.) agar (Sigma-Aldrich Corp.) containing 10% FBS over 3ml of 0.5% BME agar containing 10% FBS. The cells were maintained in a 37°C, 5% CO2 incubator for 5-10 days and then their colonies were observed by microscopy.
Tumor xenografts
Athymic Balb/c nude mice (4–6-week-old males) were purchased from Beijing HFK Bioscience CO., LTD (Beijing, China). The mice were randomized into two groups. Each of the different cell lines (3×106 in 200 μl PBS) was injected subcutaneously into the right flank of the athymic Balb/c nude mice, and the tumors were measured every other day. We estimated the tumor volume (V) from their length (l), width (w), and height (h) using the following formula: V = 0.52 (l×w×h). The mice were monitored until total volume of the tumors reached 1000 mm3. The tumors were dissected and sent for IHC analysis at the Department of Pathology in Xijing Hospital. The animal experiments were performed following the protocols approved by the Laboratory Animal Center of the Fourth Military Medical University.
Immunohistochemistry staining for samples from node mice
After the mice were euthanized, the tumors were fixed in 4% formalin, routinely processed, and embedded in paraffin. Sections of 5 μm were placed on glass slides for H&E staining and IHC analysis. All the samples from node mice were diagnosed by the Department of Pathology of Xijing Hospital. The tumor sections were stained with the p-Histone H3 (S10) antibody (1:200). Images were obtained and 400× magnified using an Olympus Imaging System Microscope.
Cycloheximide treatment
HEK293T Cells which were transiently transfected pcDNA3-HA-TOPK (WT) and pcDNA3-HA-TOPK (FF) plasmids as before were treated for 15 minutes with EGF, and then the medium was supplemented with 100 μg/ml of cycloheximide (CHX; Sigma, St. Louis, USA) to block protein synthesis. The cells were collected at various time points and expression of TOPK was visualized by Western blot. Src+/+, Src-/- MEFs were also carried out in the same way.
Statistical analysis
Significant differences were determined by one-way analysis of variance (ANOVA). The values were presented as means ± SEM. All statistical tests were two sided, and P < 0.05 was considered significant (*P < 0.05, **P < 0.01).
ACKNOWLEDGMENTS
This project was supported by the national Natural Science Foundation of China (No. 81472602, 81450110449, 81570180, 81572529 & 30271302) and the Independent innovation project of Huazhong University of Science and Technology (No. 2014TS063&2014ZZGH013), and the major program of international cooperation of the Ministry of Science and Technology of People’s Republic of China (No. 2011DFA33110).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Abe Y, Matsumoto S, Kito K, Ueda N. Cloning and expression of a novel MAPKK-like protein kinase, lymphokine-activated killer T-cell-originated protein kinase, specifically expressed in the testis and activated lymphoid cells. J Biol Chem 2000; 275: 21525–21531.
2. Gaudet S, Branton D, Lue RA. Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl Acad Sci 2000; 97: 5167–5172.
3. Matsumoto S, Abe Y, Fujibuchi T, Takeuchi T, Kito K, Ueda N, Shigemoto K, Gyo K. Characterization of a MAPKK-like protein kinase PBK/TOPK. Biochem Biophys Res Commun 2004; 325: 997–1004.
4. Park JH, Nishidate T, Nakamura Y, Katagiri T. Critical roles of T-LAK cell-originated protein kinase in cytokinesis. Cancer Sci 2010; 101: 403–411.
5. Fukukawa C, Ueda K, Nishidate T, Katagiri T, Nakamura Y. Critical roles of LGN/GPSM2 phosphorylation by PBK/TOPK in cell division of breast cancer cells. Genes Chromosomes Cancer 2010; 49: 861–872.
6. Shinde SR, Gangula NR, Kavela S, Pandey V, Maddika S. PBK/TOPK and PTEN participate in CHFR mediated mitotic checkpoint. Cell Signal 2013; 25: 2511–2517.
7. Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, Ma WY, Bode AM, Dong Z. Bidirectional signals transduced by PBK/TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology 2007; 133: 219–231.
