
INTRODUCTION
Leukocytes play a central role in our innate and adaptive immunity during both physiological and pathological conditions. As a prerequisite for the elicitation of an effective immune response, these immune cells have to be positioned precisely, in a timely manner, at specific lymphoid and non-lymphoid organs. To achieve this, the leukocyte cytoskeleton is highly flexible and versatile to support rapid and drastic reorganization in response to various stimuli. As such, they are able to generate rapid gliding movements with a migratory velocity of up to 10 µm/min for dendritic cells [1] and 30 µm/min for neutrophils [2], which is almost 100-fold faster than other mesenchymal and epithelial cell types. This gliding motion endows leukocytes with extraordinary migratory capacity that can be described as amoeboid, a migratory pattern reminiscent of the amoeba, Dictyostelium discoideum [3]. During amoeboid movement, polarized leukocytes extend their plasma membrane at the leading edge in response to chemoattractants through the polymerization of filamentous actin to form a growing pseudopod. Following attachment of the pseudopod to the underlying substratum, actomyosin contraction at the cell centre transduces an internal force to propel the rigid cell nucleus forward. The trailing edge of the cell then detaches itself from the matrix, facilitating advancement of the cell [4]. Such frequent shape changes experienced by leukocytes during amoeboid migration enables them to squeeze through physically constrained regions of the extracellular matrix, circumventing the need for proteolytic degradation and therefore preserving the structural integrity of the tissue environment [5].
Adhesion-dependent amoeboid movement of leukocytes
In order to fulfil their immune functions, leukocytes have to transverse through different tissues composed of distinct extracellular matrices in the body. Depending on the topology of the tissue structures encountered, leukocytes can adopt either an adhesion-dependent or adhesion-independent form of amoeboid movement. When transmigrating across two-dimensional tissue surfaces, such as vessel walls and basement membranes, leukocytes have to rely largely on integrin-mediated adhesion for their effective migration. In this case, the integrin repertoire on the leukocyte surface determines its affinity for the matrix present in the tissue environment. For instance, as circulating blood neutrophils tether and roll along the endothelium, chemoattractant-induced activation of their G-protein-coupled receptors triggers the activation of their surface β2-integrins, αLβ2 and αMβ2, thereby promoting their attachment to the ligand, ICAM-1, expressed on the endothelial cells [6]. As a result of this integrin-mediated adhesion, neutrophils are able to arrest themselves and bind tightly to the endothelial barrier to overcome the shear stress from the flowing blood. This adhesion facilitates the subsequent extravasation of neutrophils from the vascular lumen into the interstitial tissue, hence delivering an effective immune response.
Similar to neutrophils, circulating dendritic cells (DCs) or DC precursors in the blood make use of the integrins, αLβ2 and α4β1, for their firm binding to the endothelial ligands, ICAM-1 and VCAM, respectively. This binding facilitates their trafficking into target tissues during both homeostatic and inflammatory conditions, such as experimental autoimmune encephalomyelitis [7]. On the other hand, Langerhans cells exiting the epidermis have to transverse through a basement membrane network enriched in laminin, type IV collagen and proteoglycans [8] before arriving at the dermal afferent lymphatics for further migration to the skin-draining lymph nodes. In this case, α6 integrin-mediated adhesions have been reported to be critical for attachment and subsequent transmigration across the basement membrane [9].
Adhesion-independent amoeboid movement of leukocytes
Intriguingly, leukocyte migration in a confined three-dimensional environment was found to be driven primarily by actomyosin contractions, rather than integrin-based adhesions [10]. This was demonstrated by integrin- or talin1-deficient dendritic cells, which, surprisingly, were able to migrate from the dermis to the skin-draining lymph nodes, a route which does not require crossing of any tissue barriers, in a manner indistinguishable from their wild-type counterparts. In this case, the protrusion of actin filaments is sufficient for leukocytes to initiate movement in regions of low matrix density, while myosin II-mediated contraction is only essential when leukocytes have to squeeze through dense regions of tissue interstitium. Indeed, disruption of the actin cytoskeletal network and inhibition of myosin contractile activity through latrunculin B and blebbistatin treatment, respectively, resulted in the migration arrest of leukocytes in a three-dimensional environment [10]. Such an adhesion-independent migration strategy serves to endow leukocytes with the capacity to navigate through different organs rapidly, without being restricted to predefined routes due to the limited integrin repertoire expressed on their cell surfaces.
Dynamic switching between different modes of migration supports leukocyte function
Rather than being confined to a particular migratory speed, leukocytes are capable of alternating among fast migration, slow migration and an arrest phase. This dynamic and plastic nature of their locomotion, which depends critically on the shift between adhesion-dependent and -independent migration, is tailored to support distinct functions of the leukocytes. For example, dermal dendritic cells are highly motile cells under steady state conditions, actively crawling through the dermal interstitium in an amoeboid fashion to survey the skin microenvironment for any invading pathogens. However, upon infection with the protozoan parasite Leishmania major, they undergo rapid migratory arrest at the infection site to maximise their antigen uptake capacity [11]. Similarly, antigen-specific T cells in the lymph nodes cease migration upon contact with antigen-presenting dendritic cells, forming a stable immunological synapse mediated by αLβ2-ICAM-1 interaction to facilitate full T cell activation [12]. Therefore, striking a balance between cell adhesion and migration in different leukocytes during each of their distinct functional phases plays a key role in modulating a protective immune response.
Post-translational modifications regulate cell migration
The cell migration machinery is comprised of a myriad of temporally and spatially segregated signalling and adhesion molecules that need to be orchestrated in a rapid and precise manner to support an efficient immune response. To achieve this, post-translational modifications (PTMs) are employed as ideal tools to confer dynamic and reversible regulation in migrating leukocytes. These modifications, which can either act alone or in combination, help to expand the genetic code by increasing the functional diversity of proteins. To date, more than 400 kinds of modifications have been identified [13]. Here, we summarize the major PTMs that have been reported for various cytoskeletal and adhesion-related molecules (Table 1) and highlight their importance with regards to leukocyte migration.
Table 1: Selected post-translational modification sites identified on cytoskeletal and adhesion molecules.
