
INTRODUCTION
Renal cell carcinoma (RCC) accounts for 85% of renal cancers, of which clear cell RCC (ccRCC) is the most prevalent form [1–5]. RCC responds poorly to cytotoxic drugs. Although, the prognosis for patients with this disease has recently improved due to earlier detection of RCC and expansion of treatment possibilities such as mTor inhibitors and VEGFR-targeted tyrosine kinase inhibitors [1], these treatments rarely yield complete responses. T-cell checkpoint inhibitors show promise but are not standard of care at the time of writing. Thus, RCC is still a tumor with poor clinical outcome and identification of additional cellular treatment targets is of utmost importance for improving prognosis.
CD133 is a pentaspan transmembrane glycoprotein (Prominin-1), whose specific functions are still unclear. It was initially identified as a specific marker for hematopoietic stem cells [6, 7] but more recently it has been proposed to be a stem/progenitor marker in a variety of adult tissues including normal human kidney and RCC [8–22]. However, the specificity of CD133 as a marker for stem/progenitor cells is a matter of debate (reviewed by Wu and Wu et al., [23], as it has been found on various differentiated adult epithelial cells, including renal tubular cells [10, 24–28]. Although CD133+ cells from several human tumors exhibit xenotransplantation potential in severe combined immunodeficiency (SCID) mice [8, 18, 19, 29], CD133+ cells from human RCC tissue (RCCCD133+cells) [27] fail to form tumors when transplanted independently in SCID mice but instead potentiate tumor engraftment when co-transplanted with CD133- RCC cells [29]. Higher numbers of CD133+cells in RCC tissue has been correlated with increasing tumor grade [22] and was associated with a favourable prognosis in some studies [27, 30] but not others [31]. While it is unclear whether RCCCD133+cells are true tumor stem cells and their prognostic significance is uncertain, their ability to promote tumor formation by CD133- RCC cells may contribute to the resistance of these tumors to conventional chemotherapy so that elimination of this population may improve outcomes.
Tumor necrosis factor-alpha (TNF) is a cytokine secreted by RCC with a number of tumor promoting properties [32] and inhibition or blockade of TNF has shown clinical benefit in some patients with RCC [33–35]. This seems paradoxical as TNF was first identified as a cytokine that induced hemorrhagic necrosis of experimental tumors in mice [36]. We have shown that TNF can induce both cell death and proliferation in ccRCC cells in organ culture [37], and the relative expression and ligation of TNF receptor (TNFR) 1 and TNFR2 may be a key determinant of which pathways dominate. Specifically, ligation of TNFR1 promotes cell death whereas ligation of TNFR2 more typically induces cell cycle entry. Since the low rate of proliferation of RCCCD133+cells may be protecting them from chemotherapy, we have investigated the effects of TNFR2 on RCCCD133+cells and asked whether such pre-treatment would induce proliferation and thereby render them more susceptible to cell cycle-dependent chemotherapeutic drugs [38].
RESULTS
Enumeration and characterization of CD133+cells in NK and ccRCC
Immunostaining on sections of NK demonstrated rare interstitial CD133+cells (NKCD133+) (mean <3%) within the cortex, consistent with a previous report [10]. In comparison, sections of ccRCC contained a noticeably higher frequency of RCCCD133+cells (RCCCD133+), which increased further with tumor grade (Figure 1A panel i, quantified in B), ranging from mean ~6-8% in grade 1/2 tumors to 14-16% in grade 3/4 tumors. Combined-immunostaining for CD133 and a stem cell transcription factor, Oct4, demonstrated a strong nuclear staining of Oct4 in 8% of NKCD133+ and 12% of RCCCD133+cells (Figure 1A, panel ii). Cultures of CD133+cells isolated from either NK and ccRCC demonstrated spindle-shaped morphology and scanty cytoplasm (Figure 1C). Because the glycosylated form of CD133 expression that is recognized appears to vary with the antibody used for staining [39] and because different glycosylation patterns may be indicative with stemness, we tested different antibodies on our isolated cell populations. Four different anti-CD133 monoclonal antibodies (W6B3C1, AC133, ab19898, E93002) displayed concordant immunolabeling (Supplementary Figure 1) and the same cells expressed a marked signal for stem cell-associated markers [Oct4, Lin28, Nanog and Sox2) and for vimentin but lacked epithelial [cytokeratin and epithelial cell adhesion molecule (EpCAM)], endothelial [PECAM-1 (CD31)] and leukocyte [leukocyte common antigen (CD45)] markers (Figure 1E). This phenotype is most consistent with but does not absolutely establish identity as a stem cell population.
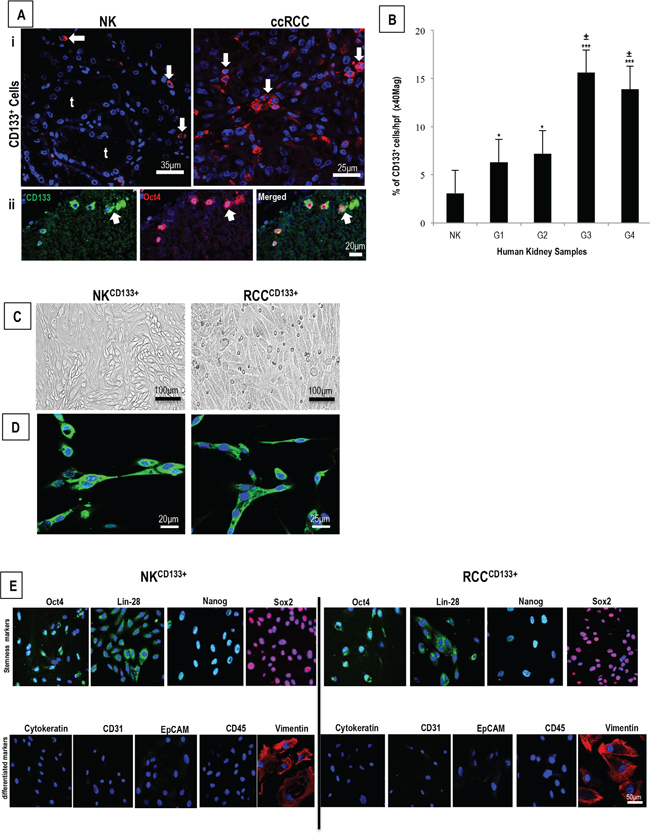
Figure 1: A. Representative confocal image of ccRCC grade 3 show a significantly larger number of CD133+cells as compared to NK (
Expression of TNF and TNFRs and responses to TNF by RCCCD133+ and NKCD133+cells
RCCCD133+cells were examined for protein expression of TNF and TNFRs at the basal level (untreated UT; 0h) and after treatment with wtTNF at various time points (3,6 & 18h). TNF and TNFRs were negligible in UT cultures, with TNFR2 expression detected at 3, 6 and 18h, with a higher level of expression at 18h. In comparison, TNF and TNFR1 expression were detected only in 18h cultures. (Figure 2A). Corresponding organ cultures of ccRCC demonstrated different levels of TNF (21+1.1%), TNFR1 (6.0+0.5%) and TNFR2 (28+1.2%) in CD133+cells. There were also a few CD133- cells within the interstitial tissue that were positive for TNF (25.0+0.2%), TNFR1 (2.0+0.1%) or TNFR2 (18.0+0.5%) (Figure 2B). Treatment with wtTNF increased TNFR2 protein expression in both cultured cell types, but the effect was more pronounced in RCCCD133+cells with a higher expression detected at 18h (15.2+0.8%) compared to 6h (8.1+0.4%) or 3h (6.0+0.5%). wtTNF also induced TNF (4.4+0.2%) and TNFR1 protein expression (3.0+0.1%) at 18h but not at 6 or 3h. Interestingly, R2TNF induced TNFR2 but not TNF or TNFR1 while R1TNF induced TNFR1 but not TNF or TNFR2 (data not shown). qRT-PCR of similar cultures demonstrated induction of TNF mRNA expression by wtTNF; increases were noted but did not reach significance in NKCD133+cells treated with R1TNF or R2TNF compared to UT cultures (***p<0.0001). In comparison, all three treatments significantly induced TNF mRNA expression in RCCCD133+cells (***p<0.0001 vs UT). Notably, wtTNF and R1TNF (but not R2TNF) induced TNFR1 mRNA expression, with a more pronounced expression demonstrated in RCCCD133+cells and induced by wtTNF (***p<0.0001) than by R1TNF (┼p<0.05) vs UT cultures. TNFR2 mRNA expression was induced by wtTNF and R2TNF (not R1TNF) (***p<0.0001 vs UT cultures) with a higher level of expression in RCCCD133+ compared to NKCD133+cells (+p<0.05) (Figure 2C).
