
Introduction
The SWI/SNF complexes act as evolutionarily conserved chromatin remodeling complexes which utilize energy from ATP hydrolysis to regulate nucleosome sliding, and histone insertion/ejection, resulting in transcriptional activation or repression [1]. In mammals, there are multiple variants of the SWI/SNF complexes, including the BRM-associated proteins (BAF) complex, and the polybromo-containing BAF (PBAF) complex. Each complex consists of ATPases (BRM or BRG1), shared core subunits (SNF5, BAF155, and BAF170), and variant subunits, which are unique to each complex (such that ARID1A or ARID1B is unique to BAF complex, while BAF180, BAF200, and BRD7 are unique to PBAF complex) [2-5]. Notably, various forms of cancers harbor frequent inactivating mutations of several subunits of the SWI/SNF complexes, strongly suggesting that the SWI/SNF complexes function as tumor suppressors in cancer development [6-11]. In addition, the SWI/SNF complex-mediated chromatin remodeling has also been shown to play critical roles in mammalian development, tissue maintenance, and stem cell homeostasis [12-14]. However, it remains less understood how these different subunits of the SWI/SNF complexes contribute to tumor suppression and tissue maintenance in tissue/context-dependent manners.
As mentioned above, BAF180 functions as a variant subunit of the PBAF complex. BAF180 consists of two bromo-adjacent homology (BAH) domains, which mediate protein-protein interactions, and six tandem bromodomains, which recognize mono-acetylated lysine residues on histones [15, 16]. It has been proposed that BAF180 mainly functions to regulate the assembly of the PBAF complex, and/or recruit the PBAF complex to specific loci and regulate the transcription of specific targets [15]. Inactivating mutations of BAF180 are frequently observed in renal cancer and intrahepatic cholangiocarcinomas [10, 11], suggesting BAF180 is a tumor suppressor in these cancers. However, the exact role and mechanism of BAF180 in tumor suppression remains obscure, and in some cases, controversial. For example, while some studies showed that knockdown of BAF180 promoted cell proliferation [17, 18], consistent with its proposed tumor suppressor function, another study reported growth suppression phenotype in BAF180 knockdown cells [19]. Since these studies were conducted in different cell lines with potentially different knockdown efficiencies, it is likely that BAF180 may play a context and cell-lineage specific function in the regulation of cell proliferation. It also highlights the necessity to further clarify its function using a genetically defined complete KO system as used in this study. Finally, the roles of BAF180 in tumor suppression and tissue maintenance in vivo remain to be addressed by genetically engineered mouse models.
Cellular senescence, the state of permanent cell cycle arrest, represents an important mechanism in both tumor suppression and tissue maintenance [20-22]. Either oncogene activation or tumor suppressor loss can induce premature senescence, which then serves as a failsafe mechanism to restrict tumor development. On the other hand, it has been shown that senescence promotes stem cell aging, leading to impaired tissue maintenance and repair [22, 23]. Senescence can be regulated by multiple pathways, most notably the p53/p21 and p16/Rb pathways [24, 25]. Senescence is also associated with altered chromatin structures characterized by senescence-associated heterochromatic foci [26]. However, how BAF180-involved chromatin remodeling regulates senescence and senescence-associated tumor suppression and stem cell aging remain largely unknown.
Here, in the course of studying the cellular and organismal functions of BAF180 via genetically defined mouse models, we showed that conditional deletion of BAF180 led to cell cycle arrest and premature cellular senescence in primary MEFs, and decreased stem cell number and function in mouse hematopoietic system. An elevated p21 level was detected in BAF180 deficient cells. We confirmed that BAF180 directly binds to p21 promoter and negatively regulates its expression. Deletion of p21 could rescue cell cycle arrest and premature cellular senescence in BAF180 deficient MEFs and hematopoietic stem cell (HSC) depletion observed in BAF180 deficient mouse. Our results suggest that BAF180 regulates transcription of p21 thereby enabling cells to proliferate and maintaining stem cell homeostasis.
Results
BAF180 deletion triggers premature cellular senescence in MEFs
To study the function of BAF180 in the adult mice as well as in primary mouse embryonic fibroblasts (MEFs), and to circumvent the embryonic lethality phenotype associated with germline nullizygosity [27], we employed a conditional somatic knockout strategy by crossing BAF180L/L mice [19] with the tamoxifen inducible Cre deleter strain Rosa26-CreERT2.
