
Introduction
Clinical responses to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in solid tumors lacking activating EGFR mutations are rare [1-3]. A complete “exceptional” response to EGFR TKI when EGFR is wild type suggests that there are additional genetic contributors that mediate EGFR TKI sensitivity [4, 5]. We recently reported the case of a patient with locally advanced head and neck squamous cell carcinoma (HNSCC) who experienced a near-complete histologic response after receiving 13 days of neoadjuvant erlotinib treatment [6]. Whole exome sequencing of the pre-treatment tumor revealed a MAPK1E322K somatic mutation, which was further implicated in mediating erlotinib sensitivity by preclinical studies.
MAPK1 encodes ERK2, which will be referred to as MAPK1, a component of the mitogen activated signaling (MAPK) pathway downstream of RAS, RAF and MEK. The MAPK1E322K hotspot mutation, which causes constitutive activation of ERK2 [7, 8], occurs in approximate 1.3% of HNSCC [9, 10] and in 8% of cervical squamous cell carcinomas [11].
We reported that HNSCC cells harboring an endogenous MAPK1E322K mutation (HSC-6) or HNSCC cells hemizygous for wild type MAPK1 (FaDu) transfected with mutant MAPK1E322K demonstrated enhanced EGFR phosphorylation and activation of downstream signaling [6, 7, 12]. FaDu cells expressing exogenous mutant MAPK1 demonstrated increased senescence to erlotinib compared with those expressing exogenous wild type MAPK1 or vector control [6]. These findings suggest potential crosstalk between mutant MAPK1 and EGFR signaling pathways. However, the molecular mechanism underlying this crosstalk remains unknown.
Previous studies demonstrated ERK activity results in the production of the EGFR ligand amphiregulin (AREG) in airway epithelial cells [13] [14]. More recently, MAPK1 specifically and not ERK1 was reported to be required for AREG production in HNSCC cells [15]. Increased AREG levels have been associated with enhanced response to EGFR TKIs in EGFR wild-type cancer cell lines and patient tumors [16, 17]. We previously reported that increased secretion of AREG in HNSCC is critical for EGFR crosstalk and transactivation [18]. The present study was undertaken to test the hypothesis that MAPK1E322K increases sensitivity to erlotinib through enhanced AREG-EGFR activation in HNSCC.
Results
MAPK1E322K is associated with increased secretion of AREG in HNSCC cells
We previously reported that HSC-6 cells harboring endogenous MAPK1E322K expressed higher basal level of p-EGFR (Y1068) compared to wild-type MAPK1 (MAPK1WT) Cal33. In addition, FaDu cells engineered to express exogenous MAPK1E322K expressed high basal level of p-EGFR (Y1068) compared to FaDu cells exogenously expressing MAPK1WT or vector control, suggesting that the activating MAPK1 mutation likely causes a feedback activation of EGFR in HNSCC cells. We determined that MAPK1E322K HSC-6 cells were more sensitive to erlotinib than MAPK1WT Cal33 cells as assessed by senescence assay (Figure 1A). Representative pictures of senescence staining are presented in Figure S1. Following treatment with erlotinib, P-EGFR and P-MAPK levels were reduced in both Cal33 and HSC-6 cells (Figure 1B), demonstrating inhibition of EGFR under experimental conditions. We previously reported that FaDu cells engineered to exogenously express MAPK1E322K were more sensitive to erlotinib than FaDu cells engineered to express MAPK1WT or vector control using this same senescence assay [6].
We hypothesized that MAPK1E322K activated EGFR through enhanced EGFR ligand secretion. To test this hypothesis, we first measured the release of several EGFR autocrine ligands in HNSCC cells including AREG, TGF-α, EGF and HB-EGF (Table S1). We found that endogenous MAPK1E322K HSC-6 cells secreted higher AREG levels compared with MAPK1WT Cal33 cells (Figure 1C). To assess AREG secretion in the same genetic background we tested FaDu cells engineered to exogenously express MAPK1E322K, MAPK1WT or vector-control and found that cells expressing MAPK1E322K had increased AREG secretion (Figure 1D). These results suggest that MAPK1E322K may be involved in an autocrine loop of AREG production leading to increased p-EGFR via ligand-dependent activation. Expression of exogenous MAPK1WT also appeared to modestly increase AREG secretion, suggesting that overexpression of wild type MAPK1 may also participate in this process, albeit to a lesser degree than MAPK1E322K.
To determine if the effects of MAPK1E322K were specific to AREG, we also examined secretion of other autocrine EGFR ligands in the conditioned media of Cal33, HSC-6 and FaDu engineered cells. Unlike AREG, MAPK1E322K did not induce detectable secretion of TGF-α, EGF or HB-EGF, which were below the limits of assay detection for all samples (Table S1), thus excluding the possible involvement of these other EGFR ligands.