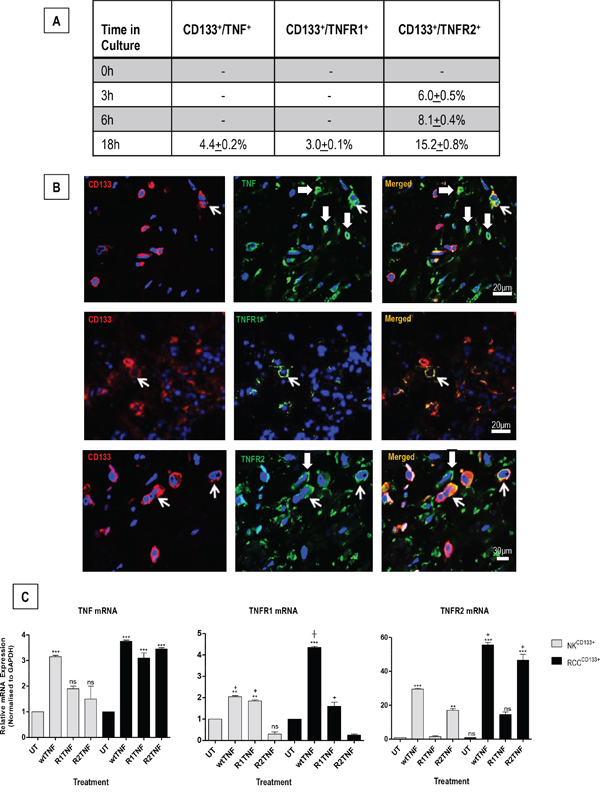
Figure 2: A. CD133+ cells isolated from RCC (RCCCD133+) were quantified for expression of TNF or TNFR1 or TNFR2 at basal level (0h) and after treatment with wtTNF at various time points (3, 6 & 18h). TNFR2 expression is detected in cells at 3, 6 and 18h, with a higher level of expression after 18h. In comparison, expression for TNF and TNFR1 is only detected after 18h cultures. B. Representative images of corresponding ccRCC organ cultures show staining of TNF or TNFR1 or TNFR2 in CD133+cells (open arrows), with a few CD133-cells also positive for TNF or TNFR2 (shaded arrows). C. qRT-PCR analysis of RCCCD133+ and NKCD133+cells at day 7 following treatment with wtTNF, R1TNF and R2TNF show mRNA expression normalized to control samples value of 1 for each cell type. Error bars represent the mean ± SEM, ***p<0.0001 vs UT; **p<0.001 vs UT; +p<0.05 vs R2TNF; ┼p<0.05 vs R1TNF, ns-not significant analyzed by ANOVA and Bonferonni post hoc; of data from at least 3 independent experiments with similar results.
We next assessed the effect of wtTNF, R1TNF and R2TNF on the induction of cell death and cell cycle activation in RCCCD133+ and NKCD133+cells. wtTNF or R1TNF each induced increased cell death in RCCCD133+cells as compared to UT or R2TNF-treated cultures [~27+0.9% by wtTNF and ~19+0.7% by R1TNF], positive for TUNEL in a time-dependent manner, with a marked increase in death in 18h cultures (Figure 3A and 3B and Supplementary Table 1). These effects were less pronounced in NKCD133+cells, in which wtTNF induced ~12+0.2% and R1TNF-induced ~6+0.2% cell death. Some RCCCD133+/TUNEL+ cells with fragmented nuclei were TNFR1+ (but not TNFR2+) with wtTNF inducing ~13+0.2% and R1TNF~6.6+0.2% (Figure 3C and Supplementary Table 2). Because R1TNF-induction of cell death in RCC organ cultures has been positively correlated with caspase activation [37], we examined similar cultures positive for TUNEL and TNFR1 for expression of cleaved caspase-3Asp175 by immunofluorescence (IF) and flow cytometry (FACS). 77% of the RCCCD133+cells treated with wtTNF that stained positive for TUNEL also stained positive for cleaved caspase 3Asp175 by FACS. However, only 14% of wtTNF-treated TUNEL positive NKCD133+cells stained positive for cleaved caspase 3Asp175 (Supplementary Figures 2A and 2B). These data suggest that the majority of TNF-induced cell death in RCCCD133+cells occurs via caspase-3 activation.
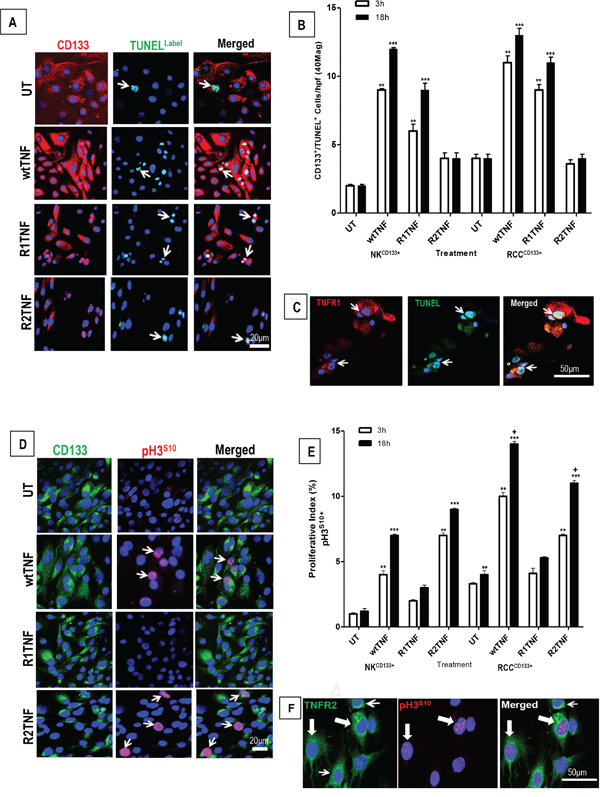
Figure 3: Representative confocal images of tissue-enriched CD133+cells from ccRCC and NK (RCCCD133+ and NKCD133+) immunostained for CD133 followed by TUNEL assay. A. Untreated (UT) and R2-TNF-treated cultures show a rare signal for TUNEL (arrows). In contrast, cultures treated with wtTNF and R1TNF show an increased number of TUNEL+nuclei (arrows). B. Quantification of RCCCD133+/TUNEL+ and NKCD133+/TUNEL+ in 10 random fields of view at x40 magnification show a time-dependent increase in the number of positive cells, more pronounced in 18h than 3h cultures, with C. some RCCCD133+/TUNEL+cells are also positive for TNFR1 (red) D. RCCCD133+cells immunostained for pH3S10 (marker of cell cycle) show a rare pH3S10 positive nuclei in untreated (UT) and in R1TNF-treated cultures, while wtTNF and R2TNF-treated cultures show a strong signal (arrows). E. Percentage of RCCCD133+/pH3S10+cells in 10 random fields of view at x40 magnification, presented as proliferative index, show a time-dependent increase in proliferative cells, more pronounced in 18h cultures. F. Some proliferative RCCCD133+/pH3S10+cells are also positive for TNFR2 (green) (shaded arrows) with a few non-poliferative cells (pH3S10-) positive for TNFR2 (open arrows). Nuclei stained with Hoechst-33342. Bars=mean + SEM, ***p<0.0001; **p<0.001 ; +p<0.05 signify statistically significant difference between treatments, data are from at least 3 independent experiments with similar results, ns-not significant.
