
Introduction
Progesterone (P4) and estradiol (E2) are major ovarian steroid hormones crucial for the development and homeostasis of the female reproductive tract (i.e., uterus, cervix, and vagina) [1]. P4 and E2 function through progesterone receptor (PR) and estrogen receptor (ER), respectively. PR and ER are ligand−dependent transcription factors belonging to the nuclear receptor superfamily [1]. ERα and ERβ are encoded by Esr1 and Esr2, respectively [2]. ERα is the major ER subtype in the female reproductive tract of mice and humans [2-4]. ERα is composed of several distinct functional domains, N−terminal activation function 1 (AF1), DNA−binding domain (DBD), a hinge region, and C−terminal ligand−binding domain containing the AF2. ERα activates its target gene expression by binding to estrogen response elements (EREs) through its DBD (classical mechanism) or other transcription factor (e.g., AP-1 and Sp-1) binding sites through protein−protein interactions (tethering mechanism) [2]. The Pgr gene coding for PR is a well−known ERα target gene in the female reproductive tract [1, 5]; however, the mechanism of Pgr activation by ERα is poorly understood. Reporter assays using isolated regulatory sequences of the PGR gene have implicated both classical and tethering mechanism in the transcriptional activation of PGR in MCF7 breast cancer cells [6-9]. However, it is unclear whether it is relevant to the normal female reproductive tract and whether both pathways are important in the natural chromatin context.
During the female sexual cycle in humans and rodents, an E2 surge promotes and a P4 surge inhibits epithelial cell proliferation in the reproductive tracts [10]. The uterine and vaginal epithelia consist of columnar and squamous epithelial cells, respectively. The cervical epithelium, however, is composed of columnar and stratified squamous epithelial cells [11]. The hormone−mediated regulation of epithelial cell proliferation in the reproductive tracts involves crosstalk between the stromal and epithelial compartment [12]. Epithelial ERα is dispensable for E2−induced proliferation of uterine columnar epithelial cells [13]. Stromal ERα is required for squamous epithelial cell proliferation in the cervix and vagina [14]. Epithelial PR is dispensable for P4−induced suppression of epithelial cell proliferation in the uterine tissue recombinants derived from neonatal mice [15]. P4 suppresses apoptosis in the uterine epithelium, which is mediated by stromal PR [16]. While PR is expressed in the cervical stroma and epithelium [17], knowledge on the function of PR and effects of P4 in the cervix is limited.
Using knock−in mice expressing mutant ERα lacking activities of DBD, AF1, or AF2, we show that all three domains are required for upregulation of Pgr in the cervical epithelium and vagina. We also show that AF1 is dispensable for upregulation of Pgr in the cervical and uterine stroma. Unlike the uterus, P4 promotes epithelial apoptosis in the cervix and vagina. P4 inhibits E2−induced cervical and vaginal squamous cell proliferation. Using epithelial Pgr−deficient mice, we demonstrate that epithelial PR is required for P4−induced apoptosis and suppression of cell proliferation in the cervical and vaginal epithelium. Our results are the first to show different functions and regulation of epithelial PR in the cervix, vagina, and uterus under the same hormonal condition.
Results
Histological features of the female reproductive tract lacking epithelial PR
To interrogate a role of epithelial PR in responses of the female reproductive tract to P4, we made use of Wnt7a−Cre/Pgrf/f mice (referred to as Pgred/ed hereafter; ed, epithelial deletion). In these mice, PR expression was ablated in the epithelium, but not stroma, of the cervix, vagina, and uterus (Figure 1A). To characterize epithelial PR functions under the same hormonal condition, we treated ovariectomized mice with E2 and P4 for 7 days. Seven−day treatment with E2 was required for the entire cervical epithelia to reach the full thickness (Supplementary Figure S1). Regions of the uterus, cervix, and vagina that were analyzed throughout the study are shown in Figure 1B. The cervical epithelia of vehicle−treated Pgrf/f and Pgred/ed mice were thin (13.2 μm in average) and composed of 2−3 cell layers (Figure 1C). E2 increased the height of the cervical epithelium in Pgrf/f (65.8 ± 4.2 μm) and Pgred/ed mice (66.6 ± 2.9 μm) (Figure 1C). The cervical epithelium height in E2+P4−treated Pgrf/f (57.5 ± 5.0 μm) and Pgred/ed mice (60.7 ± 4.6 μm) was not significantly different from genotype−matching mice treated with E2 alone. The nucleus/cytoplasm ratio (0.66 ± 0.03) of cervical suprabasal cells in E2−treated Pgrf/f mice was significantly smaller than the ratio (1.09 ± 0.06) in E2+P4−treated mice (P = 0.05). A similar difference was observed in Pgred/ed mice [0.66 ± 0.02 (E2) vs. 1.03 ± 0.01 (E2+P4); P = 0.05]. Hydropic degeneration was prominent in the cervix of E2+P4−treated Pgrf/f but not Pgred/ed mice (Figure 1C). E2 induced hyperplasia and keratinization in the vaginal epithelium of Pgrf/f and Pgred/ed mice (Figure 1D). In the vaginal epithelium of E2+P4−treated Pgrf/f mice, keratinization was absent and hydropic degeneration was observed (Figure 1D). Also found was infiltration of K14−negative non−epithelial cells (Supplementary Figure S2), which are likely neutrophils [18]. These P4−induced histologic changes in the vaginal epithelium were absent in E2+P4−treated Pgred/ed mice (Figure 1D). E2 induced hyperplasia in the uterine epithelium, which was reversed by P4 in both Pgrf/f and Pgred/ed mice (Figure 1E). Phenotypes described in this study are summarized in Table 1.
Table 1: Summary of P4-induced phenotypes in the female reproductive tract 1

1Shown are phenotypes in epithelial PR–sufficient (Pgrf/f) and –deficient (Pgred/ed) mice treated with E2 and E2+P4. 2Nucleus to cytoplasm ratio. ++, high; +, low; −, rare/absent.