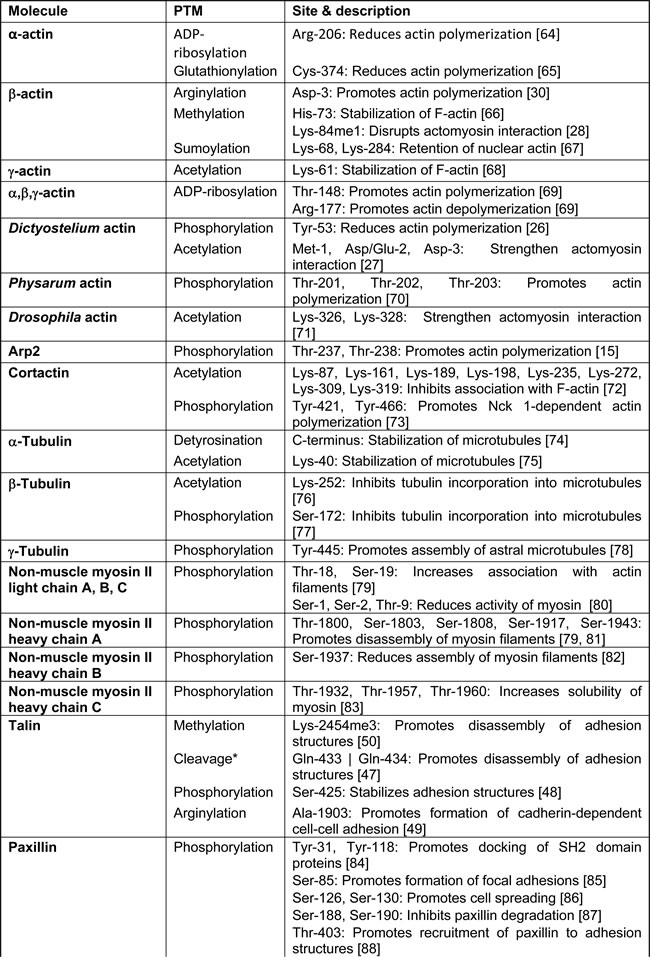
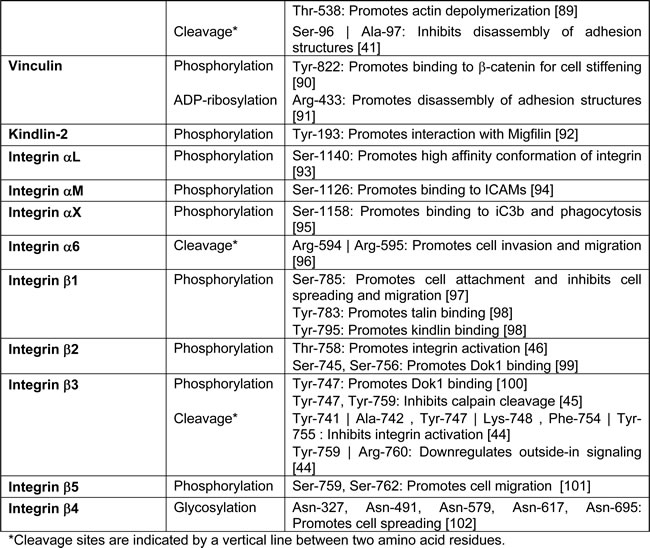
Regulation of actin dynamics
The early stage of leukocyte migration initiated by actin filament protrusion at the pseudopod is largely dependent on the interplay between actin polymerization and depolymerization. Such processes are regulated by a family of actin binding proteins including profilin, cofilin, and the actin-related protein (Arp) 2/3 complex. Arp2 is subjected to phosphorylation at Thr-237 and Thr-238 by Nck-interacting kinase (NIK) [14], which stabilizes the Arp2/3 complex in an active conformation required for its actin-nucleating activity [15]. Upon stimulation, activated Arp2/3 initiates nucleation of new actin filaments as branches on the sides of older actin filaments. Once nucleation occurs, profilin-bound ATP actin is incorporated onto the barbed ends of the new actin filaments, resulting in the growth of actin filaments that push the cell membrane forward and promote pseudopod extension. Extracellular stimulation also leads to the release of active cofilin from its inhibitory membrane binding through hydrolysis of PIP2 [16, 17]. Active cofilin at the tip of the leading edge in the proximity of the plasma membrane generates severed actin filaments that are then used as free barbed ends for further actin polymerization [18]. Meanwhile, the activated serine/ threonine kinase, LIM kinase, can also inactivate cofilin through Ser-3 phosphorylation, which probably slows down the rate of filament disassembly behind the leading edge. This inactive cofilin accelerates the dissociation of ADP•G-actin-cofilin and increases the concentration of free ATP•G-actin monomers, making them available for filament elongation at the tip of the leading edge [19, 20]. Interestingly, branches nucleated by Arp2/3 on new actin filaments generated through severing are ten times more stable than those nucleated on old actin filaments [18, 21], which could be the primary mechanism driving the formation of the dendritic actin network at the leading edge. Conversely, during migration arrest or directional change, pseudopods have to be retracted. Under these conditions, the absence of activating signals causes the un-phosphorylated Arp2/3 complex to remain inactive and the type 1 (PP1) and type 2A (PP2A) serine/threonine phosphatases to dephosphorylate and activate cofilin [22], thus increasing contractility at the cell rear through F-actin depolymerization [23].
Apart from the modifications on actin binding proteins, which can indirectly help regulate actin properties, actin itself is also susceptible to many PTMs. Phosphorylation is one of the most prominent modifications found on actin and it is capable of exerting both positive and negative regulatory influences on polymerization. For example, phosphorylation of actin at Thr-201, Thr-202 and Thr-203 by actin-fragmin kinase is known to inactivate the fragmin-actin complex, a complex which promotes the formation of short actin filaments, thereby facilitating the elongation of actin [24, 25]. On the other hand, phosphorylation at Tyr-53 can reduce the rate of nucleation and inhibit the elongation of actin filaments [26]. Thus, different levels of regulation can be imposed through the addition of a single moiety. Moreover, various acetylation sites modified by different acetyltransferases and deacetylases have also been reported on actin. Notably, acetylation of actin at the N-terminus, which has been identified on all kinds of actin, neutralizes a positive charge to strengthen its binding to myosin during the actomyosin ATPase cycle [27], whereas mono-methylation at Lys-84 disrupts the interaction between actin and myosin II [28]. Such interaction plays a critical role in regulating the contractility of the cell, which helps to control migration and other cellular functions. In addition, arginylation of β-actin at Asp-3 by arginyltransferase (Ate1) is known to be necessary for actin polymerization and hence the maintenance of a normal cellular cytoplasmic architecture [29]. In the absence of arginylation, actin filaments undergo aggregation and sequestration at the cell centre, resulting in the collapse of the leading edge, which consequently causes a migration defect in the cell [30]. Taken together, these findings demonstrate the importance of post-translational modifications in the regulation of actin dynamics, which is the key determinant underlying pseudopod formation during cell migration.