wtTNF and R2TNF (but not R1TNF) induced increased pH3S10 signal, a marker of cell proliferation, and this was more pronounced in RCCCD133+cells (wtTNF ~17%, R2TNF ~13%) compared to NKCD133+cells (wtTNF ~7%, R2TNF ~5%) (Figures 3D and 3E). Combined-immunostaining for pH3S10 and TNFR2 demonstrated a strong signal for TNFR2 in some proliferating RCCCD133+cells (wtTNF~8% and R2TNF~6% vs 5% and 3% in NKCD133+cells)(Figure 3F). To further confirm TNF-induction of cell cycle entry, similarly treated cultures were subjected to IF with an alternative marker of cell proliferation, PCNA [40] (Figure 4A). FACS analysis of similar cultures for PCNA expression were concordant with IF findings with wtTNF and R2TNF (but not R1TNF) showing a marked expression, more pronounced in RCCCD133+ compared to NKCD133+cells (Figure 4B). Similar effects of wtTNF and R2TNF were demonstrated by cell viability assays (Figures 4C and 4D) in a time-dependent manner with increase viability in cultures at day 4 vs day 2 and in RCCCD133+ vs NKCD133+cells. These data are consistent with the interpretation that TNF induces cell death of CD133+ normal and malignant renal cells through TNFR1 and cell cycle entry through TNFR2 in RCCCD133+cells. The ability of wtTNF but neither of the muteins to increase TNF staining is unexplained but all three treatments induced TNF mRNA in isolated RCCCD133+cells.
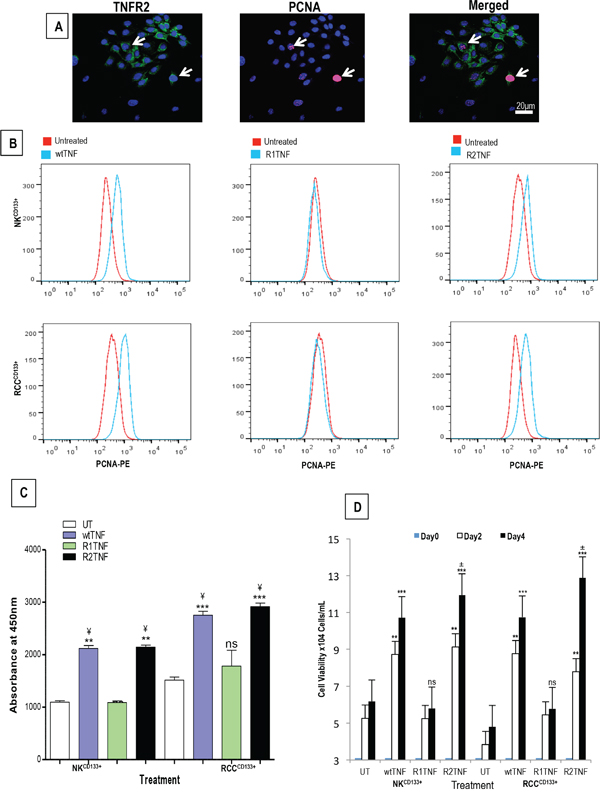
Figure 4: A. Representative confiocal images of combined staining for TNFR2 and proliferative nuclear antigen (PCNA) showing co-staining in some RCCCD133+ cells (arrows). B. Representative flow cytometry analysis of similar cultures from ccRCC and NK (RCCCD133+ and NKCD133+) show a high expression of PCNA induced by wtTNF- or R2TNF-treatment as compared to R1-TNF-treated and control cultures (UT) after day 4. Quantification of two cell viability assays; CCK-8 C. and trypan blue exclusion assay D. are concordant with flow cytometry showing an increase cell viability in cultures treated with wtTNF- and R2TNF as compared to UT and R1TNF-treated cultures. Error bars represent the mean + SEM, ***p<0.0001 vs UT; **p<0.001 vs UT, ¥p<0.05 vs R1TNF; ±p<0.05 vs Day 2 (R2TNF), ns-not significant; data of at least 3 independent experiments with similar results.
TNFR2 sensitizes RCCCD133+cells to killing by Cyclophosphamide (CP)
Having established that signling through TNFR2 does promote entry of RCCCD133+ into cell cycle, we next sought to determine whether R2TNF sensitizes RCCCD133+ or NKCD133+cells to killing by CP. Cells were exposed to either CP alone, R2TNF alone, or CP followed by R2TNF (CP+R2TNF) or to R2TNF followed by CP (R2TNF+CP), and then analyzed by staining with FITC--conjugated Annexin-V/Propidium Iodide (PI) and examined by FACS. In comparison with UT cultures, R2TNF+CP (***p<0.001), CP alone and CP+R2TNF (*p<0.05) all induced some death in RCCCD133+cells with the highest level of cell death induced by R2TNF+CP compared to treatment with CP alone (+p<0.01) or CP+R2TNF (*p<0.01). R2TNF+CP also resulted in death of NKCD133+ but a significant level of death was observed in only 1 out of 3 experiments, indicating that RCCCD133+cells are more sensitive to the cytotoxic effects of CP than are NKCD133+ (Figure 5). The percentage of dead RCCCD133+ and NKCD133+cells is presented in Supplementary Table 3. Moreover, representative images of FITC-Annexin-V/PI and phase-contrast microscopy are shown in Supplementary Figures 3A and 3B. These data indicate that signaling through TNFR2, probably by inducing proliferation, sensitizes RCCCD133+cells to killing by CP.