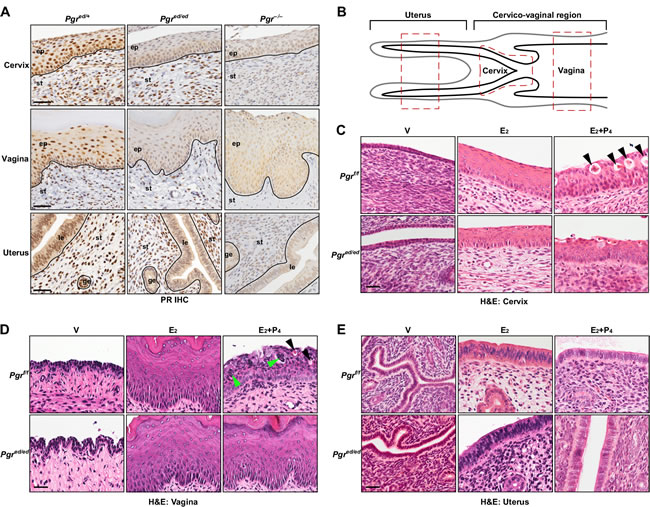
Figure 1: Histology of female reproductive tracts in epithelial PR−deficient mice treated with E2+P4. A. Epithelium−specific deletion of PR. Serial sections of the cervix, vagina, and uterus were stained for PR (brown). Nuclei were counterstained with hematoxylin. Black lines separate cervical/vaginal squamous epithelium (ep) and uterine luminal (le)/glandular epithelium (ge) from stroma (st). Scale bar, 50 μm. B. Schematics of murine female reproductive tracts. All analyses were carried out with red−boxed areas of each tissue. Note that a region between the cervix and uterus is not annotated because there is no histological boundary. C.-E. Ovariectomized mice were treated with female sex hormones as indicated and paraffin sections of the cervix C., vagina D., and uterus E. were subjected to H&E staining. Black arrowheads in C. and D. indicate cells undergoing hydropic degeneration. Green arrowheads in D. indicate non−epithelial cells. Representative images of each group are shown (n = 3-5). V, vehicle; E2, estradiol; P4, progesterone, Scale bar, 30 μm.
P4 fails to inhibit epithelial cell proliferation in the cervix and vagina, but not uterus of Pgred/ed mice
P4 inhibits epithelial cell proliferation in the female reproductive tract [15, 19]. We sought to determine whether epithelial PR is required for this effect. We analyzed expression of cell proliferation marker Ki67 to measure cell proliferation. Less than 1% of cervical epithelial cells were proliferative in vehicle−treated Pgrf/f and Pgred/ed mice (Figure 2A-2B). E2−induced cervical epithelial cell proliferation was not different between the two genotypes. Compared to genotype−matching mice treated with E2 alone, cervical epithelial cell proliferation in the basal layer was significantly decreased in E2+P4−treated Pgrf/f but not Pgred/ed mice (Figure 2A-2B). Similar phenotypes were observed in the vaginal epithelium of Pgrf/f and Pgred/ed mice (Figure 2C-2D and Table 1). In the uteri of both Pgrf/f and Pgred/ed mice, E2−induced epithelial cell proliferation was significantly inhibited by P4 (Figure 2E-2F). In both genotypes, however, stromal cell proliferation was increased in the uteri of E2+P4−treated mice compared to E2−treated mice (Figure 2F). Similar results were obtained in the columnar epithelium of the cervix (data not shown). These results indicate that, under the same hormonal condition, epithelial PR is required for P4−induced suppression of cell proliferation only in the cervical and vaginal squamous epithelium.
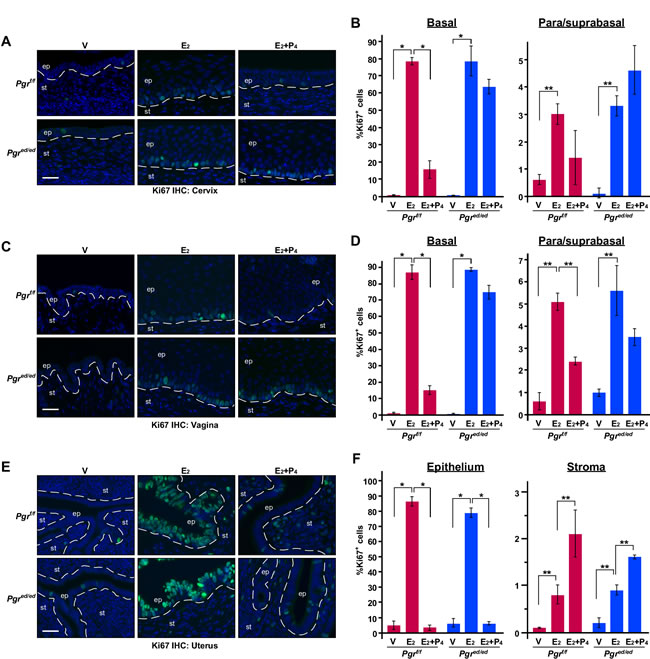
Figure 2: Epithelial PR is required for P4 to inhibit epithelial cell proliferation in the cervix and vagina, but not uterus. Ovariectomized mice were treated with female sex hormones as indicated. Paraffin sections of the cervix A., vagina C., and uterus E. were stained for Ki67 (green). Nuclei were stained with Hoechst 33342 (blue). Scale bars, 30 μm. Results were quantified as described in Materials & Methods. Quantified results of the cervix B., vagina D., and uterus F. are shown as mean ± S.E.M (n = 3-5 per group). *P ≤ 0.01, **P ≤ 0.05.
Epithelial PR is required for proper differentiation of cervical and vaginal squamous epithelial cells
Histological features observed in the cervical and vaginal epithelium (see Figure 1C-1D) suggested that P4 regulates differentiation of squamous cells. Thus, we carried out Alcian blue staining and immunohistochemistry for cytokeratin 10 (K10), squamous differentiation marker [20]. Alcian blue staining is typically used to detect cervical acidic mucins, which are expressed by differentiated cells [21]. As expected, E2 induced expression of K10 in the suprabasal layer in the cervical epithelia of Pgrf/f and Pgred/ed mice (Figure 3A). K10 expression was reduced in the cervical epithelia of E2+P4−treated Pgrf/f and Pgred/ed mice, indicating that epithelial PR is dispensable for K10 suppression by P4. Alcian blue staining was prominent in the cervix of Pgrf/f mice treated with E2+P4, but absent in identically treated Pgred/ed mice (Figure 3B). This result indicates that P4−promoted Alcian blue staining is dependent upon epithelial PR. P4 inhibited E2−induced K10 expression and increased Alcian blue staining in the vaginal epithelium of Pgrf/f but not Pgred/ed mice (Figure 3C-3D and Table 1). These results indicate that epithelial PR is required for P4−mediated regulation of squamous cell differentiation in the vagina to the greater extent than in the cervix.