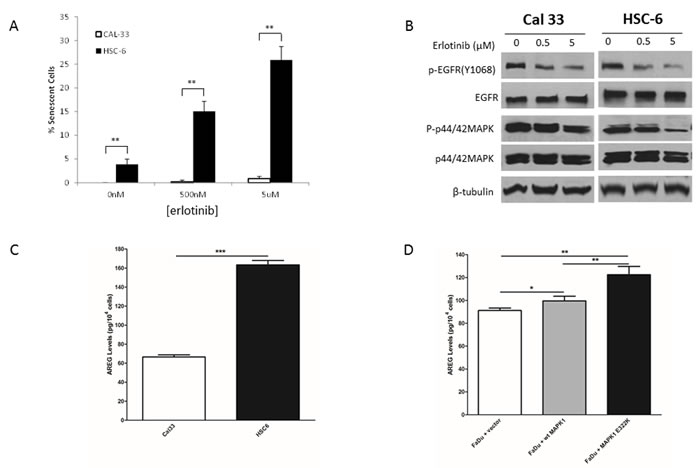
Figure 1: MAPK1E322K was associated with increased secretion of AREG and enhanced response to erlotinib in HNSCC cells. A. Following 48 h treatment with erlotinib (500 nM or 5 µM) or vehicle control, HSC-6 cells had enhanced erlotinib-induced senescence compared to Cal33 cells. B. Cal33 and HSC-6 cells were treated for 48 h treatment with erlotinib (500 nM or 5 µM) or vehicle control and cell lysates were analyzed by immunoblotting. P-EGFR levels were diminished in both cell lines following erlotinib treatment. p-MAPK levels demonstrated modest reduction in Cal-33 cells and pronounced reduction in HSC-6 cells following erlotinib treatment. C. MAPK1E322K HSC-6 cells secrete higher levels of AREG compared to MAPK1WT Cal33 cells. D. Expression of exogenous MAPK1E322K increased AREG secretion compared with vector-control or wild type transfected MAPK1 in FaDu (MAPK1-hemizygous wild type) engineered cells (n = 3). Similar results were obtained with triplicate wells in three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
MAPK1 inhibition decreases AREG secretion
To determine the role of MAPK1E322K in AREG secretion, HNSCC cells were treated with a dual MAPK1 (ERK2) and ERK1 kinase inhibitor, VX-11e [19]. Ribosomal protein S6 kinase (RSK1) is a MAPK1 (ERK2)-specific downstream substrate [19, 20]. As a pharmacodynamic measure of VX-11e inhibition, we evaluated the effects on RSK1 phosphorylation. As shown in Figures 2A and 2B, VX-11e inhibited phosphorylation of RSK1 in HNSCC cells. To test our hypothesis that MAPK1 inhibition would decrease AREG secretion, we treated cells with VX-11e (0.5 µM) or DMSO vehicle control and collected the conditioned media. We found that AREG levels were substantially decreased following treatment with VX-11e in Cal33, HSC6 and FaDu engineered cells (Figures 2C and 2D). HNSCC cells harboring the MAPK1E322K mutation had the largest decrease in AREG levels following VX-11e treatment. These results indicate that AREG production in HNSCC cells is dependent upon MAPK signaling and that the MAPK1E322K-induced AREG autocrine loop was significantly attenuated by MAPK1 inhibition. These findings implicate MAPK1E322K in the enhanced production of AREG in HNSCC.
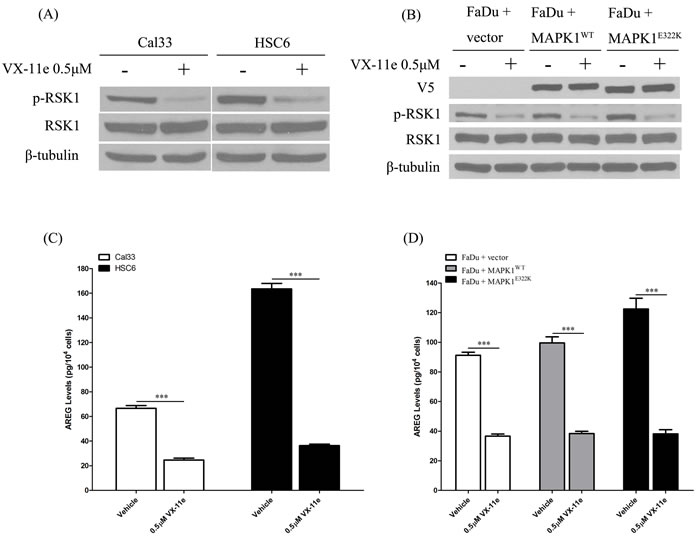
Figure 2: MAPK1 inhibition decreased AREG secretion in HNSCC, especially in the cells harboring the MAPK1E322K mutation. A. HNSCC cells were treated with DMSO vehicle or VX-11e (0.5 µM) for 24 h and cell protein extracts analyzed. VX-11e inhibited phosphorylation of RSK1 in Cal33 and HSC-6. B. VX-11e inhibited phosphorylation of RSK1 in FaDu engineered cells. C. For quantification of secreted AREG, cells were pretreated with DMSO vehicle or VX-11e (0.5 µM) for 2 hours, washed with 1× PBS, then treated for 24 h. Conditioned media was collected and assayed by ELISA. VX-11e decreased AREG secretion in Cal33 and HSC-6 cells as well as FaDu engineered cells D.. The extent of AREG decrease in endogenous MAPK1E322K HSC-6 and FaDu cells engineered to express MAPK1E322K was higher than respective endogenous MAPK1WT Cal33 and FaDu cells with vector or wild type MAPK1 transfection. (n = 3. *p < 0.05, **p < 0.01). Similar results were obtained with triplicate wells in three independent experiments.
In addition to pharmacologic inhibition of MAPK with VX-11e, we also examined AREG secretion levels in endogenous MAPK1E322K HSC-6 cells and endogenous MAPK1WT Cal33 cells upon MAPK1 knockdown by siRNA. As shown in Figure 3, MAPK1 siRNA efficiently reduced total MAPK1 (ERK2) expression levels and led to a reduced secretion of AREG compared to the non-targeting siRNA control. The decrease in AREG production following knockdown was greater in MAPK1E322K HSC-6 cells than MAPK1WT Cal33 cells.
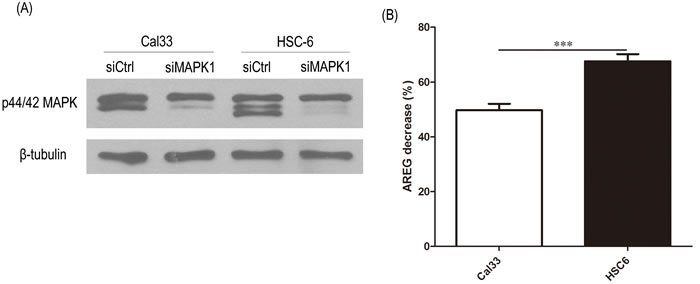
Figure 3: siRNA knockdown MAPK1 significantly decreased secretion of AREG in HSC-6 cells expressing mutant MAPK1E322K compared with Cal33 cells expressing MAPK1WT. 48 hours after transfection with control or MAPK1-targeting siRNA, CAL33 and HSC-6 cells were trypsinized and seeded into 48-well plates. 24 hours later supernatants were analyzed by ELISA and protein isolates analyzed. A. MAPK1 siRNA reduced total MAPK1 expression levels compared with non-targeting control siRNA. B. MAPK1 knockdown by siRNA reduced secretion of AREG to a greater extent in MAPK1E322K HSC-6 cells than in MAPK1WT Cal33 cells. Similar results were obtained with triplicate wells in three independent experiments.