Regulation of actomyosin contraction
Following extension and attachment of the pseudopod to the underlying substrate, actomyosin-mediated contraction of the cell body has to occur to generate forward locomotion. Here, reversible phosphorylation of the motor protein, myosin II, plays an essential role in controlling force generation during migration. Unphosphorylated myosin is inactive and exists in a compact, assembly-incompetent conformation through head-head and head-tail interactions [31]. Upon phosphorylation of Thr-18 and Ser-19 on the regulatory light chain by myosin light chain kinase (MLCK), myosin unfolds to adopt an assembly-competent conformation, thereby increasing its association with actin filaments and also its ATPase activity for the initiation of contraction. This regulation can be counteracted by protein phosphatase 1, which acts to dephosphorylate myosin. In dendritic cells, the distribution of phosphorylated myosin to either the cell front or rear gives rise to fluctuating migratory speeds, which serves to couple their antigen uptake function with their migratory capacity [32, 33]. Several phosphorylation sites have also been identified on the C-terminal region of the myosin heavy chain, including Ser-1917 and Ser-1943. Unlike those found on the regulatory light chain, phosphorylation sites on the heavy chain are associated with the disassembly of myosin filaments. Hence, by making use of phosphorylation switches, myosin is able to modulate its actin-binding and contractile activities to control cell migration.
Regulation of cell polarization
Following actomyosin contraction, the trailing edge of the leukocyte, termed the uropod, detaches itself from the underlying substratum and undergoes retraction to facilitate forward movement of the cell. Even though contraction of the uropod is dispensable for migration, it has been implicated in promoting shape changes associated with migration through constricted regions of the body. The uropod of motile leukocytes contains the microtubule organizing centre (MTOC) and an extensive network of microtubules, unlike fibroblast-like motile cells, which generally do not form a uropod and place the MTOC between the nucleus and the leading edge [34]. Microtubules, polymers composed of α- and β-tubulin subunits, are uniquely determined by their PTMs, which most likely exert their effects by regulating the binding partners and stability of microtubules. One of the earliest PTMs identified on microtubules was the acetylation of Lys-40 on the β-tubulin subunit. Even though this highly conserved modification is known to be associated with stable and long-lived microtubules, it remains enigmatic how this deeply buried acetyl group within the luminal face of the microtubule can bring about such properties [35]. Interestingly, acetylated microtubules are frequently distributed around the MTOC in the uropod of activated T cells, although their function remains unknown [36]. On the other hand, deacetylation of Lys-40 by histone deacetylase 6 (HDAC6) has been associated with the decreased stability of microtubules and hence an enhanced turnover of adhesion structures [37]. Indeed, disassembly of microtubules that radiate towards the uropod has been reported to enhance the motility of neutrophils [38]. Although many other post-translational modification sites have been mapped out on tubulin, their functional roles in mediating leukocyte migration remain poorly understood.
Regulation of cell adhesion
During integrin-dependent migration, the protruding pseudopod needs to be stabilized by the formation of new adhesions at the leading edge. Although fast-migratory cell types, like leukocytes, do not usually form visible adhesion structures to mediate their rapid gliding movements, larger adhesion structures may be induced by stimuli to cause the arrest of their migration [39]. Since the dynamic assembly and disassembly of adhesion structures is pivotal to the balance between migration and adhesion, many of these adhesion-related molecules are heavily modified by PTMs. Paxillin is one such focal adhesion-associated adaptor molecule that serves to recruit various cytoskeletal proteins for the transmission of downstream signalling. Phosphorylation of two sites in paxillin (Tyr-31 and Tyr-118), mediated by Src, function to facilitate docking of downstream SH2-containing signalling molecules that subsequently recruit focal adhesion kinase (FAK) to promote the turnover of adhesion structures. Therefore, cells are able to control their adhesion dynamics by regulating the relative proportion of unphosphorylated and phosphorylated paxillin [40]. In addition, paxillin is also susceptible to calpain-mediated proteolysis, which generates an inhibitory paxillin fragment that impairs the turnover of adhesion structures and hence impedes cellular migration [41]. Indeed, macrophages experiencing an increase in calpain-mediated cleavage of paxillin due to a deficiency in CD45, a tyrosine phosphatase, suffer from defects in their cell spreading and migration programmes [42].
Integrins are transmembrane receptors that help to link the extracellular matrix with the actin cytoskeleton. Regulation of these proteins during cell migration and adhesion occurs either through direct post-translational modification or through the PTM of various adaptor proteins such as paxillin, talin, and vinculin. Calpain-mediated cleavage of the integrin cytoplasmic domain has been shown to downregulate outside-in signalling and hence reduce cell adhesion and spreading [43, 44]. This proteolysis, however, can be antagonized by the phosphorylation of the integrin β3 cytoplasmic domain at Tyr-747 and Tyr-759 [45]. Furthermore, the interaction of the integrin β2 cytoplasmic domain with either filamin or talin, which share overlapping binding sites, but possess opposing functions, is modulated through phosphorylation of Thr-758 by protein kinase C (PKC) [46]. This modification favours the binding of talin over filamin, thereby promoting the activation of integrins and their downstream signalling pathways. Such binding of talin to the integrin cytoplasmic tail can, in turn, be antagonized by calpain-mediated proteolysis of talin [47], resulting in the formation of separate head and rod domains. The head domain of talin, which remains integrin-bound, can be ubiquitinated by the E3 ligase, Smurf1, and subsequently degraded, leading to the disassembly of adhesion structures. However, phosphorylation of the head domain by Cdk5 can inhibit the binding of Smurf1 and stabilize focal adhesions [48]. In addition to the head and tail domain, calpain-mediated proteolysis can also generate a 70kDa talin fragment that has been reported to be arginylated on Ala-1903. This modified talin fragment is found to be essential for the formation of cadherin-dependent cell-cell adhesion [49]. More recently, we have identified a talin methylation site on Lys-2454 that plays a critical role in the regulation of leukocyte migration [50]. Methylated talin exhibits reduced binding affinity for F-actin, which promotes the disassembly of adhesion structures and facilitates cellular migration. Collectively, these data demonstrate how adhesion dynamics and cell migration can be intricately regulated by an extensive array of post-translational modifications. In the following paragraphs, we will focus on the enzyme that mediates talin methylation and the functional implications of methylated talin.