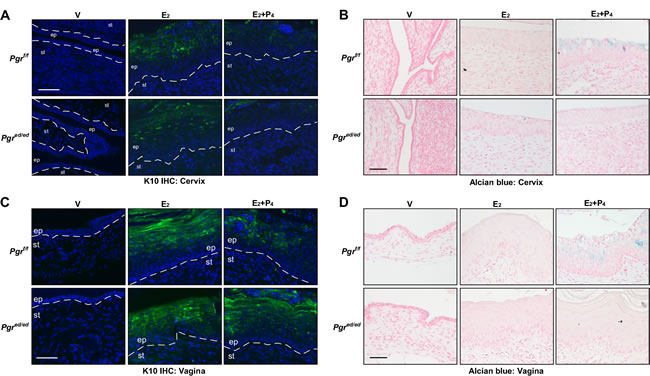
Figure 3: Epithelial PR is required for P4−mediated regulation of differentiation in the cervical and vaginal epithelium. Reproductive tracts were isolated from female mice that were ovariectomized and then treated as indicated. Representative images of each group are shown (n = 3). A. Epithelial PR is dispensable for suppression of K10 expression by P4 in the cervical epithelium. Cervical sections were stained for K10 (green) and nuclei were stained with Hoechst 33342 (blue). Dotted lines separate epithelium (ep) and stroma (st). Scale bar, 50 μm. B. Alcian blue staining is increased by P4 in an epithelial PR−dependent manner in the cervical epithelium. Cervical sections were stained with Alcian blue. Nuclei were counterstained with Nuclear Fast Red. Scale bar, 50 μm. C. Epithelial PR is necessary for suppression of K10 expression by P4 in the vaginal epithelium. Vaginal sections were stained for K10 (green) and nuclei were stained with Hoechst 33342 (blue). Scale bar, 50 μm. D. Alcian blue staining is increased by P4 in an epithelial PR−dependent manner in the vaginal epithelium. Vaginal sections were subjected to Alcian blue staining. Nuclei were counterstained with Nuclear Fast Red. Scale bar, 50 μm.
Epithelial PR plays distinct roles in epithelial apoptosis in the lower and upper reproductive tracts
P4 inhibits apoptosis in the uterine epithelium [16]. We carried out TUNEL assays to determine the role of P4 and epithelial PR in regulation of apoptosis in the cervix, vagina, and uterus. A few TUNEL−positive apoptotic cells were found in the cervical epithelium of vehicle− and E2−treated Pgrf/f and Pgred/ed mice (Figure 4A-4B). Compared to E2−treated mice, the apoptotic index was increased in the cervical epithelium of E2+P4−treated Pgrf/f but not Pgred/ed mice (Figure 4A-4B). Similar results were obtained in the vaginal epithelium (Figure 4C-4D and Table 1). E2 induced apoptosis in the uterine epithelium regardless of the genotype, which was inhibited by P4 in both Pgrf/f and Pgred/ed mice (Figure 4E-4F and Table 1). These results indicate that epithelial PR is required for P4−induced apoptosis in the epithelium of lower reproductive tracts (i.e., cervix and vagina), but dispensable for P4 to inhibit apoptosis in the uterine epithelium.
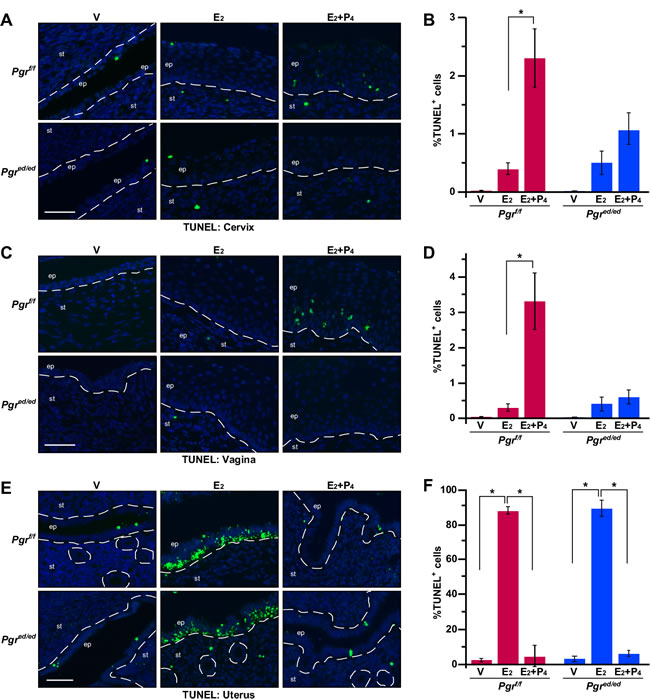
Figure 4: Epithelial PR is required for P4−mediated regulation of apoptosis in the cervix and vagina, but not the uterus. Reproductive tracts were isolated from female mice that were ovariectomized and then treated as indicated. A. P4 induces apoptosis in the cervical epithelium through epithelial PR. Cervical sections were subjected to TUNEL staining (green). Nuclei were stained with Hoechst 33342 (blue). Dotted lines separate epithelium (ep) and stroma (st). Scale bar, 50 μm. B. Results shown in A. were quantified. Results are shown as mean ± S.E.M (n = 3-5 per group). *P = 0.04. C. P4 induces apoptosis in the vaginal epithelium through epithelial PR. Vaginal sections were subjected to TUNEL staining (green). Scale bar, 50 μm. D. Results shown in C. were quantified and displayed as mean ± S.E.M (n = 3-5 per group). *P = 0.04. E. P4 prevents apoptosis in an epithelial PR−independent manner in the uterine epithelium. Uterine sections were subjected to TUNEL staining (green). Scale bar, 50 μm. F. Results shown in E. were quantified and displayed as mean ± S.E.M (n = 3 per group). *P = 0.05.