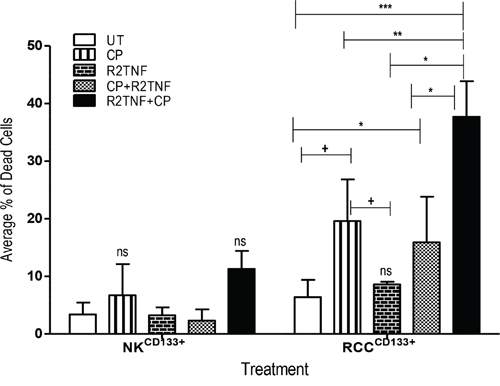
Figure 5: Flow cytometry analysis showing the effect of cyclophosphamide (CP) in cultures of tissue-enriched CD133+cells from ccRCC and NK (RCCCD133+ and NKCD133+) treated with R2TNF before and after CP or with CP alone. Quantification of average percentage of dead cells stained with Annexin V and Propidium Iodide and analyzed with flow cytometry show a strong staining for both stains in RCCCD133+ treated with R2TNF for 48h prior to treatment with CP (1.25μM) (R2TNF+CP) for a further 48h compared to untreated controls (UT) (***p<0.0001) or cultures treated with CP alone (**p<0.001) or CP+R2TNF (*p<0.01). A significant increase in both staining was demonstrated in cultures exposed to CP alone vs UT or vs R2TNF alone (+p<0.05) but not vs CP+R2TNF. R2TNF+CP also induced death in NKCD133+ but only in 1 out of 3 experiments resulting in a no significant effect. Errors bars mean + SEM, data of least 3 independent experiments with similar results.
DISCUSSION
Our rationale for studying CD133+cells in NK or RCC is their potential role as renal stem cell and tumor stem cells, respectively. In the present paper, we characterized these populations both in intact tissue and in cell culture. We have evaluated their responses to TNF, as this cytokine appears to play a complex role both in NK and ccRCC, being capable of inducing both proliferation and cytotoxic responses [37]. Analyzing responses mediated by TNFR1 and TNFR2 using muteins that selectively activate one receptor or the other can separate some of the seeming contradictions in TNF responses. Our key findings are that in intact renal tissue there is a significant increase in the numbers of CD133+cells in RCC vs NK, consistent with a prior report [27]. Furthermore, the frequency of RCCCD133+cells increased with higher tumor grade. Both NKCD133+ and RCCCD133+cells are responsive to TNF. Specifically, ligation of TNFR2 increases transcription of TNFR2 and promotes their entry into cell cycle in organ culture. The entry of RCCCD133+cells into cell cycle in response to TNFR2 signaling increased susceptibility to CP-induced cell death. R2TNF also induced proliferation of NKCD133+cells but CP was less effective in killing them as compared to RCCCD133+cells.
A key question, not fully answered by our findings, is whether the interstitial CD133+cells in the renal cortex are or are not renal stem cells. We found that CD133+cells in RCC and NK were uniformly negative for the hematopoietic stem cell markers CD31 or CD45 but were strongly and persistently positive for stem cell markers (Oct4, Sox2, Nanog and Lin28) and for vimentin [41]. These data strongly suggest that they are a primitive cell type, consistent with previous studies [27, 29]. Although, we failed to detect cytokeratin in the CD133+ cells from both the study groups, we did however, observe its expression when driven to differentiate into an epithelial lineage (RA-L unpublished data), suggesting capacity of these progenitor cells to acquire markers characteristic of fully differentiated renal epithelia. Stem cell transcription factors Oct4 and/or Sox2 have been found to bind to the P1 promoter region of the CD133 gene and ectopic Oct4 or Sox2 expression has been shown to trigger the CD133P1 activity in the lung cancer cell lines [42]. CD133 marker was initially used to identify cancer stem cells (CSCs) in brain tumor [18, 19]. CD133+stem/progenitor cells, also referred to as tumor-initiating cells, CSCs, stem-like cells [9, 20, 23, 29, 30, 43-47], have been isolated in normal and neoplastic tissue including the human kidney and RCC [9-11, 27, 29, 45, 48, 49]. Although some CD133+cells have been shown to possess self-renewal properties and clonal efficiency; current literature on the use of CD133 as the only marker to identify stem cells has been questioned since its presence has been localized in a variety of adult epithelial cells including pancreas, liver and renal tubules [10, 26, 28, 50, 51] and localize to membrane protrusions [6]. In RCC, CD133+cells have been detected in apical membrane of tumor cells in macro/microcytic regions [27] and in perinecrotic/perivascular areas [20]. Moreover, in human kidneys, CD133 immunoreactivity was localized to proximal tubules and Bowman’s capsule [50] and not restricted to stem cells. Interestingly, other groups have shown that CD133 is not only a biomarker, but also functions in cell growth, development and tumor biology [52]. Indeed, CD133 expression and post-translational modification are dynamic and reversible that is dependent on cell microenvironment and physiological regulation [53].
The tumorigenic potential of CD133+cells depends on the tissue of origin. For example, in human brain, CD133+cells are capable of self-renewal and recapitulate the original tumor when transplanted in SCID mice [19]. Similarly, CD133+cells from primary lung tumors show higher tumorigenic potential than their CD133−counterparts and are able to reproduce the original tumor in SCID mice [8]. Conversely, in RCC, undifferentiated CD133+cells express low level of Oct4, have a low proliferative rate [27] and fail to form tumors independently in SCID mice but support tumor growth when co-transplanted with tumor cells [29]. These reports indicate that CD133+cells are a heterogeneous cell population with varied function(s).
Recruitment of CD133+ pro-angiogenic cells in cancer has been considered to play an important role in resistance to vascular growth factor (VEGF) neutralizing antibody such as bevacizumab, possibly due to failure to inhibit their differentiation into endothelial cell types [54]. Moreover, CD133 gene polymorphism have been associated with lower overall survival rate in patients treated with bevacizumab in metastatic colorectal cancer treated [55]. These studies generally have as their premise that VEGF as a tumor angiogenic factor. Neutralization of VEGF may play an additional role in RCC as these cells, like endothelium, express VEGF receptor 2 (VEGFR2) and proliferate in response to its autocrine production. Our prior studies of malignant tubular epithelial cells in ccRCC organ culture indicated that signalling through TNFR2 induced ligand-independent transactivation of VEGFR2 and that blockade of VEGFR2 kinase activity inhibited TNFR2-induced cell cycle entry [37, 56, 57].
Tumor cells and tumor microenvironment may play a vital role in regulating self-renewal properties of CD133+cells in RCC [29]. Inflammation and hypoxia are characteristic features of neoplasm and facilitates tumor growth. Cancer-associated inflammation is characterized by expression of cytokines, influx of leukocytes, tissue remodeling and angiogenesis [58–60]. Cytokines have pro-tumor properties. TNF is secreted by RCC [37, 61], and increase stemness properties in RCC in 2D cultures [62]. TNF targets the quiescent/slow-cycling stem cells and promotes PI3K/AKT-driven expansion in melanoma by preventing their asymmetrical self-renewal [63]. These data strongly suggest that TNF, present in the tumor microenvironment, may influence the behaviour of CD133+cells in RCC and potentiate their self-renewal properties. In our preliminary studies, we have demonstrated TNF-induced upregulation of stem cell transcription genes (Oct4, Nanog and Lin28) in RCCCD133+cells (RA-L, unpublished data), consistent with a prior report by Ueda et al [64]. Hypoxia has been shown to influence the expression of CD133 in RCC [65], as well as in lung cancer, pancreatic cancer and glioma cells [66, 67]. We have previously reported that hypoxia both increases TNF expression and TNFR2 induction and expression of a stem cell marker Lin28 in c-kit+ cardiac stem cells that then enter cell cycle [68]. Collectively, these findings suggest that a transient exposure of RCCCD133+cells to TNF and/or hypoxia may imprint their long-lasting molecular and/or cellular changes with functional consequences.