Treatment of BAF180 L/L, Rosa26-CreERT2 MEFs with 4-hydroxytamoxifen (4-OHT) led to near complete loss of BAF180 protein in 4 days (Figure 1A and 1B). Deletion of BAF180 did not affect the expression level of SWI/SNF ATPase subunits BRG1 or BRM (Figure 1B). Analyses of multiple pairs of matched BAF180 WT and KO primary MEFs showed that loss of BAF180 in primary MEFs resulted in delayed cell proliferation and cell cycle arrest in G0/G1 phase with reduced S phase index (Figure 1C and 1D). Compared to BAF180 WT MEFs at the same passage, BAF180 KO MEFs exhibited an enlarged and flattened senescence-like morphology with significantly increased senescence associated β-galactosidase activity (Figure 1E and 1F). Correspondingly, BAF180 KO MEFs exhibited a significantly decreased cumulative population doublings with serial passages. While we could routinely obtain immortalized WT MEFs by 3T3 protocol, it was much more difficult to obtain immortalized BAF180 KO MEFs (Figure 1G for one representative BAF180 WT/KO MEF pair).
The cell cycle arrest of BAF180 deficient cells did not appear to be due to an altered DNA damage response (DDR), since gamma-H2AX and 53BP1 staining did not reveal consistent differences of DNA damage foci among different BAF180 WT/KO MEF pairs (Figure S1A and S1B). In addition, analysis of metaphase spreads showed that ablation of BAF180 did not affect chromosomal abnormalities or telomere erosion (Figure S1C) Together, our data reveal that BAF180 deletion induces premature senescence in primary MEFs, and suggest that it is less likely that DDR and genomic instability represent the prominent underlying mechanism for BAF180 deletion-induced premature cellular senescence.
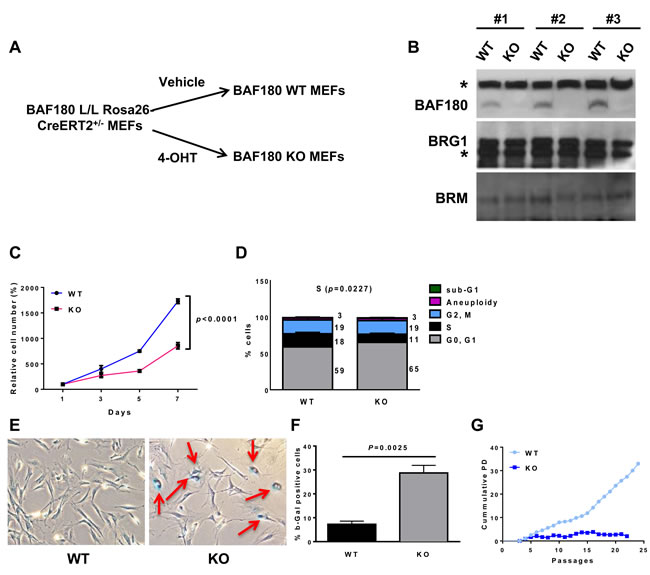
Figure 1: BAF180 depletion triggers premature cellular senescence in MEFs. A. A schematic representation of BAF180 deletion in mouse embryonic fibroblasts. B. Immunoblotting of BAF180, BRG1, and BRM in BAF180 WT and KO MEFs. The asterisk represents a nonspecific band. C. Growth curves of BAF180 WT and KO MEFs. Cells were seeded in triplicate into 12 well plates at a density of 2 X 104 at passage 4 and cell number was determined by a crystal violet staining method (means±SEM, n = 3). D. Cell cycle status of passage 3 MEFs analyzed by BrdU incorporation and PI staining. Numbers represent the percentage of cells in each cell cycle (Aneuploidy/G2, M/S/G0, G1 from top to bottom; means±SEM, n = 4). E. Representative images of senescence associated β-galactosidase staining. Cells were plated at passage 4 and cultured for 4 days before staining. F. The percentage of β-gal positive cells (means±SEM, n = 5). G. Cumulative population doublings of BAF180 WT and KO MEFs starting from passage 4.