ERα DBD and AF2 are required for upregulation of Pgr in the cervix and vagina
ERα regulates transcription of Pgr in the vagina and uterus [5]. We found that E2 increased the Pgr mRNA level significantly in the cervix of Esr1+/+ but not Esr1−/− mice (Figure 5A). Similarly, PR protein was readily detected in cervical tissue extracts and paraffin sections derived from Esr1+/+ but not Esr1−/− mice (Figure 5B-5C). These results demonstrate that ERα activates transcription of Pgr in the cervix. To understand a mechanism of transcriptional activation of Pgr by ERα, we made use of knock−in mouse models expressing ERα mutant defective for DBD, AF1, or AF2 (Figure 5D). The AA allele expresses an ERα mutant (E207A/G208A) that lacks the ERE−binding activity [22]. The AF2 allele expresses ERα harboring L543A/L544A substitution mutations in the helix 12, which abrogates the AF2 function [23]. The neo allele has a neo cassette in the exon 2, which is designed to abrogate expression of ERα [24]. This allele, however, expresses truncated ERα proteins lacking the AF1 domain due to cryptic splicing events [25, 26]. PR expression was barely detectable in cervical tissue extracts obtained from Esr1AA/− and Esr1AF2/AF2 mice (Figure 5B). Similarly, E2 failed to induce PR expression in the cervix of Esr1AA/− and Esr1AF2/AF2 mice (Figure 5C). Albeit reduced compared to Esr1+/+ mice, the PR level was increased in cervical tissue extracts from Esr1neo/neo mice compared to Esr1−/−, Esr1AA/−, and Esr1AF2/AF2 mice (Figure 5B). Upregulation of PR expression in the cervix of Esr1neo/neo mice was mostly restricted in the stroma (Figure 5C). E2 did not activate Pgr expression in the vagina of Esr1−/−, Esr1AA/−, Esr1AF2/AF2, and Esr1neo/neo mice (Supplementary Figure S3). PR expression patterns were similar in the cervix and vagina of Esr2+/+ and Esr2−/− mice (Supplementary Figure S4), indicating that ERβ is not required for upregulation of Pgr. To determine whether stroma−epithelium cross talk is involved in upregulation of Pgr by E2−ERα, we used Wnt7a−Cre/Esr1f/f (referred to as Esr1ed/ed hereafter). ERα expression was efficiently ablated in epithelial but not stromal cells in the cervix and vagina of Esr1ed/ed mice (Figure 5E). While E2 induced PR expression in the cervical stroma of Esr1f/f and Esr1ed/ed mice, E2 failed to do so in the cervical epithelium of Esr1ed/ed mice (Figure 5F). Similar results were obtained from the vagina (Figure 5G). These results suggest that, in the cervix and vagina, E2−induced upregulation of Pgr is cell−autonomous and mediated commonly by ERα DBD (i.e., ERE−binding) and AF2.
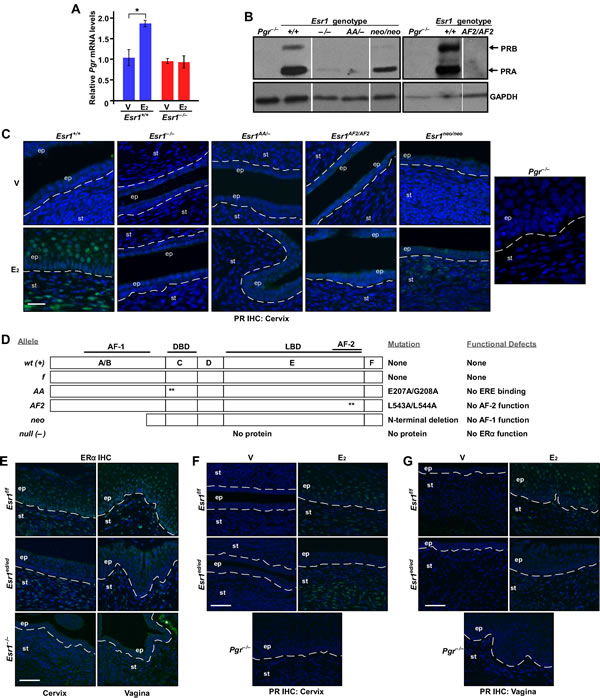
Figure 5: Mechanisms of upregulation of Pgr by ERα in the cervix and vagina. A. Pgr is upregulated by E2−ERα in the cervix. Mice were ovariectomized and treated with E2 for 24 hr. The relative levels of Pgr mRNA were compared by qPCR. The value from vehicle−treated Esr1+/+ cervix was set at 1. Results are shown as mean ± S.E.M (n = 3). *P = 0.05. B. Upregulation of PR expression is absent in the cervix of Esr1AA/− and Esr1AF2/AF2 mice. Ovary−intact mice of indicated genotypes (8-12 weeks of age) were treated with E2 for 3 days to synchronize mice at the estrus. Expression of PR in the cervix was determined by immunoblot. Pgr−/− tissue extracts were used as negative control. GAPDH was used as loading control. Results were taken from separate gels indicated by black boxes. Intervening lanes in a same gel were removed, which was indicated by vertical white lines. Representative results from more than 2 mice per genotype are shown. C. E2−induced PR expression is absent in the cervix of Esr1AA/− and Esr1AF2/AF2 mice. Ovariectomized mice (n = 3 per group) were treated with vehicle or E2 for 7 days. Cervical tissue sections were stained for PR (green). Nuclei were stained with Hoechst 33342 (blue). Dotted lines separate epithelium (ep) and stroma (st). Scale bar, 25 μm. D. Summary of ERα proteins expressed by Esr1 alleles used in the study. Asterisks denote locations of alanine substitution mutations. AF, activation function; DBD, DNA−binding domain; LBD, ligand−binding domain. E. Esr1 is deleted in the cervical and vaginal epithelium. Cervical and vaginal sections (n= 3 per tissue) were stained for ERα (green). Nuclei were stained with Hoechst 33342 (blue). Scale bar, 50 μm. F.-G. Epithelial ERα is required for upregulation of epithelial Pgr in the lower reproductive tracts. Mice (n = 3 per group) were ovariectomized and treated with vehicle or E2. Tissue sections of the cervix F. and vagina G. were stained for PR (green). Nuclei were stained with Hoechst 33342 (blue). Scale bar, 50 μm.