In summary, our data confirm that CD133+cells are present in larger numbers in ccRCC than in NK and that they respond to TNFR2 signaling, which promotes their proliferation. While the specific role of RCCCD133+cells in renal tumours remain unclear, they likely have pathogenetic signficance. TNF, signaling via TNFR2, not only causes these cells to proliferate, it also increases their senstivity to cell cycle-sensitive chemotherapy. Therefore, priming these cells with a TNFR2 agonist may enhance the efficacy of chemotherapy for ccRCC.
MATERIALS AND METHODS
Antibodies, cytokines, and reagents
Recombinant human TNF (cat~201-TA), mouse anti-TNFR1 (cat~MAB225) and mouse anti-TNFR2 (MAB226) all from R&D Systems, Abington, UK, rabbit anti-TNFR1 (cat~ab19139) (Abcam, Cambridge, UK), rabbit anti-TNFR2 (cat~sc-7862 H202, Santa Cruz Biotechnology, Heidelberg, Germany), mouse anti-PECAM/CD31 (cat~M0823), mouse anti-cytokeratin (cat~M0821 clone MNF116) and mouse anti-CD45 (cat~M0701), mouse anti-Vimentin Clone V9 (cat~M0725) all from Dakocytomation, Ely, UK, rabbit anti-Vimentin (D21H3)(Cell Signaling Technology Inc, UK), rabbit anti-CD133 (E90032, Source Bioscience, Nottingham, UK); rabbit anti-CD133 (cat~ab19898, Abcam), mouse anti-CD133/1-W6B3C1 (cat~130-092-395) and mouse anti-CD133/1-AC133 (cat~130-090-422) both from Miltenyi Biotec Ltd, Surrey, UK, rabbit anti-human EpCAM (GTX113091, Source Bioscience); mouse anti-Sox2 (cat~MCA5660T) and mouse anti-Nanog (cat~MCA5657T, Bio-Rad Laboratories Ltd, Hertfordshire, UK), Hoechst-33342 (Thermo Fisher Scientific, Paisley, UK); rabbit anti-Lin28 (cat~ab46020), rabbit anti-Oct4 (cat~ab19857), mouse anti-phosphorylated Histone H3S10 (pH3S10) (cat~ab14955), rabbit anti-phosphorylated Histone H3S10 (pH3S10)(cat~ab5176), anti-TNF-alpha (cat~ab6671) all from Abcam, mouse anti-proliferative cell nuclear antigen (PCNA, cat~MAB424 clone PC10, Millipore, Hertfordshire, UK). Cleaved caspase-3Asp175 (cat~9661, Cell Signaling Technology, Leiden, The Netherlands). Tissue Dissociation Kit (cat~130-095-929), MS columns (cat~130-042-201), C-Tubes (cat~130-093-237) were from Miltenyi Biotec. TUNEL label (dUTP-FITC) (cat~11767291910), Terminal transferase enzyme (TdT) (cat~03333566001), deoxynuclease-1 (DNase-1) and Annexin-V-FLUOS Staining Kit all from Roche Diagnostics, Mannheim, Germany. Recombinant mutations of the TNF sequence which enable the mutated protein (“mutein”) to bind selectively to either of the TNFR subtypes; TNFR1 (R1TNF) and TNFR2 (R2TNF) [37, 40, 69]. Cyclophosphamide was from Sigma-Aldrich, St. Louis, MO.
Tissue collection
Experiments using human tissue were performed with informed consent of patients and approval of the local ethics committee and Cambridge University hospital Tissue Bank. RCC tissue obtained from radical nephrectomy specimens was immediately excised from tumors that grossly appeared to be ccRCC. This tumor classification was later verified by routine histological assessment of paraffin-wax embedded samples. Non-clear cell histological tumor types (e.g. papillary, chromophobe, and collecting duct) [70] were excluded and only ccRCC, graded according to the four-tiered Fuhrman nuclear grading system [71] and pathologically staged based on the TNM classification [37, 72] were used. Tissue samples from 40 patients were collected and scored as Fuhrman grade 1 (n=10), Fuhrman grade 2 (n=10), Fuhrman grade 3 (n=12), and Fuhrman grade 4 (n=8). In parallel, adjacent non-tumor kidney (NK) (n=20), categorized histologically as normal kidney cortex was collected remote from the tumor site. All samples were either fixed overnight at 4°C in 4% formaldehyde in 0.1M phosphate buffer pH 7.6 and paraffin wax-embedded for immunofluorescence or snap-frozen in isopentene-cooled in liquid nitrogen. Parallel-unfixed fresh samples were processed for organ culture experiments.
Kidney organ cultures
Organ cultures were performed as previously described [37]. In brief, duplicate 1 mm3 fragments of tissue from ccRCC grade 1 and NK (n=5 per study group) were immersed in M199 medium containing 10% heat-inactivated fetal calf serum (FCS), antibiotics and 2.2mM glutamine. Cultures were either left in media alone (untreated controls; UT) or treated with TNF (10ng/mL) or R1TNF or R2TNF (1µg/mL) [37, 40, 73] at various time points (0, 3, 6 and 18h). Cultures were harvested, fixed in 4% formaldehyde and embedded in paraffin-wax.
Processing, isolation, and culture of CD133+cells from ccRCC and nontumor adjacent kidney (NK)
ccRCC and NK (n=10 per study group) were enzymatically digested as previously reported [68]. In brief, tissue was digested into single cell suspension using Tissue Dissociation Kit on a GentleMACs Disassociator and incubated on a MACsMix rotator (Miltenyi) for 30 min at 37oC, centrifuged at 300g for 5 min, passed through 40µm strainer to remove cell clumps. Single cell suspensions were cultured in nondifferentiating expansion medium [29] [containing 60% low-glucose Dulbecco’s modified Eagle’s medium-LG (DMEM-LG), 2% FCS, 40% MCDB-201, 1x insulin-transferrin-selenium, 1x linoleic acid 2-phosphate, 10-9 M dexamathasone, 104M ascorbic acid 2-phosphate, 10 ng/mL epidermal growth factor (EFG), 10 ng/mL platelet derived growth factor, and 1000 units/mL lymphocyte inhibitory factor, 100U penicillin, 1000U streptomycin] all from Invitrogen, Paisley, United Kingdom. Enrichment of CD133+cells from ccRCC and NK (referred to as RCCCD133+ and NKCD133+) was achieved by magnetic cell sorting system according to the manufacturer's instructions (Mitenyi). A total of 6 x108 cells were magnetically labeled with CD133 microbeads and passed through the MACS column on MiniMACS Separator. The magnetically labeled CD133+cells were eluted from magnetic column as the positively selected cell fraction. Enriched population of RCCCD133+ and NKCD133+cells (~6x105 cells/mL) were seeded in T25 flasks and transferred to eight-well glass slide chambers (Thermo-scientific, UK). Once confluent, cells were either UT or treated with wtTNF or R1TNF or R2TNF for 3 and 18h.