BAF180 KO mice exhibit defective lymphocytes differentiation, decreased hematopoietic stem/progenitor cells with aging, and impaired repopulation capability
Regulation of cell proliferation and senescence is an important mechanism for maintaining tissue homeostasis [22, 23]. Our aforementioned data thus prompted us to further investigate the impact of BAF180 loss in adult mice. BAF180 L/L, Rosa26-CreERT2 and control littermate BAF180 L/L (or BAF180+/+, Rosa26-CreERT2) were injected with tamoxifen for 5 consecutive days at 4 weeks of age, resulting in BAF180 WT and KO mice. Surprisingly, BAF180 KO mice were grossly normal. With aging, BAF180 KO mice exhibited a decrease in the rate of body weight gain compared with WT mice (Figure S2A). BAF180 KO mice also had defects in lymphocytes development, characterized by reduction in thymus weight and cellularity (Figure S2B and S2C), defective T cell development in thymus (Figure S2D and S2E) and B cell development in bone marrow (Figure S2F and S2G). BAF180 mutations/deletions have been identified in various human cancers, most notably in renal cell carcinoma [11]. However, BAF180 KO mice, which have been followed for up to 1 year, did not develop renal cell carcinoma or other type of tumors (data not shown).
It is well known that appropriate regulation of stem cell function is critical in maintaining tissue and organ homeostasis. We thus examined the roles of BAF180 in the maintenance of HSCs, the best understood adult stem cells [28-30]. Analysis of HSC-enriched KSL cells (Lin- Sca-1+ c-Kit+) in bone marrow showed no significant difference between young BAF180 WT and KO mice (0-4 months post tamoxifen injection), yet a significant reduction in both percentage and absolute number of KSL cells was observed after 8 months of post tamoxifen injection (Figure 2A and 2B). Further analysis confirmed the same phenotype in BAF180 deficient long-term hematopoietic stem cells (LT-HSCs; CD34- Flt3- KSL) (Figure 2C and 2D).
Next we conducted competitive transplantation assay to study the impact of BAF180 deletion on HSC repopulating ability in vivo. Bone marrow cells from CD45.2+ BAF180 L/L, Rosa26-CreERT2 or littermate WT mice were mixed with CD45.1+ WT competitor bone marrow cells at 1:1 ratio, and then transplanted into lethally irradiated CD45.1+ recipient mice, followed by tamoxifen treatment at 6 weeks post-transplantation to delete BAF180 in hematopoietic system in the recipient hosts. Analysis of the peripheral blood and bone marrow revealed that BAF180 KO donor cells were capable of hematopoietic reconstitution (Figure 2E and 2F). Although BAF180 KO transplants exhibited slightly reduced reconstitution relative to WT controls, the reduction did not reach statistical significance. We then performed secondary transplantation assay by transplanting whole bone marrow cells purified from primary recipients at 16 weeks after tamoxifen injection into secondary recipients. Notably, we observed that BAF180 KO secondary transplants showed markedly diminished repopulating capability relative to WT controls (Figure 2G and 2H). Together, our results suggest that BAF180 deletion leads to impaired HSC functionality under aging or stress (serial transplantation) conditions.