ERα DBD and AF2 are required for upregulation of stromal Pgr and downregulation of epithelial Pgr in the uterus
Consistent with previously published results [13], expression of ERα was lost in the epithelium but retained in the stroma of the Esr1ed/ed uteri (Figure 6A). While stromal PR expression was increased by E2 in the Esr1f/f and Esr1ed/ed uteri, E2 decreased the PR levels in the uterine epithelium in both genotypes (Figure 6B). This result confirmed the paracrine mechanism of Pgr downregulation by ERα in the uterine epithelium [5, 27]. Stromal upregulation and epithelial downregulation of Pgr by E2 were absent in the uterus of Esr1−/−, Esr1AA/−, and Esr1AF2/AF2 mice (Figure 6C). As reported previously [5], E2 activated stromal Pgr expression but failed to downregulate epithelial Pgr in the uterus of Esr1neo/neo mice (Figure 6C). In the Esr2−/− uterus, E2 increased and decreased stromal and epithelial PR expression, respectively (Supplementary Figure S4), indicating that ERβ is not required for regulation of Pgr in the uterus. These results indicate that the function of ERα DBD and AF2 in stromal cells is required for stromal upregulation and epithelial downregulation of Pgr in the uterus.
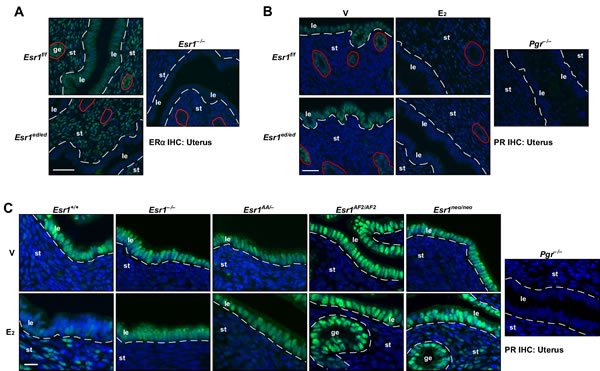
Figure 6: Mechanisms of regulation of Pgr by ERα in the uterus. A. Esr1 is deleted in the uterine epithelium. Uterine sections (n = 3) were stained for ERα (green). Nuclei were stained with Hoechst 33342 (blue). Dotted lines separate luminal epithelium (le) and stroma (st). Glandular epithelia (ge) are circumscribed by red circles. Scale bar, 50 μm. B. E2 suppresses epithelial Pgr expression in the Esr1ed/ed uteri. Mice (n = 3) were ovariectomized and treated with vehicle or E2. Uterine tissue sections were stained for PR (green). Nuclei were stained with Hoechst 33342 (blue). Dotted lines separate luminal epithelium (le) and stroma (st). Glandular epithelia (ge) are circumscribed by red circles. Note that E2 downregulates epithelial Pgr and upregulates stromal Pgr in both genotypes. Scale bar, 25 μm. C. ERα DBD and AF2 are required for Pgr regulation in the uterus. Mice were treated as described in B.. Uterine tissue sections (n = 3 per group) were stained for PR (green). Nuclei were stained with Hoechst 33342 (blue). Dotted lines are to separate luminal (le) and glandular epithelium (ge) from stroma (st). Scale bar, 30 μm.
We next analyzed binding of ERα to the Pgr gene using the previously published ERα ChIP−seq data [28, 29]. Approximately 200 kb upstream and 100 kb downstream of the Pgr transcription start site (TSS) were analyzed for the binding of wild−type ERα. While there were two E2−independent ERα−binding peaks at the 3’ end of the Pgr gene, E2 enhanced ERα binding to the intragenic regions and −72 kb and −155 kb cluster in the Esr1+/+ uterus (Figure 7A). E2 also increased RNA polII binding to the Pgr gene in the same tissue. While the −155 kb cluster was closer to AK054106, E2 did not regulate this gene (data not shown). Notably, we identified predicted half or full EREs in all ERα−binding sites (Figure 7A). Consistently, in the uterus of Esr1AA/− mice, both E2−dependent and −independent ERα binding to the Pgr locus were not observed (Figure 7A). Concordantly, E2−induced recruitment of RNA polII was absent in the Esr1AA/− uterus. We confirmed by ChIP followed by qPCR that E2 induced enrichment of ERα binding to the −62 kb region (7.1-fold) and intron 3-4 (6.2-fold) of Pgr in the uterus of Esr1+/+ but not Esr1AA/− mice (Figure 7B). These results suggest that, in the uterus, ERα activates transcription of Pgr mainly by binding to EREs.
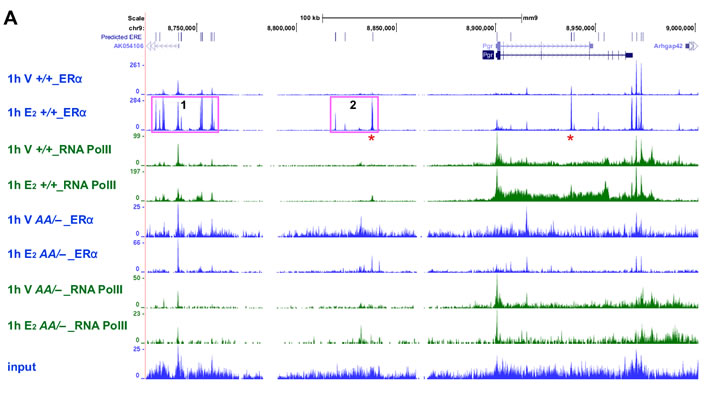
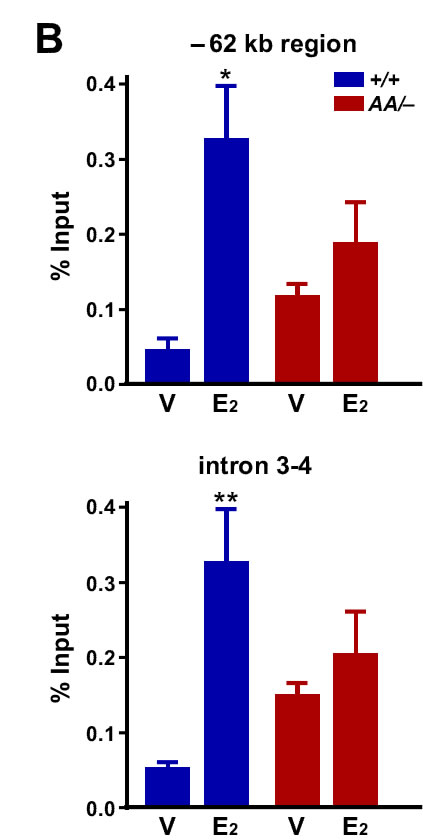
Figure 7: ERα binding to EREs correlates with RNA polII recruitment to the Pgr locus in the uterus. A. E2 induces binding of ERα and RNA polII to Pgr in the uterus of Esr1+/+ but not Esr1AA/− mice. Recruitment of ERα and RNA polII to the Pgr locus was analyzed using ChIP−seq data available in Gene Expression Omnibus (Esr1+/+, GSE36455l; Esr1AA/−, GSE56466). Vertical blue lines at the top indicate locations of predicted EREs, which were identified using position weight matrix algorithm. Two peaks validated in B. are indicated by red asterisk. Pink boxes indicate −155 kb (1) and −72 kb cluster (2). B. Validation of E2−dependent enrichment of ERα at two candidate binding regions. Mice were ovariectomized and treated with vehicle or E2. Extracted chromatin was subjected to ERα ChIP and enrichment of approximately 62 kb upstream of the Pgr TSS and intron 3-4 were quantified by qPCR. Data normalized to % input DNA is shown as mean ± S.E.M. (n = 4-5). *P = 0.001; **P = 0.0005.