Single and combined immunofluorescence (IF)
Sections of ccRCC, NK and corresponding organ cultures were immunostained for CD31 (1:50 dilution) as previously described [37, 40]. As CD133 expression has been associated with stem/progenitor cells in RCC, we co-immunostained some cultures with anti-CD133 and a panel of stemness markers (Lin28, Oct4, Nanog, Sox2) or markers for epithelial, endothelial and leukocytes (cytokeratin, CD31, EpCAM, CD45) and a mesenchymal marker (vimentin) (all used at 1:100 dilution). pH3S10 and PCNA (used at 1:500 and 1:200 dilution) were used to detect cell cycle activation and assessed in association with TNF and TNFRs. In addition, some cultures were subjected to co-immunostaining for TNFR1 and cleaved caspase-3Asp175. Antibody binding sites were detected with Northern Light-498 or -557-conjugated secondary antibodies (diluted 1:100), incubated 1h at room temperature. Hoechst-33342 (1μg/ml) was used for nuclei detection. Species-specific antiseras were used as negative controls. Slides were viewed on a Leica TCS-SPE confocal microscopy (CLSM) (Leica Microsystems, Milton Keynes, UK) and image for each fluorophore was acquired sequentially using the same constant acquisition time and settings rather than simultaneously to avoid crosstalk between channels. Images were then processed in Adobe Photoshop CS6 software.
Determination of RCCCD133+ and NKCD133+ cell death
The effect of wtTNF, R1TNF and R2TNF treatment on cell death in RCCCD133+ and NKCD133+ was assessed using TUNEL as previously described [37, 40, 74]. Briefly, RCCCD133+ and NKCD133 cells and organ cultures from each of the four treatments were incubated with FITC-dUTP-TUNEL label mix containing TdT-enzyme plus Hoechst-33342 for nuclei detection and mounted in Vectorshield mountant (Vector Laboratories, Peterborough, UK). For negative controls, TdT-enzyme was omitted and for positive controls cultures were pretreated with DNase-1 enzyme (Promega, Hampshire, UK) before TUNEL. Three experiments were performed separately. Following TUNEL, some cultures were incubated with anti-TNF and anti-TNFRs antibodies and detected using Northern Lights568-conjugated secondary antibody plus Hoechst-33342. In addition, cell suspensions from UT and wtTNF-treated cultures from both the study groups were labeled with an antibody to cleaved-caspase-3Asp175 followed by incubation with a FITC-conjugated secondary antibody and analyzed by flow cytometry (FACS) using Canto II (BD Biosciences). All slides were viewed on a CLSM.
RNA isolation and quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total cellular RNA was extracted from UT and wtTNF-, R1TNF-, R2TNF-treated cultures of RCCCD133+ and NKCD133+cells using the RNeasy Plus Mini Kit (Qiagen, Germany). 6µg RNA from each sample was reversely transcribed into cDNA and measured using Taqman Reverse Transcription Kit according to the manufacturer's instructions (ABI7700 System, Applied Biosystems, CA, USA). TaqMan_Gene Expression Assay ID: TNFR1 (Hs00533560), TNFR2 (Hs00153550), TNF (Hs99999043) were used. (GAPDH; Hs04420697) was used for normalization.
Determination of RCCCD133+ and NKCD133+ cell proliferation
The effect of wtTNF, R1TNF and R2TNF on cell proliferation was evaluated by FACS using immunostaining for anti-PCNA antibody and two different cell viability assays [cell counting kit-8 (CCK-8) and trypan blue exclusion]. RCCCD133+ and NKCD133+cells serum-depleted were seeded in 12-well culture plates for FACS (1x105cells/mL), for CCK-8 assay (3x104cells/mL) and for trypan blue staining (5×103cells/mL) in 24-well culture plates. Cultures were either left UT or treated with wtTNF, R1TNF or R2TNF for 48h and 96h. Cells were then rinsed in PBS, blocked with FcR blocking reagent (Miltenyi) and incubated with PE-conjugated antibody to PCNA- or with PE-conjugated isotype-specific antisera before examining using FACS Canto II. 10,000 events were collected per sample and the data analysed with FlowJo software. For the CCK-8, cells were treated for 48h and the WST-8 reagent (10μL/well) Sigma-Aldrich, UK was added to each well and incubated for 2h at 37°C. The absorbance optical density (OD) (proportional to the relative abundance of living cells) was measured at 450nm wavelength using a microplate reader (Bio-Rad, USA). For trypan blue exclusion assay, 200µl cells were incubated in 0.4% (w/v) trypan blue solution containing 0.81% NaCl plus 0.06% (w/v) dibasic potassium phosphate for 3 min at room temperature and counted using a haemocytometer. Viable and non-viable cells were recorded separately, and the means of 3 independent cell counts from 3 separate experiments were pooled for analysis.
Determination of cytotoxic effect of cyclophosphamide (CP) by FACS
To evaluate the effect of CP following cell cycle activation of RCCCD133+ and NKCD133+cells by R2TNF, cells (0.5×105cells/well) were seeded in 24-well plates coated with human plasma fibronectin. Cells were then either left UT or treated with CP alone or R2TNF alone, or R2TNF for 48h before CP (R2TNF+CP) (to induce cell proliferation) or after (CP+R2TNF) for a further 48h. A dose response curve was initially set up to determine the optimal effect of CP using documented concentrations (1.25, 2.5 and 5µM) [75, 76] at 24h and 48h time points. No significant difference in toxicity was demonstrated between the 3 doses, although a higher number of dead cells were evident at 48h. 1.25µM CP for 48h was therefore used for all subsequent experiments. Following treatment, cells were harvested and cytotoxic effect of CP determined by Annexin V/Propidium Iodide (PI) staining using FACS. Cells were gated as fractions of non-apoptotic live cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic and necrotic cells (Annexin V+/PI+) and calculated as the average percentage of dead cells. FlowJo and GraphPad softwares were used to analyze the data, represented as mean ± SEM from at least 3 independent experiments.
Data analysis
Cell death and proliferative indices
The average number of RCCCD133+ and NKCD133+cells positive for TUNEL were counted in 10 random fields of view at x40 magnification from each treatment divided by the total cell numbers to generate the percentage of positive cells. Similarly, the numbers of RCCCD133+ and NKCD133+cells positive for pH3S10 were counted and divided by the total cell numbers to generate the % of positive cells, calculated as proliferative index (PI) for each treatment. Error bars represent mean ± SEM. Each experiment was repeated at least 3 times and the same statistically significant differences between experimental groups were observed in all three independent experiments although the absolute values varied. Microsoft Excel 2013 and GraphPad Prism 5.02 softwares were used for data processing. Statistical significance was assessed by the analysis of variance test and a p-value of <0.05 were considered significant.
ACKNOWLEDGMENTS
The authors would like to express their gratitude to The Human Research Tissue Bank who is supported by the National Institute of Health Research (NIHR)-Cambridge Biomedical Research Centre at the Cambridge University Hospitals NHS Foundation Trust for help with accessing the tissue used in this study.
CONFLICTS OF INTEREST
No competing financial interests.
GRANT SUPPORT
The National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre (RSA, JRB, AYW, PHM), Kidney Research UK (RSA, JW, SP, JRB), NIH grant R01-HL36003 (JSP), Cancer research UK grant RG75464 (TE), The Welcome Trust (NB, PHM), NSFC (national natural science foundation of China) grant 81400278 (JY), Medical Research Council Cancer Unit (SV), Belgian grants (Interuniversity Attraction Poles), Flemish grants (Methusalem grant (BOF09/01M00709) and grants from the Research Foundation Flanders and Foundation against Cancer, Ghent University grants and grants from Flanders Institute for Biotechnology (VIB) (PV).