Ezh2 regulates integrin-dependent migration of leukocytes
We have recently reported that Ezh2, a polycomb group protein and well-established chromatin modifier that regulates gene expression through the methylation of lysine 27 on histone H3 (H3K27me), is recruited to talin by Vav1 in the cytoplasm. This recruitment promotes the direct methylation of talin, which alters the binding of talin to filamentous actin and exerts a major influence on cell migration and adhesion dynamics [50]. Our initial observation was that Ezh2-deficient bone marrow-derived mature dendritic cells (DCs) exhibited significantly reduced migratory capacity on integrin ligand coated surfaces compared to control cells. While the fast migrating wild-type mature DCs barely formed any visible adhesions, Ezh2-deficient DCs spread out extensively and frequently formed large and stable adhesion structures with reduced turnover rates. Most of these cells appeared to be trapped on the culture slide and some were observed to be oscillating at an anchored position, as if fighting against the restraining forces created by their binding to the integrin ligands. This migratory defect could be rescued by the expression of wild-type or cytosolic Ezh2, but not an enzymatically inactive Ezh2 or an Ezh2 mutant harbouring mutations that disrupt interactions with the cytoskeletal-reorganization effector, Vav1. Interestingly, when Ezh2-deficient DCs were cultured on surfaces coated with low concentrations of integrin ligands, they formed neither abnormally stable adhesion structures nor spread out extensively. Since the amount of integrin ligand was limited in this case, we believe that the Ezh2-deficient DCs were able to overcome the restraining forces caused by the reduced turnover of adhesion structures and migrate in manners similar to control cells. Taken together, these results demonstrate the integrin-dependent nature of Ezh2-regulated leukocyte migration.
Ezh2-mediated talin methylation promotes adhesion turnover and cell migration
While Ezh2-deficient DCs exhibited clear defects in cell migration, no obvious differences in H3K27me3 levels or gene expression patterns were found between Ezh2-deficient DCs and control cells [50]. However, our whole proteome analysis revealed that various proteins involved in actin cytoskeletal functions were up-regulated (Gunawan et al., unpublished data). Since some of these proteins have the potential to enhance, while others prevent, cell migration or adhesion turnover, we concluded that the phenotype of Ezh2-deficient DCs is therefore unlikely to be caused by their altered expressions. Moreover, we hypothesized that Ezh2-deficiency may affect the function of an integrin proximal molecule, subsequently leading to the formation of stabilized FAs and an accumulation of proteins associated with actin/adhesion structures or the regulation of adhesion dynamics.
Indeed, we found that Ezh2 mediates the tri-methylation of talin1 at lysine (K) 2454 and further demonstrated in vitro that such methylation interferes with the binding of talin to F-actin [50]. We further determined that K2454 methylation is critical for the regulation of adhesion dynamics and cell migration by expressing wild-type talin or talin mutants in which the lysine residue was replaced with either an un-methylatable glutamine (Q) residue or with a methyl-mimicking phenylalanine (F) residue (resembles the bulkiness and hydrophobicity of a tri-methylated lysine) in control and Ezh2-deficient DCs. Under all tested conditions, expression of the methyl-mimicking talin1-K2454F mutant restored normal migration and reversed the excessive spreading phenotype of Ezh2-deficient DCs, whereas expression of the un-methylatable talin1-K2454Q mutant converted control cells into Ezh2-deficient DC-like cells (Figure 1A, 1B) [50].
These results offer convincing support for our hypothesis that Ezh2-mediated talin1 methylation is an important contributing factor for the regulation of adhesion dynamics and cell migration. Furthermore, replacing K2454 with alanine in some of our experiments resulted in trends similar to those seen with glutamine substitution, despite the effects being less pronounced [50]. Since glutamine is not only used to mimic un-methylated lysine with preserved polarity [51], but is also frequently utilized as an acetyl-mimicking residue [52], it would be very interesting to determine whether K2454 may also be subjected to acetylation in vivo. In this case, the increased binding affinity of acetylated talin for F-actin may be required for the formation of stable adhesion structures under certain physiological conditions.
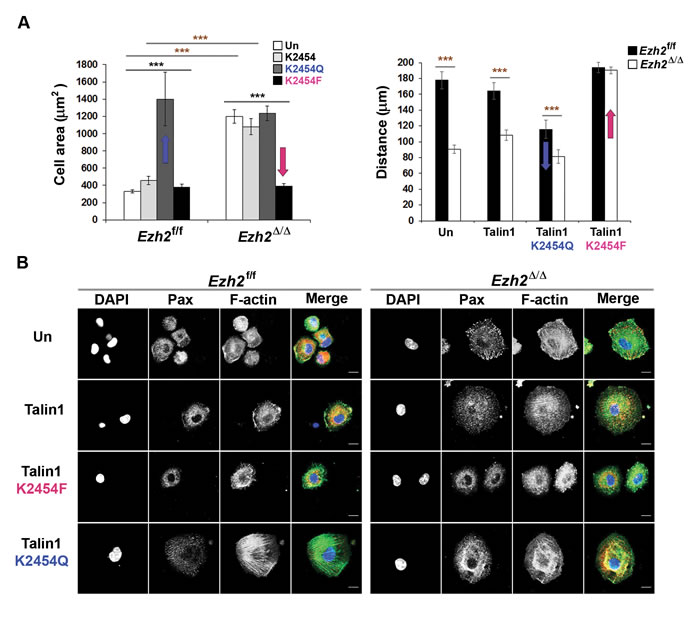
Figure 1: Tri-methyl lysine mimicking talin1 mutant promotes FA turnover and rescues excessive cell spreading and defective migration phenotypes of Ezh2-deficient DCs. A. Control and Ezh2-deficient DCs expressing GFP-talin1 variants were allowed to adhere to slides coated with VCAM-1 (20 µg/ml) for 2 h. The cell areas were visualized by GFP staining and calculated using ImageJ (left) or time-lapse images were taken every 5 min for 2 h (right). “Un” indicates untransduced control. ***P <0.0001 (black asterisks: one-way ANOVA, red asterisks: between the indicated pairs, two-tailed student’s t-test with equal variance). Data are represented as mean ± standard error of the mean (SEM) of cells pooled from 2-4 independent experiments. B. Control and Ezh2-deficient DCs expressing GFP-talin1 variants were allowed to adhere to VCAM-1 coated slides as in A for 2 h. FAs and F-actin were visualized by anti-paxillin (Pax, red) and Alexa Fluor® 647 phalloidin (pseudo-colored green), respectively. Over 90% of the cells were GFP+ expressing talin variants and the GFP staining is not shown. Scale bar, 10 µm. Technical details and original data are published in reference [50].
Integrin-dependent migration of circulating DC precursors and Langerhans cells
Integrin-dependent migration prevails during leukocyte extravasation from the bloodstream into inflamed tissues. However, this process is irrelevant for most DC subsets since they are already residing in the peripheral tissues. In this case, the migration of antigen loaded, active DCs from the periphery to the draining lymph nodes relies on actin polymerization and actomyosin contractions, processes that are independent of Ezh2 function. Accordingly, we recovered similar percentages of FITC-positive migratory dermal DCs in the skin-draining lymph nodes of control and DC-specific Ezh2-deficient mice one day following skin painting with the FITC dye, indicating that integrin-independent migration is intact in Ezh2-deficient DCs [50].