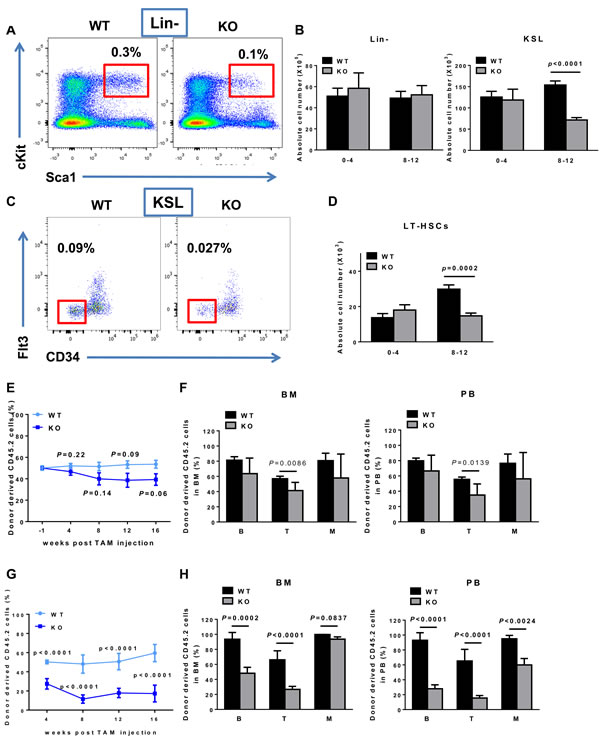
Figure 2: BAF180 KO mice exhibit decreased absolute number of HSCs and its progenitors with impaired repopulation ability. A. Flow cytometric analysis of KSL (Lin- Sca-1+ c-Kit+) cells in bone marrow of BAF180 WT and KO mice. FACS plots of Lineage negative (Lin-) cells were gated on KSL cells (Red). The numbers indicate the frequency of KSL cells in live bone marrow cells. B. Absolute numbers of lineage negative cells (X105, per 1 femur and 1 Tibia) and KSL cells (X103, per 1 femur and 1 Tibia) at 0-4 months and 8-12 months post tamoxifen injection (error bar represents means±SEM, n = 2-7; unpaired t-test). C. Flow cytometric analysis of long-term (LT)-HSCs. KSL cells were gated on CD34- Flt3- population (Red). The numbers indicate the frequency of LT-HSC in live bone marrow cells. D. Absolute number of LT-HSCs, CD34- Flt3- KSL. (means±SEM, n = 2-7). E. Percentage of donor derived cells in peripheral blood of CD45.1 recipients after competitive transplantation (n = 8-10). CD45.2 donor cells (either BAF180 WT or KO) and CD45.1 competitor cells were mixed in a 1:1 ratio and transplanted into lethally irradiated CD45.1 recipients (1X106 cells/recipient). 6 weeks post transplantation (-1 week post tamoxifen injection), blood samples were analyzed and the percentage of donor derived cells were normalized to 50%. F. Percentage of donor derived CD45.2 cells were analyzed from total bone marrow and peripheral blood at 16 weeks post transplantation. B (B cells); T (T cells); M (Myeloid cells) (WT: n = 6, KO: n = 7, 8). G. Peripheral blood chimerism of CD45.2 donor derived cells after secondary transplantation (WT n = 7, KO n = 8). Whole bone marrow cells were isolated from three primary recipients and 3X106 cells were transplanted into a secondary recipient (n = 10 recipients per group). H. Percentage of donor derived cells in hematopoietic populations 16 weeks after secondary transplantation.
BAF180 deletion induces p21 in MEFs and HSCs
The aforementioned data prompted us to further study the underlying mechanisms by which BAF180 regulate cellular senescence in MEFs and HSC homeostasis. It is well established that CDK inhibitors, up-stream regulators of p53 and retinoblastoma (Rb) pathways, play critical roles in the regulation of cellular senescence [22]. We thus compared the expression levels of several CDK inhibitors in BAF180 WT and KO MEFs. Our analysis revealed that the level of p21 was significantly induced in BAF180 deficient MEFs, whereas the expression levels of its upstream regulator p53 or other CDK inhibitors were not changed (Figure 3A and 3B). Consistent with our results from MEFs, BAF180 deletion also induced p21 expression in hematopoietic stem/progenitor cells of 8 month-old mice (Figure 3C).
Chromatin immunoprecipitation (ChIP) assay showed that BAF180 bound to p21 promoter (Figure 3D). Furthermore, BAF180 deletion in MEFs led to increased binding of modified histones associated with transcriptional activation, such as H3K9 acetylation, H3K27 acetylation and H3K4 mono-methylation, on p21 promoter (Figure 3E), suggesting that BAF180 represses p21 expression through epigenetic regulation of histone modification on p21 promoter. BAF180 deletion (by 4-OHT treatment) in SV40 large T antigen-infected (thus p53 defective) BAF180L/L, Rosa26-CreERT2 MEFs still resulted in p21 induction and reduced cell proliferation (Figure 3F and 3G), suggesting that BAF180 regulation of p21 and cell proliferation is at least partly p53-independent. Collectively, our results reveal that BAF180 represses p21 transcription, and BAF180 deletion results in increased p21 expression in both MEFs and HSCs.