MAPK1 inhibition decreases sensitivity to erlotinib in MAPK1E322K mutant HNSCC cells, while exogenous AREG restores erlotinib sensitivity
We next evaluated whether inhibition of MAPK signaling by VX-11e would decrease sensitivity to erlotinib in the HSC-6 and FaDu engineered cells expressing exogenous MAPK1E322K. We first pre-treated the cells with VX-11e for 24 hours to inhibit MAPK1E322K -driven autocrine AREG loop, then treated the cells with vehicle, VX-11e, erlotinib or a combination of VX-11e and erlotinib (0.5 or 5μM). As shown in Figures 4A and 4B, inhibition of ERK1/2 by VX-11e decreased sensitivity to erlotinib in HSC-6 and FaDu engineered cells expressing exogenous MAPK1E322K. Given that MAPK1E322K was associated with increased AREG secretion and, conversely, inhibition of MAPK1 results in reduced AREG secretion and decreased sensitivity to erlotinib, we next evaluated ability of exogenous AREG to restore erlotinib sensitivity in HSC-6 cells and FaDu engineered cells expressing exogenous MAPK1E322K under conditions of MAPK1 inhibition. We determined that AREG supplementation at 300 pg/ml, which was within the range of observed secreted AREG levels (Table S1), approximately restored AREG levels to baseline under these conditions. To test the effects of exogenous AREG on MAPK1 inhibition in HSC-6 cells and FaDu engineered cells expressing exogenous MAPK1E322K, we pretreated cells with a combination of AREG and VX-11e for 24 hours, then changed media and treated with combinations of erlotinib (0, 0.5 or 5μM), AREG and VX-11e for 48 hours. We observed that the sensitivity to erlotinib was restored by exogenous AREG in those cells under conditions of MAPK1 inhibition (Figure 4A and 4B). These same studies employing FaDu engineered control cells or FaDu cells expressing wild type MAPK1demonstrated attenuated resistance to erlotinib in the presence of VX-11e (Figure 4C and 4D) compared to MAPK1 E322K expressing cells. Collectively, MAPK1E322K drove increased secretion of AREG, which created an enhanced autocrine feedback loop involving AREG, EGFR, and ERK signaling compared with wild type MAPK1. Sensitivity to erlotinib is likely a matter of degree of dependency upon this autocrine loop.
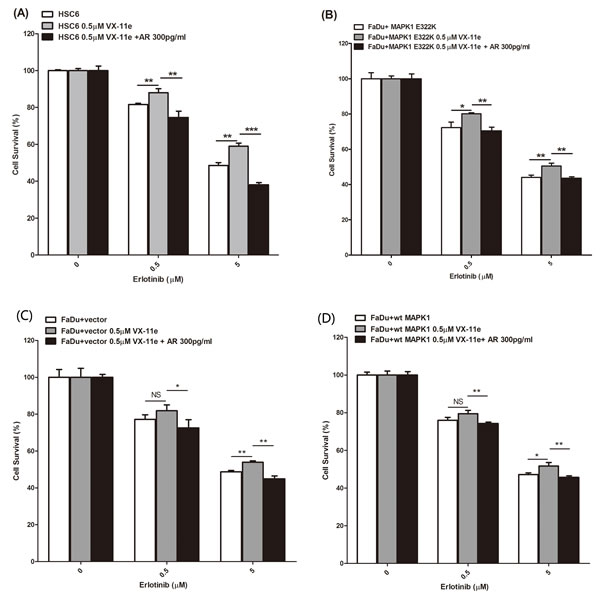
Figure 4: MAPK1 inhibition decreased sensitivity to erlotinib in MAPK1E322K mutant HNSCC cells, while exogenous AREG restored erlotinib sensitivity. HSC-6 cells and FaDu cells engineered to express MAPK1E322K were pretreated with DMSO, VX-11e (0.5 μM) or combination of VX-11e (0.5 μM) and AREG (300 pg/ml) for 24 hours. Cells were then treated with two concentrations of erlotinib or a combination of VX-11e (0.5μM) and erlotinib or the triple combination of VX-11e (0.5 μM) and AREG (300pg/ml) and erlotinib for 48 hours. Cell survival was measured by crystal violet dye extraction growth assay and plotted relative to DMSO vehicle control (erlotinib alone) or VX-11e control (combination of VX-11e and erlotinib) or VX-11e and AREG control (combination of VX-11e, AREG and erlotinib). A. MAPK1 inhibition by VX-11e decreases sensitivity to erlotinib in HSC-6 cells, while exogenous AREG restored sensitivity. B. MAPK1 inhibition by VX-11e decreased sensitivity to erlotinib in FaDu cells engineered to express MAPK1E322K, while exogenous AREG restored its sensitivity. Decreased sensitivity to erlotinib with MAPK1 inhibition was attenuated in vector control transfected FaDu cells C. and FaDu cells engineered to express MAPK1WT D. (n = 3. *p < 0.05, **p < 0.01, ***p < 0.001). Similar results were obtained with triplicate wells in three independent experiments.