However, unlike other disease models, experimental autoimmune encephalomyelitis (EAE) is one of the exceptional conditions whereby trans-endothelial migration of DCs is critical for disease progression. The extravasation of circulating DCs or DC precursors across the blood-brain barrier into the inflamed tissues of the central nervous system (CNS) requires α4β1 integrin-mediated adhesions [7] and the number of infiltrating DCs has been reported to correlate positively with disease progression [53-55]. In our recent study of EAE, DC-specific Ezh2-deficient mice that exhibited reduced disease scores, compared to control mice, were found to harbor reduced numbers of CNS infiltrating DCs and increased incidences of DCs associated with the microvasculature [50]. These migratory defects observed in Ezh2-deficient DCs in vivo are likely to be caused by mechanisms similar to those observed in vitro. Furthermore, migration of Langerhans cells (LCs) across the basement membrane requires the binding of integrin α6 expressed on LCs to the integrin ligand, laminin [9], present in the basement membrane layer. Ezh2-deficient LCs, which fail to migrate through the basement membrane under steady state and inflammatory conditions, were found to spread out extensively and form enlarged focal adhesions on laminin coated slides, resembling the phenotype of bone marrow derived DCs [50] (Loh et al., unpublished data). Since LCs have been implicated in the regulation of tolerance induction to skin sensitizers, Ezh2-regulated LC migration may be relevant for the disease progression of allergic contact dermatitis.
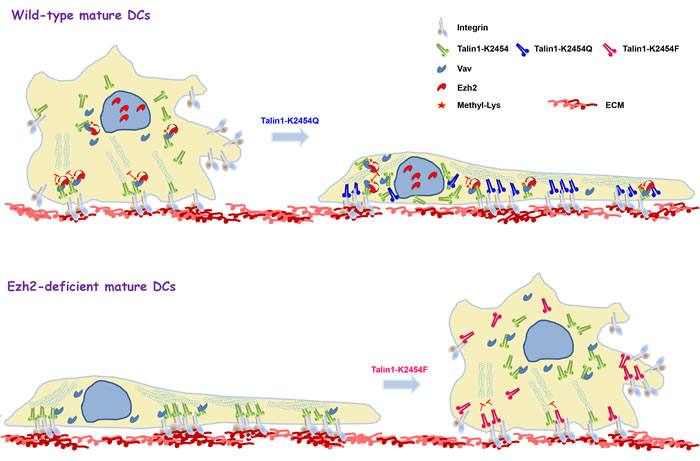
Figure 2: Schematic model for Ezh2-regulated cell adhesion and migration through direct methylation of talin1. Ezh2 is recruited to talin1 through interaction with the cytoskeletal-reorganization effector, Vav1 and mediates talin1 methylation, thereby reducing talin1 binding to F-actin and promoting adhesion turnover. Overexpression in wild-type mature dendritic cells of a mutant talin1 that was created by replacing the lysine at position 2454 with glutamine, which cannot be methylated, but preserves the polarity of the lysine (K2454Q), results in extensive cell spreading that resembles the phenotype of Ezh2-deficient mature dendritic cells (top). In contrast, overexpression in Ezh2-deficient mature dendritic cells of a methyl-mimicking talin1 mutant that was created by replacing the K2454 residue with phenylalanine (K2454F) restores normal cell spreading and migration (bottom).
Conclusions and future directions
Integrin-dependent adhesion and migration of leukocytes are critically regulated by post-translational modifications that include phosphorylation, acetylation, sumoylation, ubiquitination and proteolytic cleavage of actin cytoskeletal proteins. These PTMs are frequently amendable by a pair of enzymes that can add or remove modifications on the target proteins. Such dynamic regulation of PTMs enables temporally and spatially controlled signalling cascades and thereby ensures rapid and efficient recruitment of leukocytes in response to invading pathogens.
Recent data published by our lab expands the current role of PTMs in integrin-dependent signalling in leukocytes by suggesting a novel mechanism by which the polycomb group protein, Ezh2, promotes adhesion turnover through direct methylation of the extra nuclear protein, talin1. Interestingly, this Ezh2-mediated talin1 methylation was found to be dependent on its interaction with the cytoskeleton remodelling effector, Vav1 [50]. Subsequent work in our lab has shown that the Ezh2 interaction domain of Vav1 is highly conserved among the Vav family of proteins and that the Ezh2:Vav:talin complexes observed in leukocytes are also formed in non-hematopoietic cells (Venkatesan et al., unpublished). These results suggest that the novel mechanism described here is likely to be conserved across various cell types and may have important implications for tumor growth and metastasis. In fact, Ezh2 overexpression or gain of function mutations are known to be associated with several aggressive human solid tumour types, including prostate cancer, breast cancer, and different types of lymphomas, and are indicators of poor prognosis in patients [56-58]. These Ezh2-dependent oncogenic effects can be achieved though H3K27me3-mediated transcriptional repression of tumor suppressor genes or through promoting NF-κB target gene expression [59-63]. However, our findings suggest that Ezh2-mediated talin methylation may also promote adhesion turnover in cancer cells, resulting in altered adhesion properties that may directly contribute to the epithelial-to-mesenchymal and mesenchymal-to-amoeboid transition of metastasizing cancer cells (Venkatesan et al., unpublished). Furthermore, it is likely that Ezh2 mediates the methylation of additional proteins to modulate other cytosolic signaling events. In addition to talin, several other cytoplasmic proteins were observed to be methylated by Ezh2 in vitro (Venkatesan et al., unpublished), so more studies will now be required to determine their identities and physiological relevance.
In conclusion, PTMs play critical roles in regulating cytoskeletal/adhesion dynamics and further research in this area will lead to a better understanding of the regulation of leukocyte trafficking with implications for disease pathogenesis and cellular transformation.
ACKNOWLEDGMENTS
J.T.L. and I.S. are supported by SIgN-NTU A*STAR Graduate Scholarship and the National Research Council of Singapore (NMRC/CBRG/0057/2014), respectively. The manuscript was edited by Amy Sullivan from Obrizus Communications.
ConflictS of Interest
There is no conflict of interest.
REFERENCES
1. Gunzer M, Friedl P, Niggemann B, Brocker EB, Kampgen E and Zanker KS. Migration of dendritic cells within 3-D collagen lattices is dependent on tissue origin, state of maturation, and matrix structure and is maintained by proinflammatory cytokines. Journal of leukocyte biology. 2000; 67:622-629.
