
BACKGROUND
The treatment of non-small-cell lung cancer has been revolutionized due to development of molecularly targeted therapy in genomically defined subsets of patients. The discovery of epidermal growth factor receptor (EGFR) mutation [1] and anaplastic lymphoma kinase (ALK) gene rearrangement are classic examples of oncogenic genomic alterations (GAs) that confer sensitivity to matched targeted therapy [2]. Patients with EGFR or ALK gene alterations have experienced significant survival benefit from targeted therapy as compared to conventional chemotherapy [3, 4]. Undoubtedly, tyrosine kinase inhibitors (TKI) for EGFR and ALK are currently the standard of care for EGFR-mutant and ALK-rearranged lung cancer patients, respectively.
However, most lung adenocarcinomas lack an identifiable driver oncogene such as the above and are therefore still treated with conventional chemotherapy. Therefore, maximally identifying actionable GAs in advanced lung cancer patients is essential to improve clinical outcome. The use of comprehensive genomic profiling (CGP) enabled identification of oncogenic alterations that would previously been missed by conventional testing, as this provides simultaneous detection of alterations in multiple cancer genes with higher sensitivity and specificity. Collaborated efforts using NGS has increased the diversity and expanded the range of possible targetable alterations in lung adenocarcinomas. A recent study by the Cancer Genome Atlas (TCGA) revealed potential novel drivers such as NF1, MET, ERBB2 and RIT1 which occur in 13% of cases, and enriched in samples lacking an activated oncogene [5]. These alterations, which occurred in previously oncogene-negative subsets, may be single-gene driver events similar to EGFR and ALK.
Recently, rather than sequencing the entire genome or exome, CGP which include genes that show frequent alterations in cancer were developed to save the amount of tissue, time and effort to perform sequencing. These panels use PCR capture-based NGS assay that allow deep targeted sequencing of genes of interest from limited formalin-fixed, paraffin-embedded (FFPE) specimens [6]. Since incorporating NGS into routine oncologic practice requires accurate genomic profiling in a single assay, clinical cancer gene test may be appropriately used for clinical use.
In this study, we aimed to perform CGP on tumor specimens from patients with lung adenocarcinomas who tested negative for EGFR, KRAS and ALK previously at our institution. Our ultimate goal was to identify patients who were candidates for targeted therapy and expand treatment choices for apparently “wild type” patients.
RESULTS
Patients
Clinical characteristics of patients are shown in Table 1. This study included 51 patients (18 males and 33 females) with a median age of 58 years (range: 29-77 years). All patients had adenocarcinoma histology and the majority of patients were never-smokers (76%, n=39). Twenty-five patients (49%) were stage 4 at the time of diagnosis. All patients had underwent EGFR/KRAS/ALK screening by conventional sequencing and FISH and were found to be triple-negative.
Table 1: Clinicopathologic findings of patients (N=51)
N |
% |
|
|---|---|---|
Gender |
||
Male |
18 |
35 |
Female |
33 |
65 |
Age |
||
Median (range) |
58 (29-77) |
|
Pathology |
||
Adenocarcinoma |
51 |
100 |
Stagea |
||
I |
5 |
10 |
II |
8 |
16 |
III |
13 |
25 |
IV |
25 |
49 |
Smoking |
||
Never |
39 |
76 |
Former or current smoker |
12 |
24 |
aClinical stage at the time of initial diagnosis was determined according to the 7th American Joint Commission on Cancer guidelines
Genomic alterations
Tumors were sequenced with a median coverage of 564x and a total of 190 known and 601 unknown individual GAs were identified. One or more GAs were uncovered by CGP from 94% (n= 48/51) of patients, with average of 3.7 alterations (range 0-10) per patient (Supplementary Table S1).
Non-synonymous base substitutions comprised 50% (80/190) of the detected alterations (Figure 1): 15% (29/190) were insertions or deletion, and 3% (5/190) splice site mutation. TP53 was the most commonly mutated gene (30%, n=24/80) among non-synonymous base substitutions, followed by KRAS (10%, n=8/80) and EGFR (10%, 8/80). Insertions or deletions commonly involved TP53 (17%, 5/29) and ERBB2 (14%, 4/29), and splice site mutations occurred in TP53, INPP4B, ATR, and MAP2K4. Gene amplification comprised 20% (39/190) of genomic alterations, and MDM2 amplification was identified most frequently (13%, 5/39). Interestingly, MDM2 amplification was frequently found with CDK4 amplification in 3 out of 5 cases observed. Homozygous loss comprised 5% (10/190) of all genomic alterations and all 10 cases were observed with CDKN2A. Fusion genes were found in 7% (n=14/190) and most commonly involved ROS1 fusion (50%, n=7/14). Alterations in the PI3K/mTOR pathway such as PIK3CA mutation, AKT1 mutation, PIK3R2 mutation, STK11 inactivating mutation, MTOR mutation and RICTOR amplification were detected in 17 cases (33%).