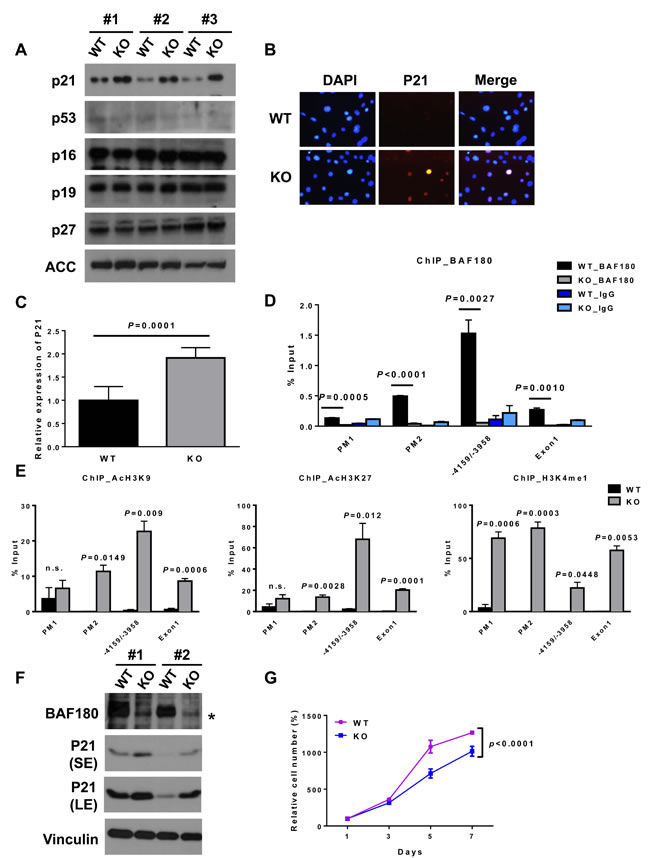
Figure 3: BAF180 negatively regulates p21 expression in MEFs and HSCs. A. Immunoblotting analysis of cell cycle regulators. Three independent MEFs per genotype were analyzed at passage 6. B. Immunofluorescence staining of p21 (Cy3) and DAPI shows elevated p21 level in BAF180 KO MEFs. C. The amount of p21 transcripts was determined by real-time PCR in purified KSL cell population (means±SD, n = 6). D. ChIP-qPCR analysis of BAF180 enrichment on p21 promoter in BAF180 WT and KO primary MEFs, showing that BAF180 can bind to p21 promoter at -5681 to -5464 (in the promoter) and -4159 to -3958 nucleotides upstream from the ATG. PM1: promoter -6858/-6610, PM2: promoter -5681 to -5464, Exon 1: +63 to +428 (means±SEM, n = 3). E. ChIP-qPCR analysis of binding of modified histones, including AcH3K9, AcH3K27 and H3K4me1, on p21 promoter in BAF180 WT and KO primary MEFs (means±SEM, n = 3). F. Immunoblotting analysis of BAF180 and p21 in two pairs of SV40 large T immortalized MEFs. The asterisk represents a nonspecific band. G. Growth curves of SV40 large T immortalized BAF180 WT and KO MEFs (means±SD, n = 3).
p21 deletion rescues cell cycle arrest and premature senescence in BAF180 deficient MEFs and HSC depletion in BAF180 deficient mice
To determine the functional relevance of BAF180 deficiency-induced p21 expression to the phenotypic defects observed in BAF180 deficient MEFs and HSCs, we crossed BAF180L/L Rosa26-CreERT2 mice with p21-/- mice, and isolated MEFs from BAF180L/L Rosa26-CreERT2 and BAF180L/L p21-/- Rosa26-CreERT2 embryos. MEFs were then treated with either vehicle or 4-OHT to obtain BAF180L/L p21+/+ (WT), BAF180-/- p21+/+ (BAF180 KO), BAF180L/L p21-/- (p21 KO), and BAF180-/- p21-/- (double KO or DKO) cells. The protein levels of p21 were increased in BAF180 KO MEFs compared to wild-type MEFs and, as expected, there was no p21 expression in p21 KO or DKO cells (Figure 4A).