2. Werr J, Xie X, Hedqvist P, Ruoslahti E and Lindbom L. beta1 integrins are critically involved in neutrophil locomotion in extravascular tissue In vivo. The Journal of experimental medicine. 1998; 187:2091-2096.
3. Barry NP and Bretscher MS. Dictyostelium amoebae and neutrophils can swim. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:11376-11380.
4. Friedl P, Borgmann S and Brocker EB. Amoeboid leukocyte crawling through extracellular matrix: lessons from the Dictyostelium paradigm of cell movement. Journal of leukocyte biology. 2001; 70:491-509.
5. Wolf K, Muller R, Borgmann S, Brocker EB and Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 2003; 102:3262-3269.
6. Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R and Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005; 106:584-592.
7. Jain P, Coisne C, Enzmann G, Rottapel R and Engelhardt B. Alpha4beta1 integrin mediates the recruitment of immature dendritic cells across the blood-brain barrier during experimental autoimmune encephalomyelitis. Journal of immunology. 2010; 184:7196-7206.
8. Fleischmajer R, Utani A, MacDonald ED, Perlish JS, Pan TC, Chu ML, Nomizu M, Ninomiya Y and Yamada Y. Initiation of skin basement membrane formation at the epidermo-dermal interface involves assembly of laminins through binding to cell membrane receptors. Journal of cell science. 1998; 111:1929-1940.
9. Price AA, Cumberbatch M, Kimber I and Ager A. Alpha 6 integrins are required for Langerhans cell migration from the epidermis. The Journal of experimental medicine. 1997; 186:1725-1735.
10. Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, Keller M, Forster R, Critchley DR, Fassler R and Sixt M. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008; 453:51-55.
11. Ng LG, Hsu A, Mandell MA, Roediger B, Hoeller C, Mrass P, Iparraguirre A, Cavanagh LL, Triccas JA, Beverley SM, Scott P and Weninger W. Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS pathogens. 2008; 4:e1000222.
12. Shakhar G, Lindquist RL, Skokos D, Dudziak D, Huang JH, Nussenzweig MC and Dustin ML. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nature immunology. 2005; 6:707-714.
13. Lothrop AP, Torres MP and Fuchs SM. Deciphering post-translational modification codes. FEBS letters. 2013; 587:1247-1257.
14. LeClaire LL, Rana M, Baumgartner M and Barber DL. The Nck-interacting kinase NIK increases Arp2/3 complex activity by phosphorylating the Arp2 subunit. The Journal of cell biology. 2015; 208:161-170.
15. LeClaire LL, 3rd, Baumgartner M, Iwasa JH, Mullins RD and Barber DL. Phosphorylation of the Arp2/3 complex is necessary to nucleate actin filaments. The Journal of cell biology. 2008; 182:647-654.
16. Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J and Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008; 87:649-667.
17. Oser M and Condeelis J. The cofilin activity cycle in lamellipodia and invadopodia. J Cell Biochem. 2009; 108:1252-1262.
18. Ichetovkin I, Grant W and Condeelis J. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr Biol. 2002; 12:79-84.
19. Bravo-Cordero JJ, Oser M, Chen X, Eddy R, Hodgson L and Condeelis J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr Biol. 2011; 21:635-644.
20. Tania N, Prosk E, Condeelis J and Edelstein-Keshet L. A temporal model of cofilin regulation and the early peak of actin barbed ends in invasive tumor cells. Biophys J. 2011; 100:1883-1892.
21. Mahaffy RE and Pollard TD. Kinetics of the formation and dissociation of actin filament branches mediated by Arp2/3 complex. Biophys J. 2006; 91:3519-3528.
22. Ambach A, Saunus J, Konstandin M, Wesselborg S, Meuer SC and Samstag Y. The serine phosphatases PP1 and PP2A associate with and activate the actin-binding protein cofilin in human T lymphocytes. European journal of immunology. 2000; 30:3422-3431.
23. Mseka T and Cramer LP. Actin depolymerization-based force retracts the cell rear in polarizing and migrating cells. Curr Biol. 2011; 21:2085-2091.
24. Gettemans J, De Ville Y, Waelkens E and Vandekerckhove J. The actin-binding properties of the Physarum actin-fragmin complex. Regulation by calcium, phospholipids, and phosphorylation. The Journal of biological chemistry. 1995; 270:2644-2651.
25. Furuhashi K and Hatano S. Control of actin filament length by phosphorylation of fragmin-actin complex. The Journal of cell biology. 1990; 111:1081-1087.
26. Liu X, Shu S, Hong MS, Levine RL and Korn ED. Phosphorylation of actin Tyr-53 inhibits filament nucleation and elongation and destabilizes filaments. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:13694-13699.
27. Abe A, Saeki K, Yasunaga T and Wakabayashi T. Acetylation at the N-terminus of actin strengthens weak interaction between actin and myosin. Biochemical and biophysical research communications. 2000; 268:14-19.
28. Li MM, Nilsen A, Shi Y, Fusser M, Ding YH, Fu Y, Liu B, Niu Y, Wu YS, Huang CM, Olofsson M, Jin KX, Lv Y, Xu XZ, He C, Dong MQ, et al. ALKBH4-dependent demethylation of actin regulates actomyosin dynamics. Nature communications. 2013; 4:1832.
29. Saha S, Mundia MM, Zhang F, Demers RW, Korobova F, Svitkina T, Perieteanu AA, Dawson JF and Kashina A. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Molecular biology of the cell. 2010; 21:1350-1361.
30. Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR, 3rd, Mogilner A, Zebroski H and Kashina A. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 2006; 313:192-196.
31. Jung HS, Komatsu S, Ikebe M and Craig R. Head-head and head-tail interaction: a general mechanism for switching off myosin II activity in cells. Mol Biol Cell. 2008; 19:3234-3242.
32. Chabaud M, Heuze ML, Bretou M, Vargas P, Maiuri P, Solanes P, Maurin M, Terriac E, Le Berre M, Lankar D, Piolot T, Adelstein RS, Zhang Y, Sixt M, Jacobelli J, Benichou O, et al. Cell migration and antigen capture are antagonistic processes coupled by myosin II in dendritic cells. Nature communications. 2015; 6:7526.
33. Faure-Andre G, Vargas P, Yuseff MI, Heuze M, Diaz J, Lankar D, Steri V, Manry J, Hugues S, Vascotto F, Boulanger J, Raposo G, Bono MR, Rosemblatt M, Piel M and Lennon-Dumenil AM. Regulation of dendritic cell migration by CD74, the MHC class II-associated invariant chain. Science. 2008; 322:1705-1710.