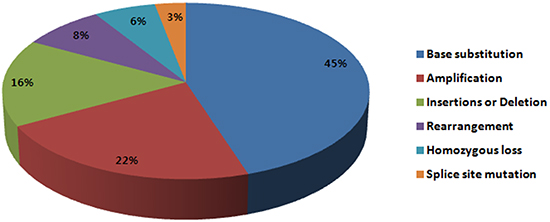
Figure 1: Frequency of genomic alterations identified in this study.
Of note, EGFR (n=8) and KRAS (n=8) mutations, which were previously not detected by conventional sequencing, were identified and were mutually exclusive. EGFR mutations were enriched in tumors from females (P = 0.03). Of 8 EGFR mutations, 3 were not previously covered by the primer in the detection kit (M137I, Y891C, D770A). Of 8 KRAS mutations, 2 were not previously covered by the primer in the detection kit (Q61H, K179N). Mutations in tumor suppressor genes including STK11 (n=4), NF1 (n=2), RB1 (n=2), CDKN2A (n=4) were observed. Mutations in chromatin modifying genes ARID1A (n=2), MLL2 (n=2) were also observed.
Clinically relevant genomic alterations
Based on National Comprehensive Cancer Network (NCCN) guidelines, we identified actionable genomic alterations with a matched targeted agent. Sixteen patients (39%) had previously known driver alterations: BRAF mutation [n=1], EGFR mutation [n=8], ERBB2 mutation [n=4], MET amplification [n=1], KIF5B-RET rearrangement [n=2], CCDC6-RET rearrangement [n=1], CD74-ROS1 rearrangement [n=1], EZR-ROS1 rearrangement [n=5], and SLC34A2-ROS1 rearrangement [n=1].
In addition, genomic alterations for which targeted therapy could be considered in clinical trials were discovered in 14 patients (27%). These include the following alterations and the corresponding therapy: NF1 mutation (MEK inhibitor, NCT01885195), KRAS mutation (MEK inhibitor, NCT00890825) CDKN2A loss and CDK4 amplification (CDK4/6 inhibitor, NCT01237236), MDM2 amplification (MDM2 inhibitor, NCT01877382) and PIK3CA mutation (PI3K inhibitor, NCT01570296).
Seven patients with ROS1 rearrangements were enrolled in an ongoing trial (NCT01964157) and received ceritinib, a ROS1 inhibitor. CD74-ROS1, EZR-ROS1, SLC34A2-ROS1 rearrangements were identified and detailed characterization of ROS1 rearrangements are outlined in Figure 2. Of note, one patient had ROS1 gene without kinase domain fused with SLC34A2.
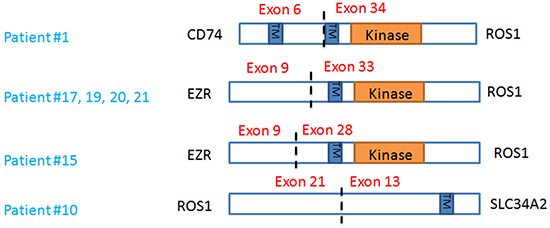
Figure 2: Characterization of ROS1 rearrangements.
DISCUSSION
In this study, we found that 16 of 51 (31%) patients harbored clinically actionable alterations that were not previously discovered in the routine clinical testing. This finding indicates an important opportunity to use targeted therapeutic strategies which will affect overall survival of these patients. In fact, 7 patients with ROS1 rearrangement received ceritinib and 6 patients showed objective radiologic responses.
In recent years, the treatment of lung adenocarcinoma has been advanced by the development of multiple targeted therapies against receptor tyrosine kinases, RAS and RAF pathways. Still, lung adenocarcinomas without known oncogenes are treated with platinum-double chemotherapy as the first-line treatment. It is well-known that platinum-based chemotherapy has generated a plateau in the overall response rate (25-35%), time to progression (4-6 months) and the median overall survival (8-10 months). Our analysis of examining oncogene-negative lung adenocarcinomas expands the range of possible targetable alterations that may be applied to patients who have no other options but chemotherapy. For example, there were 16 patients with previously known driver alterations, and vemurafenib, crizotinib, afatinib, cabozantinib could be used for BRAF V600E mutation, MET amplification, ROS1 rearrangements, HER2 mutations and RET rearrangements, respectively.