REFERENCES
1. Coppin C, Kollmannsberger C, Le L, Porzsolt F, Wilt TJ. Targeted therapy for advanced renal cell cancer (RCC): a Cochrane systematic review of published randomised trials. BJU Int. 2011; 108: 1556-1563.
2. Ivan M, Kaelin WG, Jr. The von Hippel-Lindau tumor suppressor protein. Curr Opin Genet Dev. 2001; 11: 27-34.
3. Karakiewicz PI, Trinh QD, Bhojani N, Bensalah K, Salomon L, de la Taille A, Tostain J, Cindolo L, Altieri V, Ficarra V, Schips L, Zigeuner R, Mulders PF, Valeri A, Descotes JL, Mejean A, et al. Renal cell carcinoma with nodal metastases in the absence of distant metastatic disease: prognostic indicators of disease-specific survival. Eur Urol. 2007; 51: 1616-1624.
4. Linehan WM, Zbar B. Focus on kidney cancer. Cancer Cell. 2004; 6: 223-228.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62: 10-29.
6. Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997; 90: 5013-5021.
7. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997; 90: 5002-5012.
8. Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, Camerini T, Mariani L, Delia D, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009; 106: 16281-16286.
9. Bussolati B, Brossa A, Camussi G. Resident stem cells and renal carcinoma. Int J Nephrol. 2011; 2011: 286985.
10. Bussolati B, Bruno S, Grange C, Buttiglieri S, Deregibus MC, Cantino D, Camussi G. Isolation of renal progenitor cells from adult human kidney. Am J Pathol. 2005; 166: 545-555.
11. Bussolati B, Dekel B, Azzarone B, Camussi G. Human renal cancer stem cells. Cancer Lett. 2013; 338: 141-146.
12. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005; 65: 10946-10951.
13. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008; 15: 504-514.
14. Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007; 132: 2542-2556.
15. Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, Invernici G, Parati E, Alessandri G, La Porta CA. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007; 43: 935-946.
16. Neuzil J, Stantic M, Zobalova R, Chladova J, Wang X, Prochazka L, Dong L, Andera L, Ralph SJ. Tumour-initiating cells vs. cancer 'stem' cells and CD133: what's in the name? Biochem Biophys Res Commun. 2007; 355: 855-859.
17. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007; 445: 111-115.
18. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003; 63: 5821-5828.
19. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004; 432: 396-401.
20. Varna M, Gapihan G, Feugeas JP, Ratajczak P, Tan S, Ferreira I, Leboeuf C, Setterblad N, Duval A, Verine J, Germain S, Mongiat-Artus P, Janin A, Bousquet G. Stem cells increase in numbers in perinecrotic areas in human renal cancer. Clin Cancer Res. 2015; 21: 916-924.
21. Ward HH, Romero E, Welford A, Pickett G, Bacallao R, Gattone VH, 2nd, Ness SA, Wandinger-Ness A, Roitbak T. Adult human CD133/1(+) kidney cells isolated from papilla integrate into developing kidney tubules. Biochim Biophys Acta. 2011; 1812: 1344-1357.
22. Zhang Y, Sun B, Zhao X, Liu Z, Wang X, Yao X, Dong X, Chi J. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J Surg Oncol. 2013; 108: 414-419.
23. Wu Y, Wu PY. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev. 2009; 18: 1127-1134.
24. Florek M, Haase M, Marzesco AM, Freund D, Ehninger G, Huttner WB, Corbeil D. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res. 2005; 319: 15-26.
25. Immervoll H, Hoem D, Sakariassen PO, Steffensen OJ, Molven A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer. 2008; 8: 48.
26. Karbanova J, Missol-Kolka E, Fonseca AV, Lorra C, Janich P, Hollerova H, Jaszai J, Ehrmann J, Kolar Z, Liebers C, Arl S, Subrtova D, Freund D, Mokry J, Huttner WB, Corbeil D. The stem cell marker CD133 (Prominin-1) is expressed in various human glandular epithelia. J Histochem Cytochem. 2008; 56: 977-993.
27. Kim K, Ihm H, Ro JY, Cho YM. High-level expression of stem cell marker CD133 in clear cell renal cell carcinoma with favorable prognosis. Oncol Lett. 2011; 2: 1095-1100.
28. Lardon J, Corbeil D, Huttner WB, Ling Z, Bouwens L. Stem cell marker prominin-1/AC133 is expressed in duct cells of the adult human pancreas. Pancreas. 2008; 36: e1-6.
29. Bruno S, Bussolati B, Grange C, Collino F, Graziano ME, Ferrando U, Camussi G. CD133+ renal progenitor cells contribute to tumor angiogenesis. Am J Pathol. 2006; 169: 2223-2235.
30. Costa WH, Rocha RM, Cunha IW, Fonseca FP, Guimaraes GC, Zequi Sde C. CD133 immunohistochemical expression predicts progression and cancer-related death in renal cell carcinoma. World J Urol. 2012; 30: 553-558.
31. D'Alterio C, Cindolo L, Portella L, Polimeno M, Consales C, Riccio A, Cioffi M, Franco R, Chiodini P, Carteni G, Mirone V, Longo N, Marra L, Perdona S, Claudio L, Mascolo M, et al. Differential role of CD133 and CXCR4 in renal cell carcinoma. Cell Cycle. 2010; 9: 4492-4500.
32. Szlosarek P, Charles KA, Balkwill FR. Tumour necrosis factor-alpha as a tumour promoter. Eur J Cancer. 2006; 42: 745-750.
33. Lee CP, Patel PM, Selby PJ, Hancock BW, Mak I, Pyle L, James MG, Beirne DA, Steeds S, A'Hern R, Gore ME, Eisen T. Randomized phase II study comparing thalidomide with medroxyprogesterone acetate in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006; 24: 898-903.
34. Harrison ML, Obermueller E, Maisey NR, Hoare S, Edmonds K, Li NF, Chao D, Hall K, Lee C, Timotheadou E, Charles K, Ahern R, King DM, Eisen T, Corringham R, DeWitte M, et al. Tumor necrosis factor alpha as a new target for renal cell carcinoma: two sequential phase II trials of infliximab at standard and high dose. J Clin Oncol. 2007; 25: 4542-4549.
35. Daliani DD, Eisenberg PD, Weems J, Lord R, Fueger R, Logothetis CJ. The results of a phase II randomized trial comparing 5-fluorouracil and 5-fluorouracil plus alpha-interferon: observations on the design of clinical trials for androgen-independent prostate cancer. J Urol. 1995; 153: 1587-1591.
36. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975; 72: 3666-3670.
37. Al-Lamki RS, Sadler TJ, Wang J, Reid MJ, Warren AY, Movassagh M, Lu W, Mills IG, Neal DE, Burge J, Vandenebeele P, Pober JS, Bradley JR. Tumor necrosis factor receptor expression and signaling in renal cell carcinoma. Am J Pathol. 2010; 177: 943-954.
38. Shapiro GI, Harper JW. Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest. 1999; 104: 1645-1653.
39. Hermansen SK, Christensen KG, Jensen SS, Kristensen BW. Inconsistent immunohistochemical expression patterns of four different CD133 antibody clones in glioblastoma. J Histochem Cytochem. 2011; 59: 391-407.
40. Al-Lamki RS, Wang J, Vandenabeele P, Bradley JA, Thiru S, Luo D, Min W, Pober JS, Bradley JR. TNFR1- and TNFR2-mediated signaling pathways in human kidney are cell type-specific and differentially contribute to renal injury. FASEB J. 2005; 19: 1637-1645.