34. Sanchez-Madrid F and Serrador JM. Bringing up the rear: defining the roles of the uropod. Nature reviews Molecular cell biology. 2009; 10:353-359.
35. Howes SC, Alushin GM, Shida T, Nachury MV and Nogales E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Molecular biology of the cell. 2014; 25:257-266.
36. Cabrero JR, Serrador JM, Barreiro O, Mittelbrunn M, Naranjo-Suarez S, Martin-Cofreces N, Vicente-Manzanares M, Mazitschek R, Bradner JE, Avila J, Valenzuela-Fernandez A and Sanchez-Madrid F. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Molecular biology of the cell. 2006; 17:3435-3445.
37. Tran AD, Marmo TP, Salam AA, Che S, Finkelstein E, Kabarriti R, Xenias HS, Mazitschek R, Hubbert C, Kawaguchi Y, Sheetz MP, Yao TP and Bulinski JC. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. Journal of cell science. 2007; 120:1469-1479.
38. Yoo SK, Lam PY, Eichelberg MR, Zasadil L, Bement WM and Huttenlocher A. The role of microtubules in neutrophil polarity and migration in live zebrafish. Journal of cell science. 2012; 125:5702-5710.
39. Huttenlocher A and Horwitz AR. Integrins in cell migration. Cold Spring Harbor perspectives in biology. 2011; 3:a005074.
40. Zaidel-Bar R, Milo R, Kam Z and Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. Journal of cell science. 2007; 120:137-148.
41. Cortesio CL, Boateng LR, Piazza TM, Bennin DA and Huttenlocher A. Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. The Journal of biological chemistry. 2011; 286:9998-10006.
42. St-Pierre J and Ostergaard HL. A role for the protein tyrosine phosphatase CD45 in macrophage adhesion through the regulation of paxillin degradation. PloS one. 2013; 8:e71531.
43. Pfaff M, Du X and Ginsberg MH. Calpain cleavage of integrin beta cytoplasmic domains. FEBS letters. 1999; 460:17-22.
44. Xi X, Bodnar RJ, Li Z, Lam SC and Du X. Critical roles for the COOH-terminal NITY and RGT sequences of the integrin beta3 cytoplasmic domain in inside-out and outside-in signaling. The Journal of cell biology. 2003; 162:329-339.
45. Xi X, Flevaris P, Stojanovic A, Chishti A, Phillips DR, Lam SC and Du X. Tyrosine phosphorylation of the integrin beta 3 subunit regulates beta 3 cleavage by calpain. The Journal of biological chemistry. 2006; 281:29426-29430.
46. Takala H, Nurminen E, Nurmi SM, Aatonen M, Strandin T, Takatalo M, Kiema T, Gahmberg CG, Ylanne J and Fagerholm SC. Beta2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood. 2008; 112:1853-1862.
47. Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR and Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nature cell biology. 2004; 6:977-983.
48. Huang C, Rajfur Z, Yousefi N, Chen Z, Jacobson K and Ginsberg MH. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nature cell biology. 2009; 11:624-630.
49. Zhang F, Saha S and Kashina A. Arginylation-dependent regulation of a proteolytic product of talin is essential for cell-cell adhesion. The Journal of cell biology. 2012; 197:819-836.
50. Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH, Li LY, La Win MK, Yau YH, Guo T, See PC, Yamazaki S, Chin KC, Gingras AR, Shochat SG, Ng LG, et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nature immunology. 2015; 16:505-516.
51. Huq MD, Tsai NP, Khan SA and Wei LN. Lysine trimethylation of retinoic acid receptor-alpha: a novel means to regulate receptor function. Mol Cell Proteomics. 2007; 6:677-688.
52. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV and Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325:834-840.
53. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ and Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005; 11:328-334.
54. Whartenby KA, Calabresi PA, McCadden E, Nguyen B, Kardian D, Wang T, Mosse C, Pardoll DM and Small D. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc Natl Acad Sci U S A. 2005; 102:16741-16746.
55. McMahon EJ, Bailey SL and Miller SD. CNS dendritic cells: critical participants in CNS inflammation? Neurochem Int. 2006; 49:195-203.
56. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA and Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002; 419:624-629.
57. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA and Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:11606-11611.
58. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010; 42:181-185.
59. Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, Kleer CG, Varambally S and Chinnaiyan AM. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008; 27:7274-7284.
60. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN and Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer cell. 2011; 19:86-100.
61. Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, Daignault S, Al-Mulla F, Keller E, Bonavida B, de la Serna I and Yeung KC. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer research. 2012; 72:3091-3104.
62. Yoo KH and Hennighausen L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Biol Sci. 2012; 8:59-65.
63. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC and Yu Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011; 43:798-810.
64. Terashima M, Yamamori C and Shimoyama M. ADP-ribosylation of Arg28 and Arg206 on the actin molecule by chicken arginine-specific ADP-ribosyltransferase. European journal of biochemistry. 1995; 231:242-249.
65. Dalle-Donne I, Giustarini D, Rossi R, Colombo R and Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free radical biology & medicine. 2003; 34:23-32.
66. Nyman T, Schuler H, Korenbaum E, Schutt CE, Karlsson R and Lindberg U. The role of MeH73 in actin polymerization and ATP hydrolysis. Journal of molecular biology. 2002; 317:577-589.
67. Hofmann WA, Arduini A, Nicol SM, Camacho CJ, Lessard JL, Fuller-Pace FV and de Lanerolle P. SUMOylation of nuclear actin. The Journal of cell biology. 2009; 186:193-200.
68. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ and Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006; 23:607-618.
69. Lang AE, Schmidt G, Schlosser A, Hey TD, Larrinua IM, Sheets JJ, Mannherz HG and Aktories K. Photorhabdus luminescens toxins ADP-ribosylate actin and RhoA to force actin clustering. Science (New York, NY). 2010; 327:1139-1142.
70. Furuhashi K, Hatano S, Ando S, Nishizawa K and Inagaki M. Phosphorylation by actin kinase of the pointed end domain on the actin molecule. The Journal of biological chemistry. 1992; 267:9326-9330.
71. Viswanathan MC, Blice-Baum AC, Schmidt W, Foster DB and Cammarato A. Pseudo-acetylation of K326 and K328 of actin disrupts Drosophila melanogaster indirect flight muscle structure and performance. Frontiers in physiology. 2015; 6:116.
72. Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS and Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007; 27:197-213.
73. Oser M, Mader CC, Gil-Henn H, Magalhaes M, Bravo-Cordero JJ, Koleske AJ and Condeelis J. Specific tyrosine phosphorylation sites on cortactin regulate Nck1-dependent actin polymerization in invadopodia. Journal of cell science. 2010; 123:3662-3673.