A similar study was recently published which identified actionable GAs in lung adenocarcinomas using broad, hybrid, capture-bsed NGS [7]. In this study, NGS identified actionable GAs in 65% of tumors from never or light smokers deemed wild type by extensive non-NGS testing. Never or light smokers (≤15 pack years) were selected to enrich the diagnostic yield for potential drivers, but this approach may miss important driver oncogenes such as BRAF and KRAS mutations which are enriched in tumors from patients with a history of smoking. A recent study of using molecularly matched therapy in lung cancer has shown that patients whose genomic alterations were matched to a targeted therapy lived significantly longer than those who received non-targeted therapies [8]. In this study, Kris et al. identified a driver oncogene in 64% of tumors in which no such genomic alteration was initially found on non-NGS testing. Discordant results between the conventional sequencing and CGP imply that potentially clinically actionable targets may not be detected from in clinical care. Similarly, EGFR and KRAS mutations were uncovered in 16 (31%) patients in our study. Possible reasons for the discordant results are low sensitivity of detection method and intratumoral heterogeneity. Genomic alterations with low allele frequencies lead to false-negative results on conventional sequencing [9], and subclonal mutations may be heterogeneous according to biopsy sites [10].
Of note, alterations potentially predicting sensitivity to cyclin-dependent kinase (CDK) inhibitors such as CDKN2A loss, CDK4 amplification, CCND1 amplification were common in our patients, with 21 cases in total [11, 12]. Among these, the most frequently altered gene was CDKN2A homozygous loss, with 10 events. MCL1 amplifications, which preclinical studies have shown to be associated with sensitivity to CDK inhibitors, were also found in 2 patients [13]. Co-amplification of CDK4 and MDM2 were noted in 3 patients. CDK4 is located on the band q13 of chromosome 12, and this region contains MDM2, which is often amplified together in a variety of sarcomas such as liposarcoma, and rhabdomyosarcoma [14]. It is not yet known whether co-amplification of CDK4 and MDM2 are predictive biomarkers of CDK inhibitors, and functional validation in laboratory may be warranted in the future.
Genomic alterations that affect the PI3K-AKT-mTOR pathway were also frequent, with 17 cases (33%) identified. Although functional consequences of these alterations are not all clear, one patient harbored a PIK3CA E542K mutation, which is previously reported to activate the PI3K-AKT pathway and could benefit from the use of PI3K inhibitor [15].
Although TCGA group has identified NF1 to be substantially mutated in adenocarcinomas [5], there was none identified in our patients. Since the majority of patients were never-smokers, it is reasonable that NF1 mutations are differentially enriched in transversion-high tumors. In addition, MET activation through exon 14 skipping has recently been identified as unique molecular subtype of NSCLC [16]. In this study, we identified 1 patient who harbored MET mutation (S163Y) with concurrent MET genomic amplification. It is questionable that this patient may show response to c-MET inhibitor such as crizotinib.
In conclusion, we note that more comprehensive genomic characterization of the tumor reveals actionable alterations in triple-negative lung adenocarcinoma patients. All patients who received matched targeted therapy derived clinical benefit showing objective responses or evidence of tumor shrinkage. This study highlights previously unappreciated genetic alterations, enabling further refinement in sub-classification for the improved personalization of lung cancer treatment.
MATERIALS AND METHODS
Patient selection
Patients with lung adenocarcinomas who visited Yonsei Cancer Center (Seoul, Republic of Korea) between 2013 and 2015 were identified. Eligible patients had previously underwent standard molecular testing for EGFR/KRAS mutation and EML4-ALK rearrangement, and were tested triple-negative. These patients were enrolled in the prescreening part of an ongoing phase 2 trial, “An open-label, multicenter, phase 2 study of LDK378 in patients with non-small cell lung cancer harboring ROS1-rearrangement” (NCT01964157). Tissue and clinical information of patients were collected under a protocol approved by the Institutional Review Board of Severance Hospital. All patients provided written informed consent for the genetic analysis.
Conventional testing for EGFR, KRAS and EML4-ALK
EGFR and KRAS mutations were determined by PNAClamp mutation detection kits (Panagene Inc., Daejeon, Republic of Korea). A total of 40 known mutations are detected in the exons 18-21 of EGFR gene, and a total of 14 mutations are detected in the exons 2-3 of KRAS gene. Break-apart fluorescent in situ hybridization (FISH) assays (Abbott Molecular, Inc. USA) were used to screen for EML4-ALK rearrangement, and positivity was defined as ≥ 15% of break-apart signals as described previously [17].
