
INTRODUCTION
Anaplastic lymphoma kinase (ALK) [1] is an important therapeutic target in cancer, despite the function of the wild-type protein being poorly understood. While having key roles in early nervous system development [2], the levels of wild-type ALK subsequently drop off and remain at low levels for the remainder of life. Compounding ALK’s enigmatic nature, the mammalian ligand(s) responsible for its activation are still up for debate [3–6]. Germline and acquired mutations in wild-type ALK, however, are found in the childhood cancer neuroblastoma [7–10]. Furthermore, chromosomal rearrangements fusing ALK’s C-terminal kinase domain to a number of constitutively expressed genes result in malignant transformation through activation of multiple oncogenic signaling pathways [11, 12].
In NPM-ALK, identified in 1994 [13], ALK is fused to the constitutively expressed nucleophosmin gene [14]. Approximately 70% of anaplastic large cell lymphomas (ALCL) are positive for this or similar fusions [15]. In fact, it was through the discovery of this fusion that ALK was originally cloned [13]. Subsequently, in 2007, ALK was found fused to echinoderm microtubule associated protein-like 4 (EML4) yielding the fusion kinase EML4-ALK seen in approximately 3–5% of non-small cell lung cancers (NSCLC) [16, 17]. Several other ALK fusions since have been identified, including the RANBP2 (RNA binding protein 2)-ALK fusion seen in inflammatory myofibroblastic tumor (IMT, reviewed in [14]).
Since the highly successful use of imatinib and other BCR-ABL tyrosine kinase inhibitors (TKIs) against chronic myeloid leukemia [18], there have been great efforts to find inhibitors that turn off other such kinases [19]. In 2011, a mere four years after the discovery of EML4-ALK, the U.S. FDA approved the dual ALK/MET TKI crizotinib for ALK+ NSCLC. While initial response to crizotinib may be strong [20–24], patients inevitably succumb due to acquired resistance through multiple mechanisms, including kinase-domain mutations, prompting development of newer generation inhibitors (Table 1).
Table 1: Kinase domain mutations leading to acquired resistance
Cell line |
Observed mutation from this study |
Variants observed by others |
Disease observed in |
Reported resistance phenotype |
Reference |
|---|---|---|---|---|---|
SUP-CR500-2 |
I1171S |
I1171S |
NPM-ALK ALCL |
ASP-3026-R* |
[39] |
EML4-ALK NSCLC |
Crizotinib-R Alectinib-R |
[38] |
|||
I1171T |
NPM-ALK ALCL |
Crizotinib-R Ceritinib-R Alectinib-R AP26113-R ASP3026-R |
|||
EML4-ALK NSCLC |
Crizotinib-R Ceritinib-S Alectinib-R |
||||
I1171N |
NPM-ALK ALCL |
Crizotinib-R Ceritinib-R Alectinib-R AP26113-R ASP3026-S |
|||
EML4-ALK NSCLC |
Alectinib-R |
||||
Neuroblastoma |
Somatic |
||||
I1171H |
EML4-ALK NSCLC |
Crizotinib-R |
[36] |
||
SUP-LR150 |
F1174L |
F1174L |
EML4-ALK NSCLC |
Crizotinib-R Alectinib-S |
|
RANBP2-ALK IMT |
Crizotinib-R |
[56] |
|||
Neuroblastoma |
Somatic |
||||
F1174V |
NPM-ALK ALCL |
Crizotinib-S Ceritinib-S Alectinib-R AP26113-S ASP3026-R |
[60] |
||
EML4-ALK NSCLC |
Crizotinib-R Ceritinib-R |
||||
Neuroblastoma |
Somatic |
||||
F1174C |
NPM-ALK ALCL |
Alectinib-R |
[46] |
||
EML4-ALK NSCLC |
Crizotinib-R Ceritinib-R |
||||
Neuroblastoma |
Somatic |
||||
F1174I |
NPM-ALK ALCL |
Crizotinib-S Ceritnib-S Alectinib-S AP26113-S ASP3026-R |
[39] |
||
Neuroblastoma |
Somatic |
||||
F1174S |
Neuroblastoma |
Somatic |
[73] |
||
DHL1-CR500 |
R1192P |
R1192P |
Neuroblastoma |
Germline |
[10] |
R1192Q |
Uterine leiomyosarcoma |
|
[74] |
||
DHL1-LR150 |
T1151M |
T1151M |
Neuroblastoma |
Germline |
|
T1151R |
Neuroblastoma |
Germline |
[75] |
||
T1151K |
EML4-ALK NSCLC |
Crizotinib-R |
[36] |
||
1151Tins |
EML4-ALK NSCLC |
Crizotinib-R Ceritinib-R AP26113-R ASP3026-R |
|||
DHL1-CR500-2 |
G1269A |
G1269A |
NPM-ALK ALCL |
Crizotinib-R Ceritinib-S Alectinib-R AP26113-S ASP3026-R |
[64] |
EML4-ALK NSCLC |
Crizotinib-R Ceritinib-S Alectinib-R [64] AP26113-S ASP3026-R |
||||
G1269S |
EML4-ALK NSCLC |
Crizotinib-R |
Abbreviations: ALCL = Anaplastic Large Cell Lymphoma; NSCLC = Non-Small Cell Lung Cancer; IMT = Inflammatory Myofibroblastic Tumor; R = Resistant; S = Sensitive.
*Seen by ultra-deep sequencing at low frequency.
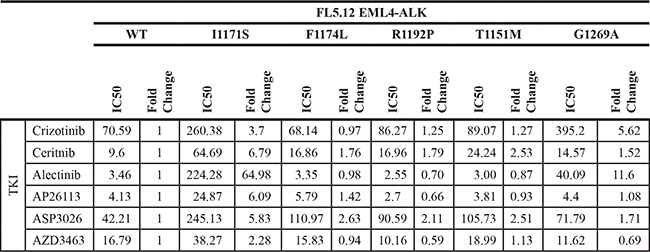
Here we assess the resistance/sensitivity profiles of mutations that arose in patient derived-cell ALCL lines continually exposed to either crizotinib or the 2nd generation ALK/IGF-1R inhibitor ceritinib (LDK378; FDA approved in 2014 for treating ALK+ NSCLC patients who failed crizotinib) [25]. Each mutation was profiled against six ALK TKIs – crizotinib, ceritinib, alectinib (which recently received FDA approval for treating ALK+ NSCLC patients who failed crizotinib [26]), AP26113 (brigatinib; a dual ALK/EGFR inhibitor in phase I/II trials that received FDA breakthrough therapy designation in 2014 [19]), ASP3026 (an ALK TKI in phase I trials [27, 28]) and AZD3463 (a dual ALK/EGFR inhibitor in preclinical development) [19, 29, 30].