Knockdown of AREG decreases EGFR-MAPK pathway activation
To further test the contribution of AREG production to erlotinib sensitivity in the setting of MAPK1E322K, we used shRNA to knockdown AREG expression. We first generated HSC-6 cells with stable green fluorescent protein (GFP) control or AREG shRNA expression by lentiviral particle transduction. As shown in Figure 5A, HSC-6 cells with AREG knockdown secreted much lower levels of AREG compare to controls. AREG knockdown in HSC-6 cells also reduced expression levels of p-EGFR (Y1068) and p-p44/42 MAPK (Figures 5B-5D). Therefore, EGFR signaling activation in cells harboring MAPK1E322K is dependent on AREG secretion.
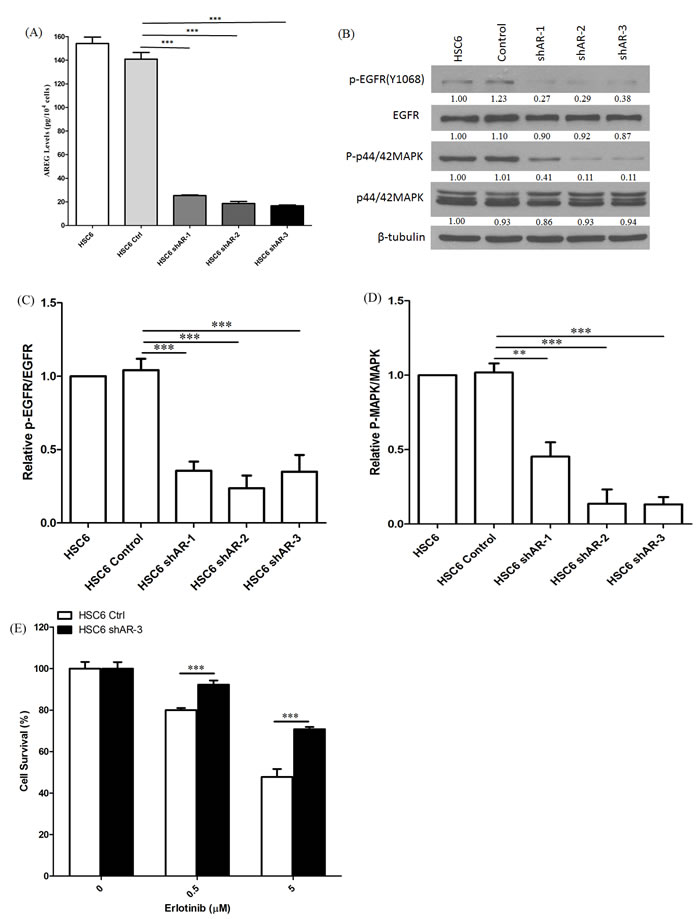
Figure 5: Knockdown of AREG reduced AREG secretion and EGFR-MAPK pathway activation. A. Conditioned media from HSC-6 cells transfected with GFP control or shAREG was analyzed by ELISA. Assays were performed in triplicate. ***p < 0.001. Similar results were obtained with triplicate wells in three independent experiments. B. AREG knockdown lead to decreased expression of P-EGFR (Y1068) and P-p42/44 MAPK in HSC-6 cells by immunoblotting. C. Densitometry analysis of EGFR phosphorylation. P-EGFR was normalized to EGFR as a loading control. Cumulative results are shown from three independent experiments. ***p < 0.001. D. Densitometry analysis of MAPK phosphorylation. P-p42/44 MAPK was normalized to p42/44 MAPK. Cumulative results from three independent experiments are shown. **p < 0.01, ***p < 0.001. E.. Depletion of AREG by shRNA decreased erlotinib sensitivity in MAPK1E322K cells. HSC-6 cells expressing GFP-control or shAREG were treated with erlotinib at the indicated concentrations. Cell survival was measured by crystal violet dye extraction growth assay and plotted relative to DMSO vehicle control. (n = 3, ***p < 0.001). Similar results were obtained in three independent experiments as well as other HSC-6 cell clones with shAREG knockdown.
AREG knockdown decreases erlotinib sensitivity in MAPK1E322K cells
To determine the contribution of AREG production to erlotinib sensitivity in MAPK1E322K mutated HNSCC models, we compared erlotinib responses in HSC-6 cells in the setting of AREG knockdown by shRNA compared with GFP-control. As shown in Figure 5E, HSC-6 cells with shAREG knockdown were significantly less sensitive to erlotinib compared to GFP-control. These data strongly indicate that MAPK1-driven AREG production mediates activation of cellular EGFR signaling and confers sensitivity to erlotinib in the setting of MAPK1E322K.
Given that MAPK1E322K increased AREG secretion and knockdown of AREG resulted in decreased erlotinib sensitivity, we next evaluated the ability of exogenous AREG to restore erlotinib sensitivity in HSC-6 cells under conditions of AREG knockdown. To test the effects of exogenous AREG under conditions of AREG knockdown, we pretreated cells with AREG for 24 hours, then treated cells with combinations of erlotinib (0, 0.5 or 5μM) and AREG for 48 hours. We compared cell viability in the presence or absence of supplemental AREG, and we observed that sensitivity to erlotinib was enhanced by exogenous AREG in HSC-6 cells under conditions of AREG knockdown (Figure S2).
Depletion of AREG expression in MAPK1E322K cells reduces tumor growth and sensitivity to erlotinib in vivo
Expression of MAPK1E322K was associated with increased secretion of AREG, and knockdown of AREG in MAPK1E322K cells resulted in an expected decrease in AREG secretion with concomitant diminished sensitivity to erlotinib in vitro. To validate these findings in vivo, we assessed the antitumor efficacy of erlotinib in endogenous MAPK1E322K HSC-6 mouse xenografts (control) and shAREG knockdown HSC-6 mouse xenografts. Consistent with our in vitro results, tumor growth was significantly suppressed in HSC-6 xenografts without AREG depletion (HSC-6-control groups) with erlotinib treatment (100 mg/kg) compared with vehicle control (p < 0.001) (Figure 6). Knockdown of AREG alone was associated with a suppression of tumor growth that was similar to that observed with erlotinib treatment of HSC-6 control xenografts (Figure 6C). The erlotinib effect was modest though significant in AREG depleted tumors (p < 0.05, Figure 6C). As shown in Figure 6C, the anti-tumor effects of erlotinib were significantly greater for HSC-6-control xenografts than HSC-6-shAR xenografts (p < 0.01), indicating that depletion of AREG decreased response to erlotinib in the setting of MAPK1E322K. Similarly, a more marked tumor weight reduction was observed upon erlotinib treatment in the HSC-6-control group versus the HSC-6-shAR group (p < 0.05, Figure 6D).