41. Moggio A, D'Armento G, Bussolati B. Efficient stem cell isolation from under vacuum preserved tissue samples. Organogenesis. 2012; 8: 71-75.
42. Iida H, Suzuki M, Goitsuka R, Ueno H. Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int J Oncol. 2012; 40: 71-79.
43. Canis M, Lechner A, Mack B, Zengel P, Laubender RP, Koehler U, Heissmeyer V, Gires O. CD133 induces tumour-initiating properties in HEK293 cells. Tumour Biol. 2013; 34: 437-443.
44. Hishikawa K, Takase O, Yoshikawa M, Tsujimura T, Nangaku M, Takato T. Adult stem-like cells in kidney. World J Stem Cells. 2015; 7: 490-494.
45. Lindgren D, Bostrom AK, Nilsson K, Hansson J, Sjolund J, Moller C, Jirstrom K, Nilsson E, Landberg G, Axelson H, Johansson ME. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am J Pathol. 2011; 178: 828-837.
46. Lu B, Chiou SH, Deutsch E, Lorico A. Cancer stem cells. J Oncol. 2011; 2011: 269437.
47. Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010; 29: 2153-2159.
48. Bussolati B, Maeshima A, Peti-Peterdi J, Yokoo T, Lasagni L. Renal Stem Cells, Tissue Regeneration, and Stem Cell Therapies for Renal Diseases. Stem Cells Int. 2015; 2015: 302792.
49. Gupta S, Verfaillie C, Chmielewski D, Kren S, Eidman K, Connaire J, Heremans Y, Lund T, Blackstad M, Jiang Y, Luttun A, Rosenberg ME. Isolation and characterization of kidney-derived stem cells. J Am Soc Nephrol. 2006; 17: 3028-3040.
50. Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, et al. Isolation and characterization of multipotent progenitor cells from the Bowman's capsule of adult human kidneys. J Am Soc Nephrol. 2006; 17: 2443-2456.
51. Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A. 1997; 94: 12425-12430.
52. Liu X, Chen L, Jiang Z, Wang J, Su Z, Li G, Yu S, Liu Z. Malignant behaviorial characteristics of CD133(+/-) glioblastoma cells from a Northern Chinese population. Exp Ther Med. 2013; 5: 65-72.
53. Feng JM, Miao ZH, Jiang Y, Chen Y, Li JX, Tong LJ, Zhang J, Huang YR, Ding J. Characterization of the conversion between CD133+ and CD133- cells in colon cancer SW620 cell line. Cancer Biol Ther. 2012; 13: 1396-1406.
54. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010; 468: 829-833.
55. Aravantinos G, Isaakidou A, Karantanos T, Sioziou A, Theodoropoulos GE, Pektasides D, Gazouli M. Association of CD133 polymorphisms and response to bevacizumab in patients with metastatic colorectal cancer. Cancer Biomark. 2015; 15: 843-850.
56. Al-Lamki RS, Mayadas TN. TNF receptors: signaling pathways and contribution to renal dysfunction. Kidney Int. 2015; 87: 281-296.
57. Wang J, Al-Lamki RS. Tumor necrosis factor receptor 2: its contribution to acute cellular rejection and clear cell renal carcinoma. Biomed Res Int. 2013; 2013: 821310.
58. Allavena P, Garlanda C, Borrello MG, Sica A, Mantovani A. Pathways connecting inflammation and cancer. Curr Opin Genet Dev. 2008; 18: 3-10.
59. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008; 66: 1-9.
60. Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008; 43: 374-379.
61. de Vivar Chevez AR, Finke J, Bukowski R. The role of inflammation in kidney cancer. Adv Exp Med Biol. 2014; 816: 197-234.
62. Zhang L, Jiao M, Wu K, Li L, Zhu G, Wang X, He D, Wu D. TNF-alpha induced epithelial mesenchymal transition increases stemness properties in renal cell carcinoma cells. Int J Clin Exp Med. 2014; 7: 4951-4958.
63. Ostyn P, El Machhour R, Begard S, Kotecki N, Vandomme J, Flamenco P, Segard P, Masselot B, Formstecher P, Touil Y, Polakowska R. Transient TNF regulates the self-renewing capacity of stem-like label-retaining cells in sphere and skin equivalent models of melanoma. Cell Commun Signal. 2014; 12: 52.
64. Ueda M, Fujisawa T, Ono M, Hara ES, Pham HT, Nakajima R, Sonoyama W, Kuboki T. A short-term treatment with tumor necrosis factor-alpha enhances stem cell phenotype of human dental pulp cells. Stem Cell Res Ther. 2014; 5: 31.
65. Sun C, Song H, Zhang H, Hou C, Zhai T, Huang L, Zhang L. CD133 expression in renal cell carcinoma (RCC) is correlated with nuclear hypoxia-inducing factor 1alpha (HIF-1alpha). J Cancer Res Clin Oncol. 2012; 138: 1619-1624.
66. Hashimoto O, Shimizu K, Semba S, Chiba S, Ku Y, Yokozaki H, Hori Y. Hypoxia induces tumor aggressiveness and the expansion of CD133-positive cells in a hypoxia-inducible factor-1alpha-dependent manner in pancreatic cancer cells. Pathobiology. 2011; 78: 181-192.
67. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, Pollack IF, Park DM. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009; 28: 3949-3959.
68. Al-Lamki RS, Lu W, Wang J, Yang J, Sargeant TJ, Wells R, Suo C, Wright P, Goddard M, Huang Q, Lebastchi AH, Tellides G, Huang Y, Min W, Pober JS, Bradley JR. TNF, acting through inducibly expressed TNFR2, drives activation and cell cycle entry of c-Kit+ cardiac stem cells in ischemic heart disease. Stem Cells. 2013; 31: 1881-1892.
69. Loetscher H, Stueber D, Banner D, Mackay F, Lesslauer W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J Biol Chem. 1993; 268: 26350-26357.
70. Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, Eble JN, Fleming S, Ljungberg B, Medeiros LJ, Moch H, Reuter VE, Ritz E, Roos G, Schmidt D, Srigley JR, et al. The Heidelberg classification of renal cell tumours. J Pathol. 1997; 183: 131-133.
71. Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982; 6: 655-663.
72. Guinan P, Sobin LH, Algaba F, Badellino F, Kameyama S, MacLennan G, Novick A. TNM staging of renal cell carcinoma: Workgroup No. 3. Union International Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 1997; 80: 992-993.
73. Van Ostade X, Vandenabeele P, Tavernier J, Fiers W. Human tumor necrosis factor mutants with preferential binding to and activity on either the R55 or R75 receptor. Eur J Biochem. 1994; 220: 771-779.
74. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992; 119: 493-501.
75. Shen K, Song N, Kim Y, Tian C, Rice SD, Gabrin MJ, Symmans WF, Pusztai L, Lee JK. A systematic evaluation of multi-gene predictors for the pathological response of breast cancer patients to chemotherapy. PLoS One. 2012; 7: e49529.
76. Zhao H, Yuan X, Li D, Chen H, Jiang J, Wang Z, Sun X, Zheng Q. Isoliquiritigen enhances the antitumour activity and decreases the genotoxic effect of cyclophosphamide. Molecules. 2013; 18: 8786-8798.