RESULTS
The ALK+ ALCL cell lines SUP-M2 and SU-DHL-1, which are highly sensitive to ALK inhibition (Figure 1A), were selected in increasing concentrations of either crizotinib or ceritinib to investigate mechanisms of acquired resistance. We previously showed resistance initially was caused by increased NPM-ALK expression in all of these selections, and that this ALK up-regulation induced TKI-dependency as drug withdrawal resulted in the death of resistant cells [31]. Individual subclones, however, were able to grow again without ALK inhibitor following prolonged passaging, leading to normalization of NPM-ALK expression. These lines were named after their respective parent lines (SUP or DHL1), the inhibitor they were grown in (CR for crizotinib resistance, LR for ceritinib (LDK378) resistance), and the top nanomolar concentration in which they were able to proliferate during selection. Despite restoration of baseline NPM-ALK expression each line still exhibited varying degrees of persistent TKI resistance. Sequencing of the ALK TKD by Sanger and deep sequencing methods had suggested second-site mutations could be driving resistance, but we did not further characterize these initial findings. For this report, we maintained resistant clones in their top TKI concentration and then twice repeated Sanger sequencing of cDNA amplified from NPM-ALK mRNA. This detected a single second-site mutation in each resistant sub-clone (Figure 1B). Two of the mutations (I1171S from SUP-CR500-2 and F1174L from SUP-LR-2) were present as single peaks in the sequencing, indicating homogeneous populations in the sub-clones following drug selections. The other three mutations (R1192P from DHL1-CR500, T1151M from DHL-LR150, and G1269A from DHL1-CR500-2) appeared together with underlying wild-type peaks, indicating heterogeneous cell populations. While some of these mutations have been observed previously in the context of ALK-fusion cancers, we characterize two novel mutations that thus far have only been observed in neuroblastoma – T1151M and R1192P – and two mutations not previously characterized in ALK+ ALCL (Table 1). Each mutation was modeled on an X-ray structure of the ALK kinase domain (Figure 1C; discussed further below).
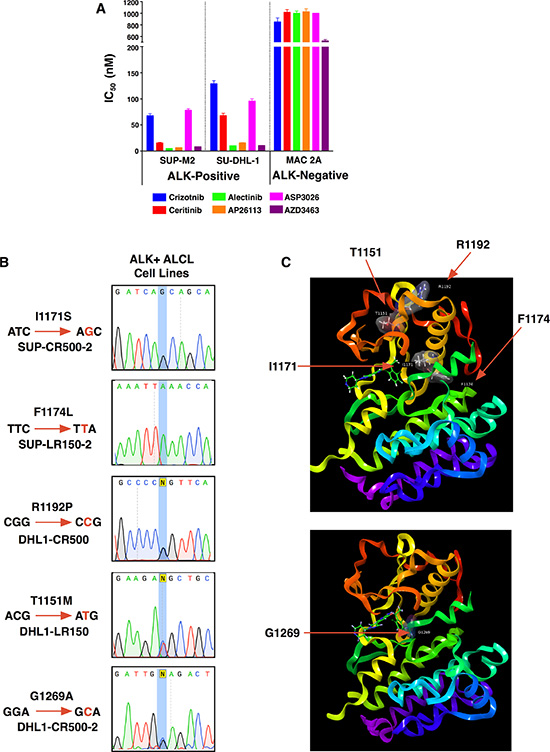
Figure 1: Acquired resistance mutations in patient-derived ALK+ ALCL cell lines. (A) IC50s of parental ALK+ ALCL cell lines (SUP-M2 and SU-DHL-1) as well as an ALK-negative ALCL line (MAC-2A). Mean ± SEM for quadruplicates. (B) Sanger sequencing identifying each resistance mutation in cell lines. (C) Location of the five mutations identified in this study with respect to the ALK kinase domain shown as ball and stick models with associated surfaces colored by atoms.
We first compared each subclone to its respective parent line for sensitivity to the TKI in which it had been selected (Figure 2A; Table 2). In all cases, subclones were significantly less sensitive, as determined by a highly significant increase in IC50, but additional factors could have arisen during selections to promote resistance. Furthermore, three of the five mutations were present in heterogeneous populations of cells also containing the wild-type NPM-ALK (as discussed above; Figure 1B). Therefore, to isolate the specific effect of each identified ALK-kinase mutation, we employed IL3-dependent FL5.12 murine pro-B cells as an independent system [32]. We generated each mutation through site-directed mutagenesis in NPM-ALK cloned into a GFP co-expressing MSCV-based vector (Supplementary Figure S1). Retroviral introduction of wild-type NPM-ALK or mutants, followed by IL3 withdrawal, transformed the FL5.12 cells from cytokine-dependence to oncogene-dependence (Figure 2B). Transformed lines withdrawn from IL3 proliferate only as 100% GFP+ clones, and dependence on NPM-ALK is further demonstrated by the failure of kinase-dead NPM-ALK or empty vector (not shown) to permit cytokine-independent growth. We then assessed sensitivity of lines transformed by each mutant to six TKIs in comparison to those transformed by wild-type NPM-ALK (Figure 2C; Supplementary Figure S2; Table 3). All five mutations exhibited significant cross-resistance to the three approved inhibitors, crizotinib, ceritinib, and alectinib. The two mutations derived from lines that had been selected in the second-generation inhibitor ceritinib, T1151M and F1174L, were pan-resistant to all five TKIs, though degree of resistance varied (discussed below). The other three mutations, from crizotinib-selected lines, remained sensitive to at least one other inhibitor. This independent system demonstrates resistance in ALK+ ALCL lines is driven at least in part by acquired ALK kinase-domain mutations, and confirms previous observations that mutations arising in response to one drug may affect multiple inhibitors of the same target.