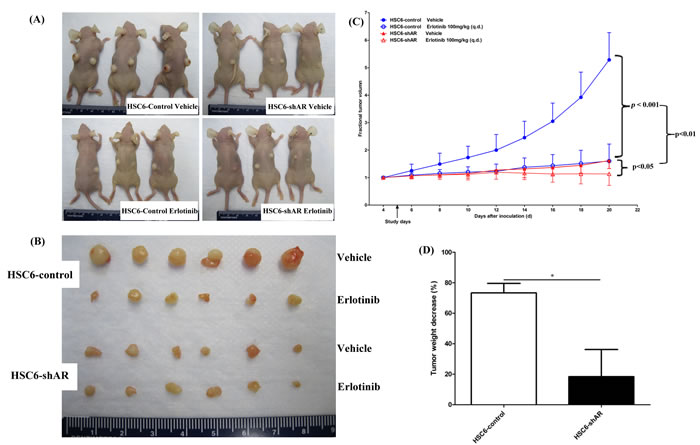
Figure 6: Depletion of AREG by shRNA decreased erlotinib sensitivity in MAPK1E322K xenografts. HSC-6 cells expressing GFP-control or AREG shRNA were selected and expanded. 2×106 cells were inoculated subcutaneously into both flanks of 5-6 week-old female nude mice (n = 6 per group). Four days after inoculation, tumor volumes were measured and the mice were divided into vehicle or erlotinib treatment groups. Erlotinib was administered at a dosage of 100 mg/kg daily by oral gavage. 10% HPBCD was administrated as vehicle control. Tumor volumes were measured every other day. A. Tumor bearing mice and B. excised tumors are shown. C. Erlotinib significantly suppressed tumor growth in HSC-6-control xenografts. (***p < 0.001). The erlotinib effect was significant but more modest in HSC-6-shAR xenografts (*p < 0.05). D. Tumor weights were significantly decreased by erlotinib in HSC-6-control xenografts to a greater degree than HSC-6-shAR xenografts (*p < 0.05).
Discussion
EGFR TKIs are effective in non-small cell lung cancer (NSCLC) patients with an activating mutation of the EGFR TK domain [21-23]. These mutations are not found in HNSCC and to date, predictive biomarkers for TKIs have been lacking in this malignancy. We recently reported a case of an HNSCC tumor harboring a MAPK1E322K somatic mutation that contributed to an exceptional clinical response to single agent erlotinib [6]. MAPK1 activity has generally been reported to be associated with resistance to EGFR TKIs in cancers other than HNSCC. MAPK1 amplification, which may also activate ERK signaling, leads to increased EGFR internalization through Thr-669, and confers EGFR TKI resistance in EGFR mutant NSCLC [24]. Similarly, in preclinical models of pancreatic cancer and lung cancer, inhibition of MAPK1 expression by siRNA or MAPK1 activity by MEK inhibitors sensitized specific cancer cell lines to erlotinib [25, 26]. Thus, the effect of two different ERK genomic alterations (point mutation or amplification of MAPK1) may have distinct clinical implications in different types of cancer. The paradoxical presence of an erlotinib sensitivity-conferring MAPK1 activating mutation in HNSCC warranted further mechanistic studies of this mutation.
We previously established that HNSCC cells harboring MAPK1E322K showed increased activation of EGFR and downstream signaling compared to MAPK1WT cells[6]. In the present study, we focused on further elucidating the mechanism underlying this exceptional erlotinib response. Our results indicate that endogenous MAPK1E322K was associated with higher secreted AREG levels compared to endogenous MAPK1WT. Exogenous expression of MAPK1E322K also increased AREG secretion compared to vector control or MAPK1WT expression in an engineered HNSCC model. Consistent with literature findings that enhanced secretion of AREG leads to hyperactivation of EGFR signaling [27, 28], our results support the hypothesis that MAPK1E322K drives increased secretion of AREG, creating an enhanced feedback autocrine loop involving AREG, EGFR and downstream signaling. This conclusion is further supported by evidence that MAPK1 targeting resulted in reduced secretion of AREG in the MAPK1E322K cell line.
AREG, a ligand of EGFR, is synthesized as a transmembrane precursor that undergoes a series of proteolytic processes to yield a mature secreted form [27]. The binding of AREG to EGFR induces autophosphorylation of the EGFR intracellular tyrosine kinase domain, which activates the downstream signaling pathways, including the MAPK pathway. Consequently, growth, proliferation, migration, or invasiveness are enhanced after the activation of EGFR [29].
Our results demonstrated that MAPK1E322K increased AREG secretion. This dependency on AREG-EGFR signaling conferred erlotinib sensitivity, while depletion of AREG reduced EGFR signaling and lead to decreased sensitivity to erlotinib in vitro and in vivo. It has been reported that autocrine ligand production can predict sensitivity to gefitinib in wild type EGFR cancers (10 NSCLC lines and 4 HNSCC lines), as gefitinib was significantly more effective at inhibiting the growth of high AREG-producing cell lines compared with the low AREG-producing cells[16]. Rogers et al. also reported that AREG secretion showed a positive correlation with sensitivity to gefitinib in a panel of 18 HNSCC cell lines [30]. A study comparing AREG expression in 24 EGFR wild type NSCLC patients found AREG expression was significantly higher in NSCLC patients who developed stable disease following gefitinib or erlotinib treatment compared with those who developed disease progression [16]. Another study in 73 WT EGFR NSCLC showed that overall survival and progression-free survival were significantly longer in AREG-positive patients compared to AREG-negative patients[17]. Exploratory molecular analyses of a phase II trial in pancreatic carcinoma, showed patients with high baseline serum AREG levels might benefit from erlotinib [31].