NGS-based comprehensive genomic profiling assay
The archived tissue samples were histologically reviewed by an experienced pathologist (H.S.S) and analyzed by CGP at Foundation Medicine. Submitted specimens underwent additional pathologic review to make sure tissue adequacy (≥ 20% tumor nuclei and ≥ 50ng of DNA) before testing. DNA was extracted from FFPE samples and quantified by a Picogreen fluorescence assay. After DNA extraction of 50-100ng, library construction and hybrid capture of 4,557 exons of 287 cancer-related genes and 47 introns of 19 genes frequently rearranged in cancer was performed. Hybrid capture libraries were sequenced to > 500x average coverage with >100x at >99% of exons using Illumina HiSeq 2500 sequencer. A customized analysis pipeline was used to process sequencing data and genomic alterations such as base substitutions, short insertions and deletions, copy number alterations and genomic rearrangements were detected and reported [6].
Data collection
Medical records of all patients and radiologic images were reviewed to evaluate demographic and clinicopathologic parameters, tumor response and survival outcome using a predesigned data collection format. Never-smokers were defined as those with a lifetime smoking-dose less than 100 cigarettes. Tumor response was assessed by a computed tomography scan in accordance with the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [18].
ACKNOWLEDGMENTS
This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI12C1440, B. C. Cho) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI13C2162 to S. Paik).
CONFLICTS OF INTEREST
SMA and JRG are employees of and have equity interest in Foundation Medicine, Inc.
REFERENCES
1. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J and Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004; 350:2129-2139.
2. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561-566.
3. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England journal of medicine. 2009; 361:947-957.
4. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, Moro-Sibilot D, Camidge DR, Mok T, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England journal of medicine. 2013; 368:2385-2394.
5. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543-550.
6. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, Iwanik K, Maillet A, Buell J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013; 31:1023-1031.
7. Drilon A, Wang L, Arcila ME, Balasubramanian S, Greenbowe JR, Ross JS, Stephens P, Lipson D, Miller VA, Kris MG, Ladanyi M and Rizvi NA. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clinical cancer research. 2015; 21:3631-3639.
8. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba, II, Varella-Garcia M, Franklin WA, Aronson SL, Su PF, Shyr Y, Camidge DR, Sequist LV, Glisson BS, Khuri FR, Garon EB, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama. 2014; 311:1998-2006.
9. Han HS, Lim SN, An JY, Lee KM, Choe KH, Lee KH, Kim ST, Son SM, Choi SY, Lee HC and Lee OJ. Detection of EGFR mutation status in lung adenocarcinoma specimens with different proportions of tumor cells using two methods of differential sensitivity. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012; 7:355-364.
10. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, Gold KA, Kalhor N, Little L, Mahadeshwar H, Moran C, Protopopov A, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014; 346:256-259.
11. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G and Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast cancer research : BCR. 2009; 11:R77.
12. Logan JE, Mostofizadeh N, Desai AJ, E VONE, Conklin D, Konkankit V, Hamidi H, Eckardt M, Anderson L, Chen HW, Ginther C, Taschereau E, Bui PH, Christensen JG, Belldegrun AS, Slamon DJ, et al. PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer research. 2013; 33:2997-3004.
13. Booher RN, Hatch H, Dolinski BM, Nguyen T, Harmonay L, Al-Assaad AS, Ayers M, Nebozhyn M, Loboda A, Hirsch HA, Zhang T, Shi B, Merkel CE, Angagaw MH, Wang Y, Long BJ, et al. MCL1 and BCL-xL levels in solid tumors are predictive of dinaciclib-induced apoptosis. PloS one. 2014; 9:e108371.
14. Khatib ZA, Matsushime H, Valentine M, Shapiro DN, Sherr CJ and Look AT. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer research. 1993; 53:5535-5541.
15. Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, Dela Vega T, Kenski DM, Bowman KK, Lorenzo M, Li H, Wu J, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer cell. 2009; 16:463-474.
16. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, Heng JC, Dahlberg SE, Janne PA, Verma S, Christensen J, Hammerman PS and Sholl LM. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. Journal of clinical oncology. 2016.
17. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, Settleman J, Kobayashi S, Mark EJ, Rodig SJ, Chirieac LR, Kwak EL, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. Journal of clinical oncology. 2009; 27:4247-4253.
18. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D and Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European journal of cancer. 2009; 45:228-247.