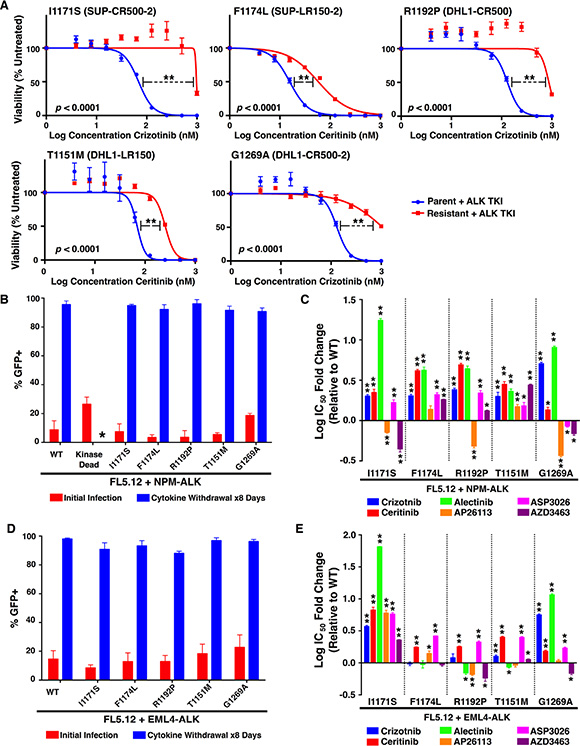
Figure 2: Resistance profiles of ALK mutations against six ALK TKIs. (A) Cell viability assays (with denoted IC50s) for each resistant line compared to the parental line from which they were derived. (B) Cellular transformation of FL5.12 cells infected with an MSCV-based vector co-expressing GFP and wild-type or mutant NPM-ALK constructs upon cytokine withdrawal. The kinase-dead mutant was unable to survive in the absence of cytokine (*). (C) IC50’s for each FL5.12 NPM-ALK construct against six ALK TKIs (see also Supplementary Figure S2). (D) Same as (B) but with mutations created in a retroviral vector containing EML4-ALK. (E) IC50’s for each FL5.12 EML4-ALK construct against six ALK TKIs (see also Supplementary Figure S4). Mean ± SEM for quadruplicates (A, C and E) or triplicates (B and D). (A, C and E) Unpaired two-tailed t-test was performed using GraphPad Prism version 6 to compare the IC50s for each mutant to their respective parental (A) or wild-type (C, E) cells. *p < 0.05, **p < 0.001.
Table 2: IC50s of each resistant line compared to the parent line from which they were derived
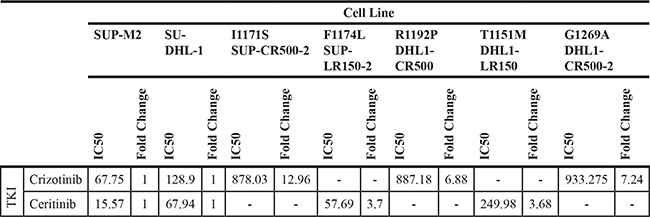
Table 3: FL5.12 NPM-ALK mutant IC50s
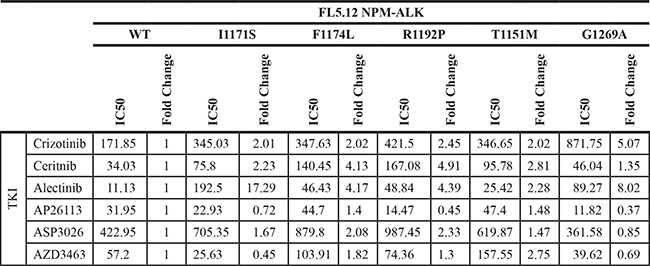
To further identify the potential clinical implications of each mutation, and to assess whether the effects of each mutation are fusion-specific, we employed an additional in vitro model. Each mutation was generated via site-directed mutagenesis in EML4-ALK, the most common ALK fusion detected in NSCLC, and cloned into the same GFP-expressing MSCV-based vector backbone described above (Supplementary Figure S3). Once again, using FL5.12 murine pro-B cells, retroviral introduction of each EML4-ALK mutant construct and subsequent IL3 withdrawal resulted in cytokine-independent, oncogene-dependent cellular transformation (Figure 2D). The resistance/sensitivity profiles of each EML4-ALK mutant versus wild-type EML4-ALK to the same panel of TKIs were assessed in the same manner as above (Figure 2E; Supplementary Figure S4; Table 4). For mutations previously observed in ALK TKI resistance models, our results were largely consistent with individual findings reported by others (Table 1). We observed some important differences, however, between the effects of particular mutations on NPM-ALK vs. EML4-ALK (compare Figure 2E and Figure 2C). In particular, R1192P and T1151M – never before detected in resistant EML4-ALK-driven cell lines or patient samples – had substantially greater effect on NPM-ALK in promoting resistance than on EML4-ALK. These findings among others highlight potentially clinically important differences mediated by ALK’s fusion partner affecting selection by tumors for particular mutations (see discussion).
Table 4: FL5.12 EML4-ALK mutant IC50s
We next assessed through immunoblotting the signaling consequences in FL5.12 cells of NPM-ALK-R1192P and T1151M, the two mutations not previously described as fusion ALK-kinase resistance mutations (Figure 3). Results suggest a particularly strong resistance phenotype of R1192P to all the inhibitors except AP26113, in line with the IC50 data. The effect of T1151M, meanwhile, was weaker, with higher concentrations of all drugs except ASP3026 overcoming its effects on ALK phosphorylation. We note that pERK levels decreased in crizotinib-treated FL5.12 cells not transfected with NPM-ALK (growing in IL3), and pERK is correspondingly more crizotinib sensitive in all the transformed lines as well. This suggests an off-target effect such as this drug’s known activity against MET [33]. Similar to our published findings, phosphorylation of ERK and AKT were overall variable in these results, providing inconsistent markers of drug potency [31]. STAT3 activation, however, (indicated by phosphorylation at Y705), which is known to be ALCL’s core survival pathway was strongly consistent with drug potency and ALK phosphorylation status [34, 35]. We therefore confirm drug-resistant ALK activation promoting ALCL’s core survival pathway mediated by NPM-ALK-R1192P and T1151M, consistent with viability data.