In contrast, increased levels of serum AREG have been correlated with a lack of benefit from gefitinib treatment in patients with advanced NSCLC [32, 33], and in an independent study AREG overexpression was reported to promote resistance to gefitinib-induced apoptosis rather than sensitivity in KRAS mutant NSCLC cell lines [34, 35]. These discrepancies may be a result of different cancer types, use of different cell lines, heterogeneous methods used to detect AREG expression, and/or differences in AREG concentrations in the local tumor microenvironment and the systemic circulation. Our results are consistent with increased AREG secretion leading to EGFR signaling-dependency and erlotinib sensitivity in wild type EGFR HNSCC.
There are limitations to this study. To our knowledge only one cell line with endogenous MAPK1E322K has been identified to date according to the literature, the Cancer Cell Line Encyclopedia and the Catalogue of Somatic Mutations in Cancer [12, 36]. The study of additional cell lines harboring this same mutation would be more powerful. The use of engineered cell lines provide the opportunity to evaluate the effects of the mutation given a specific context. However, these cells lack a natural selection history that likely accompanies an advantage-conferring mutation. Therefore, results may not exactly phenocopy cell lines harboring endogenous MAPK1E322K. Given these caveats, the mechanism of enhanced AREG secretion and increased erlotinib sensitivity that accompanies MAPK1E322K in the engineered and endogenous cell line studies is compelling. It will be important to further elucidate the mechanisms contributing to MAPK1E322K and AREG enhanced erlotinib sensitivity in order to molecularly define patients who may be candidates for EGFR TKI treatment in addition to those whose tumors harbor this mutation.
In summary, our studies revealed an essential role for MAPK1E322K in mediating erlotinib sensitivity. We identified autocrine AREG-EGFR signaling as mediating the erlotinib sensitivity resulting from MAPK1E322K. Thus, activation of AREG-EGFR signaling might confer erlotinib treatment benefit for the subset of patients with tumors harboring MAPK1E322K mutation.
Materials and Methods
Cell culture
Three HNSCC cell lines were selected based on their MAPK1 genotype: FaDu (MAPK1 hemizygous wild type), Cal33 (MAPK1 wild type (MAPK1WT)) and HSC-6 (MAPK1E322K). The HSC-6 cell line was a kind gift from Prof. Johji Inazawa (Tokyo Medical and Dental University, Japan). A set of engineered FaDu cell lines exogenously expressing GFP, MAPK1WT or MAPK1E322K were generated by retrovirus transduction. Cells were maintained in Dulbecco’s modified Eagle’s medium (Mediatech, Manassas, VA, USA) supplemented with 10% inactivated fetal bovine serum (FBS; Lonza, Walkersville, MD, USA) and 1% penicillin/streptomycin (Mediatech). Cells were incubated at 37℃ with 5% CO2.
Reagents and antibodies
Erlotinib (Tarceva™) was obtained from OSI Pharmaceuticals (Uniondale, NY). The MAPK1 inhibitor VX-11e was purchased from MedChem Express, and was purchased from Peprotech. Crystal Violet was purchased from Sigma. The following antibodies were purchased from commercial sources: anti-phospho-EGFR (Tyr1068; CST-3777s); anti-EGFR (BD Biosciences-610017); anti-p44/42 MAPK (CST-9102s); anti-phospho-p44/42 MAPK (CST-4370s); anti-phospho-Rsk1 (EMD Millipore-04-419); anti-RSK (BD Biosciences-610226); anti-V5 tag (Abcam-ab27671) and anti-beta-Tubulin (Abcam-ab6046).
Senescence assay
β-galatosidase activity at pH 6 was detected in senescent cells by light microscopy (100x) following staining using the senescence staining kit (Cell Signaling Technology, USA). Number of senescent cells and total number of cells per field were analyzed for at least 5 fields for each cell type and treatment conditions. Representative results from 3 independent experiments are presented. Representative staining pictures are provided in Figure S1.
Western blotting analysis
Western blot analyses were performed as previously described [37].
ELISAs
The concentration of secreted AREG protein in cultured media was determined using the AREG Quantikine ELISA kit (R&D Systems-DAR00). The concentrations of TGF-α (R&D Systems-DTGA00), EGF (R&D Systems-DEG00) and HB-EGF (Abcam-ab100531) were also measured in the same conditioned media using specific Quantikine ELISA kits. In brief, 2×104 cells/well were seeded in the 48-well plates and cultured overnight. Cells were then washed with 1×phosphate-buffered saline (PBS) once and incubated in 0.5 ml of complete medium for 24 h. The media were collected and centrifuged and supernatant was collected and stored at -80℃ prior to analysis. Cells were trypsinized and counted. ELISA assays were performed according to the manufacturer’s protocols. Samples were run in triplicate, and secreted AREG was normalized against standard curves and presented as pg per 1×104 cells.
Viral transduction
To generate retroviruses, plasmids encoding GFP (vector control), MAPK1WT or MAPK1E322K were transfected into Platinum-A cells using FuGENE HD Transfection Reagent (Promega, Madison, USA). Virus particles were collected 72 hours post transfection. For viral infection, targeted cells were infected on three consecutive dates with virus in fresh complete medium containing Polybrene (14 μg/ml; Sigma, St Louis, MO, USA) then replaced with fresh medium. To generate AREG knockdown in cells, HSC-6 cells were infected with AREG-targeting short-hairpin RNA (shRNA) lentiviral particles (sc-39412-V, Santa Cruz Biotechnology, USA) and selected by puromycin according to the manufacturer’s protocols. Cell colonies expressing AREG shRNA were selected and assayed by enzyme-linked immunosorbant assays (ELISA) and Western blotting.
siRNA transfection
Cal33 and HSC-6 cells were transfected with 5 uM non-targeting small interfering RNA (siRNA) (OriGene-SR30004) or MAPK1-targeting siRNA (OriGene-SR303751) using the siTran1.0 siRNA Transfection Reagent (OriGene-TT300001) according to the manufacturer’s protocol. Briefly, cells were apportioned into T25 flasks and transfected 24 hours later with 5 uM control siRNA or MAPK1-targeting siRNA. At 48 hours after transfection, cells were trypsinized and seeded into 48-well plates. ELISA quantification was performed using 24-hour conditioned media. In parallel, cells protein samples were isolated, and the efficiency of knockdown was analyzed by western blotting.