Discussion
P4 inhibits E2−promoted epithelial cell proliferation in the murine female reproductive tract [19, 30]. We showed that, under the same hormonal condition, epithelial PR was necessary for P4 to suppress E2−induced epithelial cell proliferation in the cervix and vagina, but dispensable in the uterus (Figure 2). It is shown that epithelial PR is required for P4−mediated inhibition of uterine epithelial cell proliferation [30]. This discrepancy may be due to differences in treatment design (P4 for 3 days and then E2+P4 for 1 day vs. E2+P4 for 7 days), E2 doses (50 ng vs. 1 μg), mouse genetic background (B6.SJL.129 mixed vs. FVB) and/or housing environment. We note that 7−day treatment with E2 and P4 might be pharmacologic rather than physiologic. Nonetheless, we believe our results are biologically relevant because P4−mediated inhibition of uterine epithelial proliferation during pregnancy depends on Hand2 transcription factor [31]. Expression of Hand2 depends on P4 and PR and is restricted in the uterine stroma [31].
P4 inhibited E2−induced apoptosis in the uterus of both Pgrf/f and Pgred/ed mice (Figure 4E-4F). It is consistent with that P4 prevents epithelial cell apoptosis in the tissue recombinant composed of Pgr+/+ stroma and Pgr−/− epithelium [16]. E2 induces apoptosis in the uterine epithelium of Esr1f/f and Esr1ed/ed mice [13]. These results suggest that stromal ERα promotes and stromal PR inhibits apoptosis of uterine epithelial cells. One possibility is that stromal PR activates expression of a paracrine factor(s) that promotes cell survival. PR interacts with ERα and modulates the function of ERα in breast cancer cells [32]. Another possibility is that PR interacts with ERα in uterine stromal cells and inhibits ERα−mediated expression of a pro−apoptotic secretory factor(s). In the rodent uterus, E2−induced hyperplasia is eliminated by apoptosis [33]. Perhaps, E2−induced apoptosis is to balance out E2−induced hyperproliferation of uterine epithelial cells. Consistent with this idea, the apoptotic index was correlated with the proliferative index in the uterine epithelium (Figure 2F and 4F). Unlike the uterus, P4 promoted apoptosis in the cervical and vaginal epithelium, which was dependent on epithelial PR (Figure 4A-4D). Epithelial PR was necessary for P4 to increase the ratio of nucleus to cytoplasm and suppress expression of K10 in the vagina but not cervix (Table 1). These results reveal that epithelial PR is differentially required for P4 responses of epithelial cells in the cervix, vagina, and uterus.
Using mouse genetic models, we determined that ERE binding (i.e., classical mechanism) of ERα is required for upregulation of Pgr in the cervix, vagina, and uterus (Figure 5B-5D, 6C, 7 and Supplementary Figure S3). The ERα DBD mutant (E207A/G208A) has acquired the capability to bind hormone response elements such as progesterone response element [29]. We do not believe that the gain−of−function compromises our conclusion because this ERα mutant did not bind to the Pgr locus (Figure 7). It is possible that the ChIP−seq analyses might have failed to identify weak ERα−binding sites; thus, we cannot rule out a possibility that the ERα tethering mechanism also contributes to transcription of Pgr in the female reproductive tract. Knock−in mice expressing an ERα mutant defective for the tethering mechanism would be required to test this. Reporter assays have shown that AP-1 and Sp-1 sites at the promoter downstream are required for ERα−mediated PGR activation in MCF7 breast cancer cells [7, 8]. In the uterus, strong E2−induced DBD−dependent ERα binding occurred mostly at far upstream ( > 60 kb) of the Pgr gene (Figure 7A). Genome−wide enrichment of ERα binding to promoter−distal regions has been identified [28, 34]. ERE−dependent long−range activation of PGR by ERα has been demonstrated in MCF7 cells [6]. It is likely that ERα binding to Pgr in the uterus occurs in stromal cells because epithelial ERα was not required for upregulation of stromal Pgr and downregulation of epithelial Pgr (Figure 6B). We postulate that similar long−range regulation by ERα is involved in transcriptional activation of Pgr in the cervix and vagina. The AF2 domain was also required for upregulation of Pgr by ERα in the cervix, vagina, and uterus (Figure 5B-5D, 6C and Supplementary Figure S3). Upregulation of Pgr was evident in the cervical and uterine stroma, but not vagina of Esr1neo/neo mice (Figure 5C, 6C and Supplementary Figure S3). In tissue recombinants derived from Esr1neo/neo mice, PR expression is induced by E2 in the uterine but not vaginal stroma [5]. These results indicate that (1) ERα DBD and AF2 are commonly required for activation of Pgr in the female reproductive tracts and (2) AF1 is required for upregulation of Pgr only in the vaginal and cervical epithelium and vaginal stroma. Differential requirement of ERα domains for activation of Pgr may be due to distinct chromatin structures in different tissues and cell types, which may require different coactivators [2, 35].
E2 suppressed expression of Pgr in the uterine epithelium via stromal ERα (Figure 6B), which is in agreement with other findings [5, 13]. Downregulation of Pgr in the uterine epithelium involves transcriptional repression [5, 27]. It is unlikely that the non−cell autonomous downregulation of Pgr is through a cell−cell contact because epithelial and stromal cells are physically separated by the basement membrane. Thus, we postulate that a paracrine factor(s) produced by stromal cells is responsible for repression of Pgr in the uterine epithelium. ERα DBD, AF1, and AF2 were required for downregulation of Pgr in the uterine epithelium (Figure 6C), further suggesting that ERα DBD, AF1, and AF2 are necessary for regulating expression of such a factor(s). The AP-1 site at the +745 bp region of PGR has been implicated in repression of PGR in MCF7 cells [9]. It is plausible that a paracrine factor activates AP-1, which in turn suppresses transcription of Pgr in the uterine epithelium.