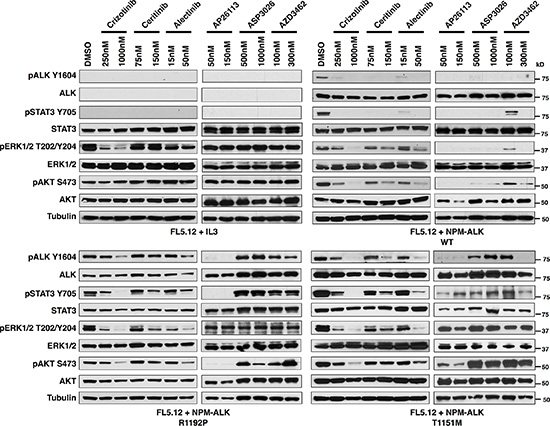
Figure 3: Activated ALK and downstream signaling is preserved in the novel ALK-fusion mutations, R1192P and T1151M. Immunoblotting for ALK and downstream signaling targets at the indicated concentrations of six ALK TKIs for FL5.12 cells (A), FL5.12 cells infected with an MSCV-based vector co-expressing GFP and wild-type NPM-ALK (B), NPM-ALK R1192P (C) or NPM-ALK T1151M (D).
DISCUSSION
Resistance to ALK-kinase inhibition in lung cancer is described in several published studies that employed cell lines, patient samples, or both. Our goal has been to understand resistance in ALK+ ALCL, which is less well studied. We previously reported resistance driven by NPM-ALK up-regulation, which also paradoxically drives dependence on continued inhibitor exposure, defining for the first time oncogene overdose by ALK over-activation [31]. Here, we examine five NPM-ALK kinase domain mutations that drive resistance in ALCL in an absence of over-expression. Two of these, R1192P and T1151M, are previously unreported as ALK TKI resistance mutations. We evaluated mutations in an independent system permitting isolation of mutation effect from other factors that may arise in ALK-dependent cells continually exposed to inhibitors. These experiments showed R1192P, T1151M, and the other mutations in NPM-ALK adversely affected the activity of multiple ALK TKIs but also pointed to inhibitors for which sensitivity is maintained or even enhanced. Interestingly, cross-validation using the lung cancer-derived EML4-ALK fusion showed both similarities and key differences in the effect of mutations on particular drugs. Our study therefore characterizes novel NPM-ALK TKI resistance mutations, providing guidance for appropriate choice of alternate therapies as more of these drugs move toward regulatory approval. It also highlights key differences in the effect of mutations on the ALK kinase domain’s sensitivity to inhibition depending on its fusion partner. For example, the fact that R1192P and T1151M have not previously been detected in resistant EML4-ALK-driven systems is not surprising since their effect on drug activity was significantly less for that kinase than for NPM-ALK. The profile of ALK TKI resistance mutations likely to arise in a tumor, therefore, is inherently shaped by ALK’s fusion partner, as should choices of alternative therapy.
I1171S
I1171S previously was seen in an in vitro accelerated mutagenesis screen in ALK+ NSCLC using increasing concentrations of crizotinib [36] and somatic mutations in this residue also have been reported in neuroblastoma [10, 37]. Subsequently, this mutation was identified in an ALK+ NSCLC patient as conferring alectinib resistance [38]. Consistent with its original identification in EML4-ALK-driven systems, we find I1171S is potently resistance-conferring to FL5.12 cells dependent on this fusion. All six TKIs tested had significantly increased IC50s when tested vs. EML4-ALK-I117S compared to wild-type (Figure 2E). In NPM-ALK, I1171S promoted resistance to crizotinib, the drug used in selecting the line in which it was isolated, ceritinib, and again pronounced resistance to alectinib. Ultra-deep sequencing previously identified I1171S at very low frequencies as a candidate ASP3026-resistant mutation in ALK+ ALCL [39], which is also confirmed in our functional system. Interestingly, however, NPM-ALK-I1171S promoted no resistance, indeed increased sensitivity compared to wild type, to both AP26113 and AZD3463. The effect of a resistance mutation conferring sensitivity to other inhibitors is similar to a recent high-profile case report in which the L1198F mutation, providing resistance to the third-generation inhibitor lorlatinib, resulted in resensitization to crizotinib [40]. The novel finding in our study is that this effect was seen for NPM-ALK but not EML4-ALK. Our data do not identify the reason for differential the effects of I1171S on AP26113 and AZD3463 (structurally similar compounds that also inhibit EGFR) between NPM-ALK and EML4-ALK. Given the FL5.12 system used is identical in all ways other than ALK’s fusion partner, the effect is likely on-target, mediated by subtle ATP binding-pocket differences between the two fusion kinases.
Several other amino acid substitutions occur at I1171 residue, all promoting resistance to both crizotinib and alectinib regardless of whether their characterization was in the context of NSCLC or ALCL (Table 1) [39, 41–46]. There are some differences in TKI resistance between our findings for I1171S and other amino acid substitutions at this residue. For instance, in lymphoma studies, I1171N and I1171T are reported to be AP26113-resistant [39, 41], and while both appear resistant to ceritinib in NPM-ALK [39, 41], they show sensitivity in EML4-ALK [43, 44, 47, 48]. Further variability exists for both I1171N and I1171T, with the former being ASP3026-sensitive [39, 42] and the latter resistant [39] in NPM-ALK.
I1171 is part of the hydrophobic regulatory spine (R-spine), along with residues C1182, F1271 (DFG motif), H1247 (HRD motif) and D1311 (F-helix), which connects the two major ALK TKD lobes. Mutations in I1171 are believed to lock ALK in its active conformation, accelerating its activation through autophosphorylation [37, 43, 49–52]. Additionally, mutations at this residue decrease stability of inhibitor binding at the DFG motif, and, in the case of at least I1171N, modify the structure of the kinase-inhibitor complex [41]. Our data suggest NPM-ALK+ ALCL patients who develop resistance due to I1171S could benefit from treatment using either AP26113 or AZD3463, as both overcome this mutation in our independent system. Other inhibitors may be necessary in the context of EML4-ALK.