Cell proliferation assay
The effects of erlotinib on cell line proliferation were determined by crystal violet dye extraction growth assay. Briefly, exponentially growing cells were seeded into 12-well plates at a low cell density of 2.5×104 cells/well in complete medium, cultured overnight and treated with indicated drugs. Crystal violet dye extraction growth assays were performed at the end of treatment. Cells were washed twice with 1× PBS, fixed with 96% ethanol solution for ten minutes at room temperature then stained with 0.1% crystal violet solution for 30 min at room temperature with gentle shaking. Stained cells were washed with distilled water 3 times. Crystal violet dye was extracted using 1% SDS and absorbance was determined at 570nm. Assays were performed in triplicate.
In vivo studies
Animal studies were conducted following a protocol approved by institutional animal care and use committee of the University of Pittsburgh. HSC-6 cells expressing GFP or AREG shRNA were selected and expanded in vitro. Then, 2 million cells were inoculated subcutaneously into each flank of 5-6 week-old female nude mice (Harlan Laboratories). Beginning at four days following inoculation, tumor volumes were measured daily by dial caliper. Tumor volumes in mm3 were calculated using the formula: tumor volume = (length×width2)/2. Tumor-bearing mice inoculated with the same cancer cells were randomized into the 2 treatment groups (3 mice, 6 tumors per group) to receive vehicle or erlotinib. Erlotinib (final concentration 10 mg/ml) was dissolved in a solvent of 10% (2-Hydroxypropyl)-β-cyclodextrin (HPBCD, Sigma-H107) in sterile water. Erlotinib was administered at a dose of 100 mg/kg by oral gavage once daily. 10% of HPBCD was administrated as vehicle control. Mice were sacrificed when the study was terminated and tumor weights were measured.
Statistical analysis
Student’s t-test was used to analyze data from ELISA, cell proliferation, immunoblotting densitometry and in vivo xenograft experiments in GraphPad Prism software package (GraphPad Prism software, San Diego, CA, USA). Data were considered significant at p < 0.05. Representative data are presented as means ± SD.
Acknowledgments
The study is supported by the National Cancer Institute Head and Neck Cancer SPORE P50 CA097190 (JRG), the National Cancer Institute K07 CA137140 (AME), and Sun Yat-Sen University 5010 Plan 2010004 (WW, YW).
Conflicts of interest
Dr. Grandis receives research support from Novartis, OSI and Bristol Meyers Squibb.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Yoshioka H, Hotta K, Kiura K, Takigawa N, Hayashi H, Harita S, Kuyama S, Segawa Y, Kamei H, Umemura S, Bessho A, Tabata M, Tanimoto M and Okayama Lung Cancer Study G. A phase II trial of erlotinib monotherapy in pretreated patients with advanced non-small cell lung cancer who do not possess active EGFR mutations: Okayama Lung Cancer Study Group trial 0705. J Thorac Oncol. 2010; 5:99-104.
2. Rizvi NA, Rusch V, Pao W, Chaft JE, Ladanyi M, Miller VA, Krug LM, Azzoli CG, Bains M, Downey R, Flores R, Park B, Singh B, Zakowski M, Heelan RT, Shen R, et al. Molecular characteristics predict clinical outcomes: prospective trial correlating response to the EGFR tyrosine kinase inhibitor gefitinib with the presence of sensitizing mutations in the tyrosine binding domain of the EGFR gene. Clin Cancer Res. 2011; 17:3500-3506.
3. Kobayashi T, Koizumi T, Agatsuma T, Yasuo M, Tsushima K, Kubo K, Eda S, Kuraishi H, Koyama S, Hachiya T and Ohura N. A phase II trial of erlotinib in patients with EGFR wild-type advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2012; 69:1241-1246.
4. Mody K, Strauss E, Lincer R and Frank RC. Complete response in gallbladder cancer to erlotinib plus gemcitabine does not require mutation of the epidermal growth factor receptor gene: a case report. BMC Cancer. 2010; 10:570.
5. Cohen EE, Lingen MW, Martin LE, Harris PL, Brannigan BW, Haserlat SM, Okimoto RA, Sgroi DC, Dahiya S, Muir B, Clark JR, Rocco JW, Vokes EE, Haber DA and Bell DW. Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR. Clin Cancer Res. 2005; 11:8105-8108.
6. Van Allen EM, Lui VW, Egloff AM, Goetz EM, Li H, Johnson JT, Duvvuri U, Bauman JE, Stransky N, Zeng Y, Gilbert BR, Pendleton KP, Wang L, Chiosea S, Sougnez C, Wagle N, et al. Genomic Correlate of Exceptional Erlotinib Response in Head and Neck Squamous Cell Carcinoma. JAMA Oncol. 2015; 1:238-244.
7. Arvind R, Shimamoto H, Momose F, Amagasa T, Omura K and Tsuchida N. A mutation in the common docking domain of ERK2 in a human cancer cell line, which was associated with its constitutive phosphorylation. Int J Oncol. 2005; 27:1499-1504.
8. Mahalingam M, Arvind R, Ida H, Murugan AK, Yamaguchi M and Tsuchida N. ERK2 CD domain mutation from a human cancer cell line enhanced anchorage-independent cell growth and abnormality in Drosophila. Oncol Rep. 2008; 20:957-962.