Our results demonstrate distinct functions of epithelial PR in the cervix, vagina, and uterus, suggesting that PR regulates unique sets of genes and pathways in these tissues. Warranted are further studies to identify PR target genes and pathways in the stroma and epithelium of these tissues. Our results also demonstrate unique mechanisms of Pgr regulation in the cervix, vagina, and uterus. Together, our results underscore the complexity of function and regulation of Pgr in the female reproductive tracts.
Materials and Methods
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to national and international guidelines. All procedures were carried out according to animal protocols approved by the University of Houston Institutional Animal Care and Use Committee.
Mice and treatments
All Esr1 and Pgr mice as well as Esr2 knockout and Wnt7a-Cre transgenic mice were described previously [13, 22-24, 36-39], and genotyped by PCR. All mouse strains except for Esr2 knockout mice (C57BL/6) were backcrossed to FVB at least 5 generations. Mice were ovariectomized at the age of 6-8 weeks and rested for 2 weeks. They were then i.p. injected with vehicle, 17β-estradiol (E2) alone, or E2 (1 μg/day) plus P4 (1 mg/day) for 7 days.
Histological staining
The female reproductive tracts were harvested 24 hours after final hormone injections and processed as described previously [19]. Hematoxylin & eosin (H&E) and Alcian blue staining were carried out as described previously [19].
Immunohistochemistry and immunoblot
Antibodies were purchased from Santa Cruz Biotechnology [ERα (H184), PR (H190) and GAPDH (V-18)], Thermo Scientific [Ki67 (clone SP6) and K10 (clone DE-K10)], BioLegend (K14), GenDEPOT (HRP−conjugated anti−rabbit/goat IgG), Life Technologies (Alexa488−conjugated anti−rabbit/mouse IgG) and Rockland Immunochemicals (biotinylated anti−rabbit IgG). For IHC, sections were deparaffinized, rehydrated, microwaved in 10 mM sodium citrate buffer (pH 6.0) and incubated with primary antibodies diluted in blocking buffers (PR, 1:200 in 5% goat serum; ERα, 1:100 in 10% goat serum and 0.5% skim milk; Ki67, 1:100 in 10% goat serum; K10, 1:50 in 5% goat serum). After extensive washes, sections were incubated with Alexa488−conjugated anti−rabbit/mouse IgG or biotinylated anti−rabbit IgG followed by ABC complex (Vector Laboratories) as described previously [19]. For immunoblot, cervical tissue homogenates were resolved in 7.5% SDS−polyacrylamide gel and proteins were transferred to PVDF membranes.
TUNEL assay
TUNEL staining was carried out using ApopTag Fluorescein in situ apoptosis detection kit (Millipore) according to the manufacturer’s instructions.
Microscopy and digital image analyses
Stained tissue sections were visualized by an Olympus BX51 or a Nikon ECLIPSE Ti microscope, and photographed with a color camera (Olympus DP73) or cooled CCD monochrome cameras (Olympus XM10 and Photometrics CoolSNAP HQ2). Digital images were acquired with imaging software (Olympus cellSens Dimension and Nikon NIS-Elements AR). For quantification, images were acquired from 5 microscopic fields per tissue with a 20X objective lens, and cells were quantified using the counting function of the NIH ImageJ software. Typical ranges of total cell count per tissue were 400 - 600 in the basal layer and 1,500 - 2,000 in the suprabasal layers.
Chromatin immunoprecipitation and quantitative real−time PCR
Chromatin was prepared from ovariectomized Esr1+/+ or Esr1AA/− uterus one hour after i.p. injection with saline or E2 (250 ng/mouse) and ERα was immunoprecipitated as described previously [29]. Candidate ERα−binding regions in the Pgr locus were amplified using qPCR as described previously [29]. Nanograms of amplimer DNA was calculated using a standard curve, and normalized to % input DNA for each sample. Primers for the −62 kb region were 5’-GGGTCGAAACTACCAGCTAAA-3’ (forward) and 5’-CAAAGGCTTGGACAAATGAA-3’ (reverse). Primers for intron 3-4 were 5’-TCTGCTCCAATGACTGTGTTC-3’ (forward) and 5’-ATCACATGCACTGAGAAGATCA-3’ (reverse). To compare expression levels of Pgr mRNA in the cervix, qPCR was carried out using a SYBR Green detection method. The relative mRNA levels were calculated using the ∆∆Ct method with Ppia mRNA as control. Primers for Pgr were 5’-CCAGCATGTCGTCTGAGAAA-3’ (forward) and 5’-GCCTGGCTCTCGTTAGGAAG-3’ (reverse). Primers for Ppia were 5’-GGGTTCCTCCTTTCACAGAA-3’ (forward) and 5’-GATGCCAGGACCTGTATGCT-3’ (reverse).
Statistical analyses
Wilcoxon rank sum test was used for analysis of cell proliferation and apoptosis. Student’s t-test or 2-way ANOVA with Fisher’s LSD was used for analysis of qPCR data.
Acknowledgments
Authors thank Dr. Roger E. Price for the help with histological analyses. Authors also thank Dr. M. David Stewart for the help with microscopy and Seung Han Baik for the help with mouse tissue processing and colony maintenance.
Conflicts of Interest
Authors have nothing to disclose.
Grant Support
The work was supported by the Cancer Prevention and Research Institute of Texas grant RP120617 (S.-H.C) and NIH grants R01 CA188646 (S.-H.C.) and R01 HD042311 (J.P.L.) and by the Division of Intramural Research of the NIH, National Institute of Environmental Health Sciences Project 1ZIAES070065 (S.C.H. and K.S.K.).
References
1. Patel B, Elguero S, Thakore S, Dahoud W, Bedaiwy M and Mesiano S. Role of nuclear progesterone receptor isoforms in uterine pathophysiology. Hum Reprod Update. 2015; 21:155-173.
2. Thomas C and Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011; 11:597-608.
3. Chung SH, Wiedmeyer K, Shai A, Korach KS and Lambert PF. Requirement for estrogen receptor alpha in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008; 68:9928-9934.