F1174L
The phenylalanine residue at position 1174, located at the end of the αC helix amongst a highly hydrophobic cluster of residues, maintains autoinhibitory interactions when ALK is in its inactive conformation [37, 49, 51] and is a mutational hotspot within the ALK kinase domain [53]. We found F1174L in a cell line grown in increasing concentrations of ceritinib. It is a frequent somatic mutation in neuroblastoma and increases ALK’s affinity for ATP, despite not directly contacting the ATP pocket. Accordingly, resistance is not due to steric hindrance of drug binding, but rather promotion of ALK’s active conformation indicated by accelerated autophosphorylation [10, 37, 54]. F1174L therefore acts as both a TKI-resistance and an activating mutation [7–10, 37, 54]. In agreement with our observations for NPM-ALK (Table 1; Figure 2C; Supplementary Figure S2), F1174L results in resistance to crizotinib in both ALK+ NSCLC and ALK+ IMT [55–57], although we did not observe a significant change in resistance in our EML4-ALK in vitro assay (p = 0.4846; Table 4; Figure 2E; Supplementary Figure S4). In contrast to previous reports in ALK+ NSCLC [56–58] along with our EML4-ALK in vitro assay, we observe resistance to alectinib, which is more in agreement with this mutation favoring ATP binding over inhibitor binding, and suggests potential differences in the conformational changes induced by a leucine substitution at this residue based on ALK’s fusion partner. Studies involving other amino acid substitutions at position 1174 support this. Whereas F1174C and F1174V are resistant to crizotinib and ceritinib in ALK+ NSCLC cell line models and patient samples [36, 47, 59], F1174V and F1174I are sensitive to both TKIs in ALK+ ALCL [39, 60].
Furthermore, despite F1174C and F1174V showing resistance to alectinib in ALK+ ALCL [46, 60], this drug is able to overcome an isoleucine substitution in the same model [39]. Additionally, while we observe a small fold-increase in the IC50 for AP26113 (1.4-fold) for this mutant compared to wild-type NPM-ALK, the increase is not statistically significant (p = 0.0513), and AP26113 has been reported to show sensitivity to both F1174V and F1174I [39, 60]. Our data for both fusions, in agreement with other studies in NPM-ALK, show ASP2036 is unable to overcome any amino acid substitutions at this location [39, 60]. Furthermore, while AP26113 may show slight efficacy in overcoming ALK+ ALCL resistance acquired by this mutation (p = 0.0513), it is predicted to be ineffective in ALK+ NSCLC, with the opposite being true for AZD3463 with respect to each disease. This further highlights the care required when considering treatment options to overcome resistance in patients, as both the specific substitution and the particular ALK-fusion may be key factors.
R1192P
The R1192P mutation, residing in the N-lobe, is one of the most frequent germline mutations in neuroblastoma [10]. In a similar fashion to both I1171 and F1174 mutations, R1192P results in accelerated autophosphorylation of ALK’s TKD [37]. In fact, this mutation is considered an exception, as most residues in the N-lobe have smaller impacts. We report here for the first time the identification of this mutation in any ALK-fusion cancer, in a cell line selected in crizotinib. Perhaps unsurprisingly, due to its ability to strongly turn on the kinase activity of ALK, all but one TKI proved to be ineffective at overcoming resistance to this mutation in NPM-ALK (Table 1; Figures 2C, 2D and 3; Supplementary Figure S2). The only exception, AP26113, may be an attractive therapeutic in the event of resistance by this mutation. The effect of this mutation was dramatically less vs. all drugs when it was introduced instead to EML4-ALK, likely explaining, as discussed above, why it has never been reported in resistant ALK+ lung cancer. Detailed structural determinations are outside the scope of this report, but it is interesting to note that all three mutations reported to activate ALK kinase rather than block drug binding to the ATP pocket – F1174L, R1192P, and T1151M (below) – have greater effect on drug activity when found in NPM-ALK than in EML4-ALK. Follow-up structural chemistry studies to this report may confirm and identify the basis for this effect.
T1151M
T1151M was identified recently in neuroblastoma patients [37] and we present here the first report of this mutation in the ALK-fusion context, arising in a cell line grown in ceritinib. An amino acid insertion, 1151Tins is well-characterized as causing cross-resistance to several ALK TKIs in ALK+ NSCLC cell lines and patients [25, 47, 61], although one study reports that alectinib is effective against it [58]. Additionally, 1151Tins was reported in neuroblastoma [62]. Finally, the amino acid substitution T1151K was also found in an accelerated mutagenesis screen in ALK+ NSCLC assessing crizotinib resistance [36].
Mutations at residue 1151, located at the N-terminal lobe of the ALK catalytic domain, alter affinity of the mutated kinase for ATP, and diminish inhibitor binding through conformational changes, despite lying some distance from the ATP pocket [47, 61]. Furthermore, mutations in this domain lead to activation of the ALK TKD, albeit more modestly than I1171, F1174, or R1192 [37]. It is therefore perhaps unsurprising that 1151Tins, T1151K, or, in our case, T1151M in NPM-ALK, cause cross-resistance to several ALK TKIs due to the mutant kinase domain strongly favoring ATP binding, as confirmed by fold-changes in IC50 and western blotting (Table 1; Figure 2C and 2D; Supplementary Figure S2). Somewhat surprising is the lack of significant change in response to AP26113 (p = 0.2090) and sensitivity to alectinib (p = 0.0169; in agreement with Kodama et al. [58] as mentioned above) when compared to wild-type EML4-ALK, further highlighted the importance of the particular amino acid substitution, as well as the potential differences in protein folding dependent upon the fusion partner in question.
G1269A
In agreement with our observations, this mutation resists crizotinib in both NPM-ALK (as expected as it was isolated from a cell line grown in crizotinib) and EML4-ALK, but is sensitive to AP26113 in both fusions [47, 63, 64]. A serine substitution at the same residue behaves similarly in EML4-ALK [36, 55]. G1269 lies in the ATP-binding pocket, making direct contact with crizotinib. Using computer modeling (Figure 1C), we observed that the dichlorofluorophenyl ring of crizotinib binds near G1269 (distance 3.49 Å between Ca G1269 and fluorine atom). G1269A causes a steric clash between the dichlorofluorophenyl ring and the alanine’s methyl group, apparently sufficient to disrupt the drug’s activities [47, 63, 65]. Much like T1151 mutations, G1269 mutations cause modest constitutive ALK tyrosine kinase activity [37].