9. Valiathan GM, Thenumgal SJ, Jayaraman B, Palaniyandi A, Ramkumar H, Jayakumar K, Bhaskaran S and Ramanathan A. Common docking domain mutation E322K of the ERK2 gene is infrequent in oral squamous cell carcinomas. Asian Pac J Cancer Prev. 2012; 13:6155-6157.
10. Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517:576-582.
11. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevino V, Vazquez-Santillan K, Guadarrama AS, Wright AA, Rosenberg MW, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014; 506:371-375.
12. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603-607.
13. Blanchet S, Ramgolam K, Baulig A, Marano F and Baeza-Squiban A. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2004; 30:421-427.
14. Chokki M, Eguchi H, Hamamura I, Mitsuhashi H and Kamimura T. Human airway trypsin-like protease induces amphiregulin release through a mechanism involving protease-activated receptor-2-mediated ERK activation and TNF alpha-converting enzyme activity in airway epithelial cells. FEBS J. 2005; 272:6387-6399.
15. Gusenbauer S, Zanucco E, Knyazev P and Ullrich A. Erk2 but not Erk1 regulates crosstalk between Met and EGFR in squamous cell carcinoma cell lines. Mol Cancer. 2015; 14:54.
16. Yonesaka K, Zejnullahu K, Lindeman N, Homes AJ, Jackman DM, Zhao F, Rogers AM, Johnson BE and Janne PA. Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res. 2008; 14:6963-6973.
17. Chang MH, Ahn HK, Lee J, Jung CK, Choi YL, Park YH, Ahn JS, Park K and Ahn MJ. Clinical impact of amphiregulin expression in patients with epidermal growth factor receptor (EGFR) wild-type nonsmall cell lung cancer treated with EGFR-tyrosine kinase inhibitors. Cancer. 2011; 117:143-151.
18. Zhang Q, Thomas SM, Xi S, Smithgall TE, Siegfried JM, Kamens J, Gooding WE and Grandis JR. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004; 64:6166-6173.
19. Chaikuad A, Tacconi EM, Zimmer J, Liang Y, Gray NS, Tarsounas M and Knapp S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol. 2014; 10:853-860.
20. Tanoue T and Nishida E. Molecular recognitions in the MAP kinase cascades. Cell Signal. 2003; 15:455-462.
21. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497-1500.
22. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J and Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129-2139.
23. Inoue A, Suzuki T, Fukuhara T, Maemondo M, Kimura Y, Morikawa N, Watanabe H, Saijo Y and Nukiwa T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol. 2006; 24:3340-3346.
24. Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, Tadi M, Rogers A, Repellin C, Capelletti M, Maertens O, Goetz EM, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012; 2:934-947.
25. Diep CH, Munoz RM, Choudhary A, Von Hoff DD and Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011; 17:2744-2756.
26. Buonato JM and Lazzara MJ. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014; 74:309-319.
27. Chen JC, Chen YJ, Lin CY, Fong YC, Hsu CJ, Tsai CH, Su JL and Tang CH. Amphiregulin enhances alpha6beta1 integrin expression and cell motility in human chondrosarcoma cells through Ras/Raf/MEK/ERK/AP-1 pathway. Oncotarget. 2015; 6:11434-11446. doi: 10.18632/oncotarget.3397.
28. Hosur V, Johnson KR, Burzenski LM, Stearns TM, Maser RS and Shultz LD. Rhbdf2 mutations increase its protein stability and drive EGFR hyperactivation through enhanced secretion of amphiregulin. Proc Natl Acad Sci U S A. 2014; 111:E2200-2209.
29. Busser B, Coll JL and Hurbin A. The increasing role of amphiregulin in non-small cell lung cancer. Pathol Biol (Paris). 2009; 57(7-8):511-512.
30. Rogers SJ, Box C, Chambers P, Barbachano Y, Nutting CM, Rhys-Evans P, Workman P, Harrington KJ and Eccles SA. Determinants of response to epidermal growth factor receptor tyrosine kinase inhibition in squamous cell carcinoma of the head and neck. J Pathol. 2009; 218:122-130.
31. Propper D, Davidenko I, Bridgewater J, Kupcinskas L, Fittipaldo A, Hillenbach C, Klughammer B and Ducreux M. Phase II, randomized, biomarker identification trial (MARK) for erlotinib in patients with advanced pancreatic carcinoma. Ann Oncol. 2014; 25:1384-1390.
32. Ishikawa N, Daigo Y, Takano A, Taniwaki M, Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, Tsuchiya E, Kohno N and Nakamura Y. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res. 2005; 65:9176-9184.
33. Masago K, Fujita S, Hatachi Y, Fukuhara A, Sakuma K, Ichikawa M, Kim YH, Mio T and Mishima M. Clinical significance of pretreatment serum amphiregulin and transforming growth factor-alpha, and an epidermal growth factor receptor somatic mutation in patients with advanced non-squamous, non-small cell lung cancer. Cancer Sci. 2008; 99:2295-2301.
34. Busser B, Sancey L, Josserand V, Niang C, Favrot MC, Coll JL and Hurbin A. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Mol Ther. 2010; 18:528-535.
35. Jeannot V, Busser B, Brambilla E, Wislez M, Robin B, Cadranel J, Coll JL and Hurbin A. The PI3K/AKT pathway promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by a deacetylase-dependent mechanism. Int J Cancer. 2014; 134:2560-2571.
36. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA and Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC). Current Protocols in Human Genetics. 2008; Chapter 10:Unit 10 11.
37. Lui VW, Boehm AL, Koppikar P, Leeman RJ, Johnson D, Ogagan M, Childs E, Freilino M and Grandis JR. Antiproliferative mechanisms of a transcription factor decoy targeting signal transducer and activator of transcription (STAT) 3: the role of STAT1. Mol Pharmacol. 2007; 71:1435-1443.