74. Kreitzer G, Liao G and Gundersen GG. Detyrosination of tubulin regulates the interaction of intermediate filaments with microtubules in vivo via a kinesin-dependent mechanism. Mol Biol Cell. 1999; 10:1105-1118.
75. Al-Bassam J and Corbett KD. alpha-Tubulin acetylation from the inside out. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:19515-19516.
76. Chu CW, Hou F, Zhang J, Phu L, Loktev AV, Kirkpatrick DS, Jackson PK, Zhao Y and Zou H. A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell. 2011; 22:448-456.
77. Fourest-Lieuvin A, Peris L, Gache V, Garcia-Saez I, Juillan-Binard C, Lantez V and Job D. Microtubule regulation in mitosis: tubulin phosphorylation by the cyclin-dependent kinase Cdk1. Mol Biol Cell. 2006; 17:1041-1050.
78. Vogel J, Drapkin B, Oomen J, Beach D, Bloom K and Snyder M. Phosphorylation of gamma-tubulin regulates microtubule organization in budding yeast. Developmental cell. 2001; 1:621-631.
79. Vicente-Manzanares M, Ma X, Adelstein RS and Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nature reviews Molecular cell biology. 2009; 10:778-790.
80. Totsukawa G, Himi-Nakamura E, Komatsu S, Iwata K, Tezuka A, Sakai H, Yazaki K and Hosoya H. Mitosis-specific phosphorylation of smooth muscle regulatory light chain of myosin II at Ser-1 and/or -2 and Thr-9 in sea urchin egg extract. Cell Struct Funct. 1996; 21:475-482.
81. Clark K, Middelbeek J, Lasonder E, Dulyaninova NG, Morrice NA, Ryazanov AG, Bresnick AR, Figdor CG and van Leeuwen FN. TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. Journal of molecular biology. 2008; 378:790-803.
82. Even-Faitelson L and Ravid S. PAK1 and aPKCzeta regulate myosin II-B phosphorylation: a novel signaling pathway regulating filament assembly. Molecular biology of the cell. 2006; 17:2869-2881.
83. Ronen D and Ravid S. Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. The Journal of biological chemistry. 2009; 284:24948-24957.
84. Petit V, Boyer B, Lentz D, Turner CE, Thiery JP and Valles AM. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. The Journal of cell biology. 2000; 148:957-970.
85. Kwak TK, Lee MS, Ryu J, Choi YJ, Kang M, Jeong D and Lee JW. Cell adhesion-dependent serine 85 phosphorylation of paxillin modulates focal adhesion formation and haptotactic migration via association with the C-terminal tail domain of talin. The Journal of biological chemistry. 2012; 287:27499-27509.
86. Cai X, Li M, Vrana J and Schaller MD. Glycogen synthase kinase 3- and extracellular signal-regulated kinase-dependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Molecular and cellular biology. 2006; 26:2857-2868.
87. Abou Zeid N, Valles AM and Boyer B. Serine phosphorylation regulates paxillin turnover during cell migration. Cell communication and signaling. 2006; 4:8.
88. Brown MC, Perrotta JA and Turner CE. Serine and threonine phosphorylation of the paxillin LIM domains regulates paxillin focal adhesion localization and cell adhesion to fibronectin. Mol Biol Cell. 1998; 9:1803-1816.
89. Romanova LY, Holmes G, Bahte SK, Kovalchuk AL, Nelson PJ, Ward Y, Gueler F and Mushinski JF. Phosphorylation of paxillin at threonine 538 by PKCdelta regulates LFA1-mediated adhesion of lymphoid cells. Journal of cell science. 2010; 123:1567-1577.
90. Bays JL, Peng X, Tolbert CE, Guilluy C, Angell AE, Pan Y, Superfine R, Burridge K and DeMali KA. Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. The Journal of cell biology. 2014; 205:251-263.
91. Simon NC and Barbieri JT. Bacillus cereus Certhrax ADP-ribosylates vinculin to disrupt focal adhesion complexes and cell adhesion. The Journal of biological chemistry. 2014; 289:10650-10659.
92. Liu Z, Lu D, Wang X, Wan J, Liu C and Zhang H. Kindlin-2 phosphorylation by Src at Y193 enhances Src activity and is involved in Migfilin recruitment to the focal adhesions. FEBS letters. 2015; 589:2001-2010.
93. Fagerholm SC, Hilden TJ, Nurmi SM and Gahmberg CG. Specific integrin alpha and beta chain phosphorylations regulate LFA-1 activation through affinity-dependent and -independent mechanisms. The Journal of cell biology. 2005; 171:705-715.
94. Fagerholm SC, Varis M, Stefanidakis M, Hilden TJ and Gahmberg CG. alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood. 2006; 108:3379-3386.
95. Uotila LM, Aatonen M and Gahmberg CG. Integrin CD11c/CD18 alpha-chain phosphorylation is functionally important. The Journal of biological chemistry. 2013; 288:33494-33499.
96. Pawar SC, Demetriou MC, Nagle RB, Bowden GT and Cress AE. Integrin alpha6 cleavage: a novel modification to modulate cell migration. Experimental cell research. 2007; 313:1080-1089.
97. Mulrooney JP, Hong T and Grabel LB. Serine 785 phosphorylation of the beta1 cytoplasmic domain modulates beta1A-integrin-dependent functions. Journal of cell science. 2001; 114:2525-2533.
98. Meves A, Geiger T, Zanivan S, DiGiovanni J, Mann M and Fassler R. Beta1 integrin cytoplasmic tyrosines promote skin tumorigenesis independent of their phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:15213-15218.
99. Gupta S, Chit JC, Feng C, Bhunia A, Tan SM and Bhattacharjya S. An Alternative Phosphorylation Switch in Integrin beta2 (CD18) Tail for Dok1 Binding. Scientific reports. 2015; 5:11630.
100. Oxley CL, Anthis NJ, Lowe ED, Vakonakis I, Campbell ID and Wegener KL. An integrin phosphorylation switch: the effect of beta3 integrin tail phosphorylation on Dok1 and talin binding. The Journal of biological chemistry. 2008; 283:5420-5426.
101. Li Z, Zhang H, Lundin L, Thullberg M, Liu Y, Wang Y, Claesson-Welsh L and Stromblad S. p21-activated kinase 4 phosphorylation of integrin beta5 Ser-759 and Ser-762 regulates cell migration. The Journal of biological chemistry. 2010; 285:23699-23710.
102. Kariya Y and Gu J. N-glycosylation of ss4 integrin controls the adhesion and motility of keratinocytes. PloS one. 2011; 6:e27084.