While we observed resistance to ceritinib in both fusions (Table 1; Figure 2C; Supplementary Figure S2), others have shown that ceritinib effectively overcomes G1269A in both ALK+ NSCLC [25, 47] and ALK+ ALCL [64] due to it stabilizing ALK’s conformational dynamics and exhibiting increased potency for this mutant over WT ALK [48]. Once again, the degree of resistance in our study was only mild (1.35-fold for NPM-ALK and 1.52-fold for EML4-ALK), albeit still statistically significant (p = 0.0018 and < 0.0001, respectively), which may explain this difference. Another discrepancy is with ASP3026, predicted to overcome resistance in NPM-ALK by our data but unable to do so in a separate study in both ALK+ NSCLC and ALK+ ALCL [64]. However, the sensitivity reported in our study is just barely significant (p = 0.0495), perhaps explaining this discrepancy. Additionally, while we and Fontana and colleagues [64] report that alectinib is unable to overcome G1269A in the context of either disease, two other studies showed a response to alectinib in vitro, in xenograft models, and in a patient harboring this mutation [58, 66]. Borderline results in vitro may therefore be less reliable predictors of responses in vivo. Indeed, others have shown some TKIs with IC50 shifts in vitro may still cause tumor regression in vivo [47]. Further trial and error, including studies in resistant patients, will be necessary to clarify the situation for some mutations. Our data suggest AZD3463 may be able to overcome G1269A in both fusion-cancers. Both ASP3026 and AZD3463 interact differently compared to crizotinib with the active site near G1269. The X-ray crystal structure of ASP3026 with the ALK kinase domain (PDB code: 2XB7) shows that the isopropylsulfone group interacts with lysine 1150 through hydrogen bonding. This bond draws ASP3026’s binding away from G1269 (distance = 5.85 Å between G1269 Ca and methylene carbon), and it is expected that ASP3026 would bind G1269A mutant in similar fashion.
The ALK kinase has several mutational hotspots, such as F1174, F1245 and R1275 [53]. The identification of two mutations not previously reported in any malignancy driven by an ALK-fusion (T1151M and R1192P) is therefore highly informative. Despite a predilection for certain mutations conferring resistance, “novel” mutations at sites other than mutational hotspots can still occur, perhaps albeit at lower frequencies, as the tumor evolves in a desperate attempt to evade death. Our findings with mutations favoring ATP-binding and dampening inhibitor-binding and/or constitutively activating ALK’s TKD, suggest newer competitive inhibitors are desperately needed to overcome acquired resistance. One such inhibitor, the first 3rd generation ALK (and ROS1) inhibitor PF-06463922 (lorlatinib), exhibits increased potency against F1174L, 1151Tins, I1171T and G1269A in preclinical EML4-ALK models [67, 68], with impressive anti-tumour activity observed in xenograft neuroblastoma models harboring F1174L [53]. Another new inhibitor, a structural analog of alectinib, JH-VIII-157-02, also shows great promise against a series of ALK resistance mutations, including G1269A, F1174L and 1151Tins [69]. Further development and screening of newer generation ALK inhibitors is highly important in overcoming resistance mutations in patients suffering from ALK-related malignancies.
MATERIALS AND METHODS
Cell lines, reagents and inhibitors
RPMI 1640 and penicillin/streptomycin (P/S) supplemented with 10% fetal bovine serum (FBS) for SU-DHL-1 and MAC 2A, or with 20% FBS for SUP-M2. Phoenix cells grown in DMEM plus 10% FBS and P/S. FL5.12 cells cultured in RPMI 1640 with 10% FBS, P/S, ± 10% WeHi-3B supernatant and murine IL3 (400 ρM, eBioscience). All lines purchased from DSMZ apart from FL5.12’s (gift from Wendel lab). Crizotinib and alectinib were purchased from Selleck Chemicals; ceritinib (LDK378) was kindly provided by Novartis.
Cell viability assays
3000 cells per well were seeded in serial dilutions of the indicated ALK TKIs. Viability was assessed after 72 hours (Cell Titer Glo®; Promega) by measuring luminescence on a BioTek Synergy HT plate reader. GraphPad Prism version 6 was used to calculate IC50s with non-linear curve-fit regression.
Computer modeling
Images in Figure 1C were created using the X-ray structure of ALK kinase domain using the Maestro software (Schrodinger Inc.)
Identification of kinase-domain mutations
RNA extraction (RNeasy® Mini Ki; QIAgen) followed by cDNA synthesis (Taqman® Reverse Transcriptase kit; Roche) was carried out two independent times for all resistant cell lines as well as their respective parental lines. The NPM-ALK fusion was PCR amplified (Primers - Forward: GTCCGCCTTCTCTCCTACCT, Reverse: TTGGCACAAAACAAAACGTG) on a BioRad T100 Thermal Cycler. The kinase domain was sequenced by Sanger sequencing. The sequences obtained for the resistant lines were aligned to their respective parental lines using the ClustalW online sequence alignment tool to identify base pair changes. The identified mutations were the same for both independent sets of sequencing.
Protein extraction, quantification and immunoblotting
As described previously [31], loading 30 μg per lane, with all primary and secondary antibodies from Cell Signal Technology, developed using autoradiograph film (GeneMate).
Site directed mutagenesis
Plasmid purification (PerfectPrep Spin Mini Kit; 5Prime) was carried out for MSCV-based -NPM-ALK and -EML4-ALK vectors mutated using site directed mutagenesis (QuikChange II XL; Agilent Technologies). Sanger sequencing was then used to confirm the presence of mutations in the ALK TKD.
Transfections, infections and cellular transformation
As described previously [31] using an MSCV-based vector co-expressing GFP plus either wild-type NPM-ALK, wild-type EML4-ALK, each NPM-ALK mutation, the same mutations in EML4-ALK, or an NPM-ALK kinase dead mutation (K210R) as a negative control. Cellular transformation was assessed by flow cytometry (Guava EasyCyte).
ACKNOWLEDGMENTS
The MIG-NPM-ALK and MIG-NPM-ALK-Kd plasmids were generously provided by Dr. Emanuela Colombo (University of Milano, Milan, Italy). The pCDH1-MCS1-EML4-ALKv3-EF1-Puro plasmid was a generous gift from Dr. Robert Doebele (University of Colorado, USA). Custom DNA Constructs (Ohio, USA) cloned the EML4-ALK ORF into the MSCV-backbone.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by the NIH/NCGI grant 1R01CA190696-01 awarded to JHS.
Authors’ contributions
Conceived and designed the experiments: ADA and JHS; Performed the experiments: ADA, LL, SSR, VG, MJG, PP, EOT and MW; Analyzed the data: ADA, LL, SSR, VG, MJG, PP, EOT, MW and JHS; Wrote the paper: ADA and JHS.