8. Oh SM, Zhu F, Cho YY, Lee KW, Kang BS, Kim HG, Zykova T, Bode AM, Dong Z. T-lymphokine-activated killer cell-originated protein kinase functions as a positive regulator of c-Jun-NH2-kinase 1 signaling and H-Ras-induced cell transformation. Cancer Res 2007; 67: 5186–5194.
9. Ayllón V, O’ connor R. PBK/ TOPK promotes tumour cell proliferation through p38MAPK activity and regulation of the DNA damage response. Oncogene 2007; 26: 3451–3461.
10. Zykova TA, Zhu F, Lu C, Higgins L, Tatsumi Y, Abe Y, Bode AM, Dong Z. Lymphokine-activated killer T-cell-originated protein kinase phosphorylation of histone H2AX prevents arsenite-induced apoptosis in RPMI7951 melanoma cells. Clin Cancer Res 2006; 12: 6884–6893.
11. Zykova TA, Zhu F, Vakorina TI, Zhang J, Higgins LA, Urusova DV, Bode AM, Dong Z. T-LAK cell-originated protein kinase (PBK/TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in RPMI7951 melanoma cells through the regulation of Prx1 peroxidase activity. J Biol Chem 2010; 285: 29138–29146.
12. O Leary PC, Penny SA, Dolan RT, Kelly CM, Madden SF, Rexhepaj E, Brennan DJ, McCann AH, Pontén F, Uhlén M, Zagozdzon R, Duffy MJ, Kell MR, et al. Systematic antibody generation and validation via tissue microarray technology leading to identification of a novel protein prognostic panel in breast cancer. BMC Cancer 2013; 13: 175.
13. Wei DC, Yeh YC, Hung JJ, Chou TY, Wu YC, Lu PJ, Cheng HC, Hsu YL, Kuo YL, Chen KY, Lai JM. Overexpression of T-LAK cell-originated protein kinase predicts poor prognosis in patients with stage I lung adenocarcinoma. Cancer Sci 2012; 103: 731–738.
14. Park JH, Lin ML, Nishidate T, Nakamura Y, Katagiri T. PDZ-binding kinase/T-LAK cell-originated protein kinase, a putative cancer/testis antigen with an oncogenic activity in breast cancer. Cancer Res 2006; 66: 9186–9195.
15. Simons-Evelyn M, Bailey-Dell K, Toretsky JA, Ross DD, Fenton R, Kalvakolanu D, Rapoport AP. PBK/TOPK is a novel mitotic kinase which is upregulated in Burkitt’s lymphoma and other highly proliferative malignant cells. Blood Cells Mol Dis 2001; 27: 825–829.
16. Singh PK, Srivastava AK, Dalela D, Rath SK, Goel MM, Bhatt ML. Expression of PDZ-binding kinase/T-LAK cell-originated protein kinase (PBK/TOPK) in human urinary bladder transitional cell carcinoma. Immunobiology 2014; 219: 469–474.
17. Zlobec I, Molinari F, Kovac M, Bihl MP, Altermatt HJ, Diebold J, Frick H, Germer M, Horcic M, Montani M, Singer G, Yurtsever H, Zettl A, et al. Prognostic and predictive value of TOPK stratified by KRAS and BRAF gene alterations in sporadic, hereditary and metastatic colorectal cancer patients. Br J Cancer 2010; 102: 151–161.
18. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29.
19. Deschoolmeester V, Baay M, Specenier P, Lardon F, Vermorken JB. A review of the most promising biomarkers in colorectal cancer: one step closer to targeted therapy. Oncologist 2010; 15: 699–731.
20. Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol 2001; 2: 467–475.
21. Chen J, Elfiky A, Han M, Chen C, Saif MW. The role of Src in colon cancer and its therapeutic implications. Clinical Colorectal Cancer 2014; 13: 5–13.
22. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–2342.
23. Penuel E, Martin GS. Transformation by v-Src: Ras-MAPK and PI3K-mTOR mediate parallel pathways. Mol Biol Cell 1999; 10: 1693–1703.
24. Xie G, Peng Z, Raufman JP. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am J Physiol Gastrointest Liver Physiol 2012; 302: 1006–1015.
25. Cartwright CA, Kamps MP, Meisler AI, Pipas JM, Eckhart W. Pp60c-Src activation in human colon carcinoma. J Clin Invest 1989; 83: 2025–2033.
26. Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60cSrc in progressive stages of human colorectal cancer. J Clin Invest 1993; 91: 53–60.
27. Aligayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer 2002; 94: 344–351.
28. Diella F, Cameron S, Gemünd C, Linding R, Via A, Kuster B, Sicheritz-Pontén T, Blom N, Gibson TJ. Phospho. ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics 2004; 5: 79.
29. Araujo J, Logothetis C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treatment Reviews 2010; 36: 492–500.
30. Kim LC, Rix U, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs 2010; 19: 415–425.
31. Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell 2000; 103: 263–271.
32. Choi HS, Choi BY, Cho YY, Mizuno H, Kang BS, Bode AM, Dong Z. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer Res 2005; 65: 5818–5827.
33. Hu F, Gartenhaus RB, Eichberg D, Liu Z, Fang HB, Rapoport AP. PBK/TOPK interacts with the DBD domain of tumor suppressor p53 and modulates expression of transcriptional targets including p21. Oncogene 2010; 29: 5464–5474.
34. Nandi AK, Rapoport AP. Expression of PDZ-binding kinase (PBK) is regulated by cell cycle-specific transcription factors E2F and CREB/ATF. Leuk Res 2006; 30: 437–447.
35. Hu F, Gartenhaus RB, Zhao XF, Fang HB, Minkove S, Poss DE, Rapoport AP. c-Myc and E2F1 drive PBK/ TOPK expression in high-grade malignant lymphomas. Leukemia Research 2013; 37: 447–454.
36. Abe Y, Takeuchi T, Kagawa-Miki L, Ueda N, Shigemoto K, Yasukawa M, Kito K. A mitotic kinase PBK/TOPK enhances Cdk1/cyclin B1-dependent phosphorylation of PRC1 and promotes cytokinesis. J Mol Biol 2007; 370: 231–245.
37. Binefa G, Rodríguez-Moranta F, Teule A, Medina-Hayas M. Colorectal cancer: From prevention to personalized Medicine. World J Gastroenterol 2014; 20: 6786–6808.
38. Tobin NP, Foukakis T, De Petris L, Bergh J. The importance of molecular markers for diagnosis and selection of targeted treatments in patients with cancer. J Intern Med 2015; 278: 545–570.
39. Ji S, Qin Y, Shi S, Liu X, Hu H, Zhou H, Gao J, Zhang B, Xu W, Liu J, Liang D, Liu L, Liu C, et al. ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res 2015; 25: 561–73.
40. Zhang J, Zhu F, Li X, Dong Z, Xu Y, Peng C, Li S, Cho YY, Yao K, Zykova TA, Bode AM, Dong Z. Rack1 protects N-terminal phosphorylated c-Jun from Fbw7-mediated degradation. Oncogene 2012; 31: 1835–44.
41. Li Y, Yang Z, Li W, Xu S, Wang T, Wang T, Niu M, Zhang S, Jia L, Li S. TOPK promotes lung cancer resistance to EGFR tyrosine kinase inhibitors by phosphorylating and activating c-Jun. Oncotarget 2016. doi: 10.18632/oncotarget.6826.
42. Lieu C, Kopetz S. The Src Family of Protein Tyrosine Kinases: A New and Promising Target for Colorectal Cancer Therapy. Clin Colorectal Cancer 2010; 9: 89–94.
43. Kang NJ, Lee KW, Kim BH, Bode AM, Lee HJ, Heo YS, Boardman L, Limburg P, Lee HJ, Dong Z. Coffee phenolic phytochemicals suppress colon cancer metastasis by targeting MEK and TOPK. Carcinogenesis 2011; 32: 921–928.
44. Kim DJ, Li Y, Reddy K, Lee MH, Kim MO, Cho YY, Lee SY, Kim JE, Bode AM, Dong Z. Novel PBK/TOPK inhibitor HI-PBK/TOPK-032 effectively suppresses colon cancer growth. Cancer Res 2012; 72: 3060–3068.
45. Matsuo Y, Park JH, Miyamoto T, Yamamoto S, Hisada S, Alachkar H, Nakamura Y. TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis. Sci Transl Med 2014; 6: 259ra145.
46. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248–254.