4. den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, Schott M, Chung L, He Q, Lambert P, Walker J, Newton MA, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A. 2015; 112:E3255-3264.
5. Kurita T, Lee KJ, Cooke PS, Taylor JA, Lubahn DB and Cunha GR. Paracrine regulation of epithelial progesterone receptor by estradiol in the mouse female reproductive tract. Biol Reprod. 2000; 62:821-830.
6. Boney-Montoya J, Ziegler YS, Curtis CD, Montoya JA and Nardulli AM. Long-range transcriptional control of progesterone receptor gene expression. Mol Endocrinol. 2010; 24:346-358.
7. Petz LN, Ziegler YS, Loven MA and Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002; 143:4583-4591.
8. Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK and Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004; 88:113-122.
9. Petz LN, Ziegler YS, Schultz JR and Nardulli AM. Fos and Jun inhibit estrogen-induced transcription of the human progesterone receptor gene through an activator protein-1 site. Mol Endocrinol. 2004; 18:521-532.
10. Chung SH. Targeting female hormone receptors as cervical cancer therapy. Trends Endocrinol Metab. 2015; 26:399-401.
11. Herfs M, Yamamoto Y, Laury A, Wang X, Nucci MR, McLaughlin-Drubin ME, Munger K, Feldman S, McKeon FD, Xian W and Crum CP. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc Natl Acad Sci U S A. 2012; 109:10516-10521.
12. Cunha GR, Cooke PS and Kurita T. Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol. 2004; 67:417-434.
13. Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR and Korach KS. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci U S A. 2010; 107:19272-19277.
14. Chung SH, Shin MK, Korach KS and Lambert PF. Requirement for Stromal Estrogen Receptor Alpha in Cervical Neoplasia. Horm Cancer. 2013; 4:50-59.
15. Kurita T, Young P, Brody JR, Lydon JP, O’Malley BW and Cunha GR. Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology. 1998; 139:4708-4713.
16. Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O’Malley BW, Isaacs JT, Dahiya R and Cunha GR. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ. 2001; 8:192-200.
17. Kurita T, Cooke PS and Cunha GR. Epithelial-stromal tissue interaction in paramesonephric (Mullerian) epithelial differentiation. Dev Biol. 2001; 240:194-211.
18. Sonoda Y, Mukaida N, Wang JB, Shimada-Hiratsuka M, Naito M, Kasahara T, Harada A, Inoue M and Matsushima K. Physiologic regulation of postovulatory neutrophil migration into vagina in mice by a C-X-C chemokine(s). J Immunol. 1998; 160:6159-6165.
19. Yoo YA, Son J, Mehta FF, DeMayo FJ, Lydon JP and Chung SH. Progesterone signaling inhibits cervical carcinogenesis in mice. Am J Pathol. 2013; 183:1679-1687.
20. Chateau D and Boehm N. Regulation of differentiation and keratin 10 expression by all-trans retinoic acid during the estrous cycle in the rat vaginal epithelium. Cell Tissue Res. 1996; 284:373-381.
21. Barker TE and Walker BE. Initiation of irreversible differentiation in vaginal epithelium. Anat Rec. 1966; 154:149-159.
22. Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ and Jameson JL. An estrogen receptor (ER)alpha deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo. Mol Endocrinol. 2002; 16:2188-2201.
23. Arao Y, Hamilton KJ, Ray MK, Scott G, Mishina Y and Korach KS. Estrogen receptor alpha AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci U S A. 2011; 108:14986-14991.
24. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS and Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993; 90:11162-11166.
25. Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O and Korach KS. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995; 9:1441-1454.
26. Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, Bayard F and Arnal JF. The AF-1 activation-function of ERalpha may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci U S A. 2002; 99:2205-2210.
27. Kurita T, Lee KJ, Cooke PS, Lydon JP and Cunha GR. Paracrine regulation of epithelial progesterone receptor and lactoferrin by progesterone in the mouse uterus. Biol Reprod. 2000; 62:831-838.
28. Hewitt SC, Li L, Grimm SA, Chen Y, Liu L, Li Y, Bushel PR, Fargo D and Korach KS. Research resource: whole-genome estrogen receptor alpha binding in mouse uterine tissue revealed by ChIP-seq. Mol Endocrinol. 2012; 26:887-898.
29. Hewitt SC, Li L, Grimm SA, Winuthayanon W, Hamilton KJ, Pockette B, Rubel CA, Pedersen LC, Fargo D, Lanz RB, DeMayo FJ, Schutz G and Korach KS. Novel DNA motif binding activity observed in vivo with an estrogen receptor alpha mutant mouse. Mol Endocrinol. 2014; 28:899-911.
30. Franco HL, Rubel CA, Large MJ, Wetendorf M, Fernandez-Valdivia R, Jeong JW, Spencer TE, Behringer RR, Lydon JP and Demayo FJ. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012; 26:1218-1227.
31. Li Q, Kannan A, DeMayo FJ, Lydon JP, Cooke PS, Yamagishi H, Srivastava D, Bagchi MK and Bagchi IC. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science. 2011; 331:912-916.
32. Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, Serandour AA, Birrell SN, Bruna A, Saadi A, Menon S, Hadfield J, Pugh M, Raj GV, Brown GD, D’Santos C, et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature. 2015; 523:313-317.
33. Sato T, Fukazawa Y, Kojima H, Enari M, Iguchi T and Ohta Y. Apoptotic cell death during the estrous cycle in the rat uterus and vagina. Anat Rec. 1997; 248:76-83.
34. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006; 38:1289-1297.
35. Jozwik KM and Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012; 12:381-385.
36. Fernandez-Valdivia R, Jeong J, Mukherjee A, Soyal SM, Li J, Ying Y, Demayo FJ and Lydon JP. A mouse model to dissect progesterone signaling in the female reproductive tract and mammary gland. Genesis. 2010; 48:106-113.
37. Goulding EH, Hewitt SC, Nakamura N, Hamilton K, Korach KS and Eddy EM. Ex3alphaERKO male infertility phenotype recapitulates the alphaERKO male phenotype. J Endocrinol. 2010; 207:281-288.
38. Ismail PM, Li J, DeMayo FJ, O’Malley BW and Lydon JP. A novel LacZ reporter mouse reveals complex regulation of the progesterone receptor promoter during mammary gland development. Mol Endocrinol. 2002; 16:2475-2489.
39. Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA and Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A. 1998; 95:15677-15682.