REFERENCES
1. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013; 13:685–700.
2. Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, Mariame B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol. 2000; 156:1711–1721.
3. Guan J, Umapathy G, Yamazaki Y, Wolfstetter G, Mendoza P, Pfeifer K, Mohammed A, Hugosson F, Zhang H, Hsu AW, Halenbeck R, Hallberg B, Palmer RH. FAM150A and FAM150B are activating ligands for Anaplastic Lymphoma Kinase. Elife. 2015; 4:e09811.
4. Murray PB, Lax I, Reshetnyak A, Ligon GF, Lillquist JS, Natoli EJ, Jr., Shi X, Folta-Stogniew E, Gunel M, Alvarado D, Schlessinger J. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci Signal. 2015; 8:ra6.
5. Stoica GE, Kuo A, Aigner A, Sunitha I, Souttou B, Malerczyk C, Caughey DJ, Wen D, Karavanov A, Riegel AT, Wellstein A. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem. 2001; 276:16772–16779.
6. Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, Wellstein A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem. 2002; 277:35990–35998.
7. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008; 455:971–974.
8. George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S Gregor VE, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008; 455:975–978.
9. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008; 455:967–970.
10. Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008; 455:930–935.
11. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008; 8:11–23.
12. Webb TR, Slavish J, George RE, Look AT, Xue L, Jiang Q, Cui X, Rentrop WB, Morris SW. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther. 2009; 9:331–356.
13. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994; 263:1281–1284.
14. Pearson JD, Lee JK, Bacani JT, Lai R, Ingham RJ. NPM-ALK: The Prototypic Member of a Family of Oncogenic Fusion Tyrosine Kinases. J Signal Transduct. 2012; 2012:123253.
15. Ferreri AJ, Govi S, Pileri SA, Savage KJ. Anaplastic large cell lymphoma, ALK-positive. Crit Rev Oncol Hematol. 2012; 83:293–302.
16. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007; 131:1190–1203.
17. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561–566.
18. Moen MD, McKeage K, Plosker GL, Siddiqui MA. Imatinib: a review of its use in chronic myeloid leukaemia. Drugs. 2007; 67:299–320.
19. Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015; 8:17.
20. Bang YJ. The potential for crizotinib in non-small cell lung cancer: a perspective review. Ther Adv Med Oncol. 2011; 3:279–291.
21. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, Salgia R, Fidias P, Engelman JA, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012; 13:1011–1019.
22. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363:1693–1703.
23. Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, Ingle AM, Ahern C, Adamson PC, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013; 14:472–480.
24. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013; 368:2385–2394.
25. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, Riely GJ, Solomon BJ, Wolf J, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014; 370:1189–1197.
26. Song Z, Wang M, Zhang A. Alectinib: a novel second generation anaplastic lymphoma kinase (ALK) inhibitor for overcoming clinically-acquired resistance. Acta Pharmaceutica Sinica B. 2015; 5:34–37.
27. Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, Saito R, Mori K, Shindou N, Soga T, Sakagami H, Furutani T, Doihara H, et al. The selective anaplastic lymphoma receptor tyrosine kinase inhibitor ASP3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice. Mol Cancer Ther. 2014; 13:329–340.
28. Ono A, Murakami H, Wakuda K, Taira T, Kenmotsu H, Naito T, Ohde Y, Endo M, Nakajima T, Takahashi T. P6.01Dramatic response to ASP-3026 in patient with highly aggressive pulmonary inflammatory myofibroblastic tumor harboring ALK rearrangement. Ann Oncol. 2015; 26:ii28.
29. Farina F, Stasia A, Gambacorti Passerini C. Developments in anaplastic large-cell lymphoma: targeting the anaplastic lymphoma kinase. Blood Lymphat Cancer. 2014; 4:69–79.
30. Vijayvergia N, Mehra R. Clinical challenges in targeting anaplastic lymphoma kinase in advanced non-small cell lung cancer. Cancer Chemoth Pharm. 2014; 74:437–446.
31. Amin AD, Rajan SS, Liang WS, Pongtornpipat P, Groysman MJ, Tapia EO, Peters TL, Cuyugan L, Adkins J, Rimsza LM, Lussier YA, Puvvada SD, Schatz JH. Evidence Suggesting that Discontinuous Dosing of ALK Kinase Inhibitors May Prolong Control of ALK+ Tumors. Cancer Res. 2015; 75:2916–2927.
32. Algate PA, Steelman LS, Mayo MW, Miyajima A, McCubrey JA. Regulation of the interleukin-3 (IL-3) receptor by IL-3 in the fetal liver-derived FL5.12 cell line. Blood. 1994; 83:2459–2468.
33. Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011; 3:S7–S19.
34. Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, Karras JG, Levy DE, Inghirami G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005; 11:623–629.
35. Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E, Todaro M, Boi M, Acquaviva A, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer cell. 2015; 27:516–532.
36. Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, Moran L, Mohemmad QK, Anjum R, Wang Y, Narasimhan NI, Dalgarno D, Shakespeare WC, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011; 78:999–1005.
37. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, Hogarty MD, London WB, Radhakrishnan R, et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer cell. 2014; 26:682–694.
38. Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, Ross JS, Stephens PJ, Miller VA. Identification of a Novel HIP1-ALK Fusion Variant in Non-Small-Cell Lung Cancer (NSCLC) and Discovery of ALK I1171 (I1171N/S) Mutations in Two ALK-Rearranged NSCLC Patients with Resistance to Alectinib. J Thorac Oncol. 2014; 9:1821–1825.
39. Mologni L, Ceccon M, Pirola A, Chiriano G, Piazza R, Scapozza L, Gambacorti-Passerini C. NPM/ALK mutants resistant to ASP3026 display variable sensitivity to alternative ALK inhibitors but succumb to the novel compound PF-06463922. Oncotarget. 2015; 6:5720–5734. doi: 10.18632/oncotarget.3122.
40. Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, Burke BJ, Deng YL, Liu W, Dardaei L, Frias RL, Schultz KR, Logan J, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. New Engl J Med. 2016; 374:54–61.
41. Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013; 11:122–132.
42. George SK, Vishwamitra D, Manshouri R, Shi P, Amin HM. The ALK inhibitor ASP3026 eradicates NPM-ALK(+) T-cell anaplastic large-cell lymphoma in vitro and in a systemic xenograft lymphoma model. Oncotarget. 2014; 5:5750–5763. doi: 10.18632/oncotarget.2170.
43. Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, Iafrate AJ, Takeuchi K, Taiji M, Okuno Y, Fujita N, Engelman JA, Shaw AT. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014; 20:5686–5696.
44. Ou SH, Greenbowe J, Khan ZU, Azada MC, Ross JS, Stevens PJ, Ali SM, Miller VA, Gitlitz B. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung cancer. 2015; 88:231–234.
45. Toyokawa G, Hirai F, Inamasu E, Yoshida T, Nosaki K, Takenaka T, Yamaguchi M, Seto T, Takenoyama M, Ichinose Y. Secondary mutations at I1171 in the ALK gene confer resistance to both Crizotinib and Alectinib. J Thorac Oncol. 2014; 9:e86–87.
46. Zdzalik D, Dymek B, Grygielewicz P, Gunerka P, Bujak A, Lamparska-Przybysz M, Wieczorek M, Dzwonek K. Activating mutations in ALK kinase domain confer resistance to structurally unrelated ALK inhibitors in NPM-ALK-positive anaplastic large-cell lymphoma. J Cancer Res Clin Oncol. 2014; 140:589–598.
47. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S, Pferdekamper AC, Li J, Kasibhatla S, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014; 4:662–673.
48. Ni Z, Zhang TC. Computationally unraveling how ceritinib overcomes drug-resistance mutations in ALK-rearranged lung cancer. J Mol Model. 2015; 21:2716.
49. Bossi RT, Saccardo MB, Ardini E, Menichincheri M, Rusconi L, Magnaghi P, Orsini P, Avanzi N, Borgia AL, Nesi M, Bandiera T, Fogliatto G, Bertrand JA. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry. 2010; 49:6813–6825.
50. Cui JJ, Tran-Dube M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, McTigue M, Grodsky N, Ryan K, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011; 54:6342–6363.
51. Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA. 2006; 103:17783–17788.
52. Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci USA. 2008; 105:14377–14382.
53. Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, Li Y, Zou HY, Lee NV, Smeal T, Lemmon MA, Mosse YP. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016; 6:96–107.
54. Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, Cole K, Toporovskaya Y, Zhao H, Carpenter EL, Christensen JG, Maris JM, Lemmon MA, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011; 3:108ra114.
55. Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, Peifer M, Weiss J, Lovly CM, Grutter C, Rauh D, Pao W, Thomas RK. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011; 17:7394–7401.
56. Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, Gray NS, Wilner K, Christensen JG, Demetri G, Shapiro GI, Rodig SJ, Eck MJ, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010; 70:10038–10043.
57. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, Capelletti M, Shimamura T, Ercan D, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011; 71:6051–6060.
58. Kodama T, Tsukaguchi T, Yoshida M, Kondoh O, Sakamoto H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014; 351:215–221.
59. Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, Casey C, He J, Ali SM, Klempner SJ, Miller VA. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014; 9:549–553.
60. Ceccon M, Mologni L, Giudici G, Piazza R, Pirola A, Fontana D, Gambacorti-Passerini C. Treatment Efficacy and Resistance Mechanisms Using the Second-Generation ALK Inhibitor AP26113 in Human NPM-ALK-Positive Anaplastic Large Cell Lymphoma. Mol Cancer Res. 2015; 13:775–783.
61. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012; 4:120ra117.
62. Chand D, Yamazaki Y, Ruuth K, Schonherr C, Martinsson T, Kogner P, Attiyeh EF, Maris J, Morozova O, Marra MA, Ohira M, Nakagawara A, Sandstrom PE, et al. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis Model Mech 2013; 6:373–382.
63. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012; 18:1472–1482.
64. Fontana D, Ceccon M, Gambacorti-Passerini C, Mologni L. Activity of second-generation ALK inhibitors against crizotinib-resistant mutants in an NPM-ALK model compared to EML4-ALK. Cancer Med. 2015; 4:953–965.
65. Huang Q, Johnson TW, Bailey S, Brooun A, Bunker KD, Burke BJ, Collins MR, Cook AS, Cui JJ, Dack KN, Deal JG, Deng YL, Dinh D, et al. Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to crizotinib. J Med Chem. 2014; 57:1170–1187.
66. Yoshimura Y, Kurasawa M, Yorozu K, Puig O, Bordogna W, Harada N. Antitumor activity of alectinib, a selective ALK inhibitor, in an ALK-positive NSCLC cell line harboring G1269A mutation: Efficacy of alectinib against ALK G1269A mutated cells. Cancer Chemother Pharmacol. 2016.
67. Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, Engstrom LD, He M, Hoffman J, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014; 57:4720–4744.
68. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, Tang RW, Wang H, Tsaparikos K, Wang J, Timofeevski S, Katayama R, Dinh DM, et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer cell. 2015; 28:70–81.
69. Hatcher JM, Bahcall M, Choi HG, Gao Y, Sim T, George R, Janne PA, Gray NS. Discovery of Inhibitors That Overcome the G1202R Anaplastic Lymphoma Kinase Resistance Mutation. J Med Chem. 2015; 58:9296–9308.
70. Schönherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J. 2011; 440:405–413.
71. Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer cell. 2011; 19:679–690.
72. Carén H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. 2008; 416:153–159.
73. Martinsson T, Eriksson T, Abrahamsson J, Caren H, Hansson M, Kogner P, Kamaraj S, Schonherr C, Weinmar J, Ruuth K, Palmer RH, Hallberg B. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res. 2011; 71:98–105.
74. McDuff FK, Lim SV, Dalbay M, Turner SD. Assessment of the transforming potential of novel anaplastic lymphoma kinase point mutants. Mol Carcinog. 2013; 52:79–83.
75. Bourdeaut F, Ferrand S, Brugieres L, Hilbert M, Ribeiro A, Lacroix L, Benard J, Combaret V, Michon J, Valteau-Couanet D, Isidor B, Rialland X, Poiree M, et al. ALK germline mutations in patients with neuroblastoma: a rare and weakly penetrant syndrome. Eur J Human Genet. 2012; 20:291–297.
76. Kodama T, Tsukaguchi T, Satoh Y, Yoshida M, Watanabe Y, Kondoh O, Sakamoto H. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther. 2014; 13:2910–2918.