As expected, BAF180 KO MEFs exhibited decreased, while p21 KO MEFs showed increased, proliferation rates compared with WT MEFs (Figure 4B). Notably, p21 deletion fully rescued the proliferation defects observed in BAF180 KO MEFs, such that the proliferation rate of DKO MEFs was indistinguishable from that of p21 KO MEFs (Figure 4B), which was further corroborated by the cell cycle profiling analysis (Figure 4C). Full rescue of premature senescence in BAF180 KO MEFs was also observed in DKO MEFs (Figure 4D and 4E). Together, our results strongly suggest that BAF180 deletion in MEFs induces p21-dependent checkpoints, resulting in cell cycle arrest and premature senescence.
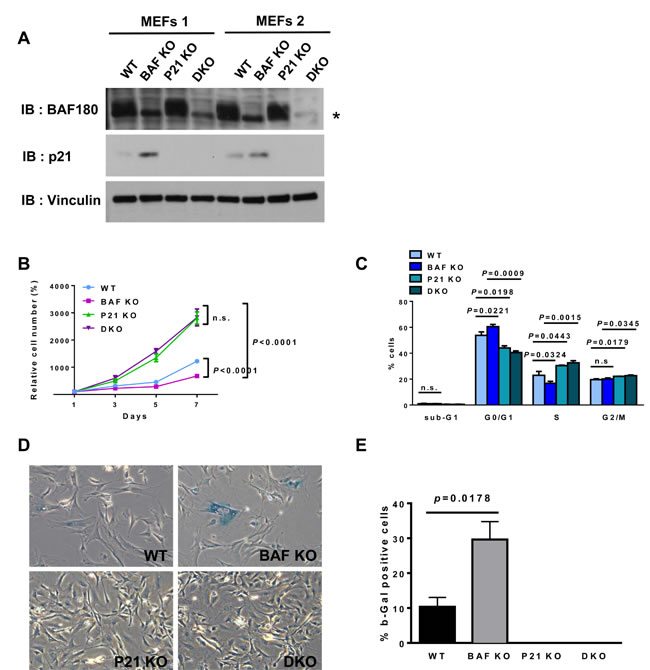
Figure 4: p21 deletion rescues premature senescence in BAF180 KO MEFs. A. Immunoblotting analysis of BAF180 and p21 in passage 6 MEFs. An increased p21 level was observed in BAF180 KO MEFs. Two independent MEFs lines were used for each genotype. The asterisk represents a nonspecific band. B. Growth curves of wild-type, BAF180 KO, p21 KO, double KO (DKO) MEFs at passage 4 (n = 2, 3). C. Cell cycle status of MEFs analyzed by BrdU incorporation and PI staining at passage 3 (means±SD, n = 2, 3). D. Representative images of senescence associated β-galactosidase staining at passage 4 (n = 2, 3). E. The percentage of β-gal positive cells (means±SD, n = 2, 3). There was no blue stained cell in p21 KO and DKO MEFs at passage 4.
Since BAF180 deletion resulted in HSCs depletion and defects in stem cell self-renewal with increase in p21 expression, we examined whether deletion of p21 in BAF180 KO mice could rescue HSC defects, as observed in MEFs. We observed that deletion of p21 did not affect lineage negative cell population (Figure 5A) but improved absolute number of KSL cells in 8-10 month-old BAF180 KO mice (Figure 5B). p21 depletion also significantly enhanced LT-HSC population in BAF180 KO mice in both absolute cell number and frequency (Figure 5C and 5D).
To analyze if loss of p21 also rescued self-renewal defects in BAF180 deficient HSCs, we performed in vitro colony formation assay from single CD34-CD150+ KSL cells. BAF180 deficient CD34-CD150+ KSL cells formed less colonies compared with WT cells, with most notable reduction in high potential proliferating colony forming cells (HPP-CFCs) larger than 2 mm in diameter. Notably, p21 deletion or BAF180/p21 double deletion significantly increased the population of HPP-CFCs ( > 2mm). In conclusion, our data suggests that BAF180 deletion results in decreased number and function of hematopoietic stem/progenitor cells, and this defect is at least partly caused by elevated level of p21 in hematopoietic system.
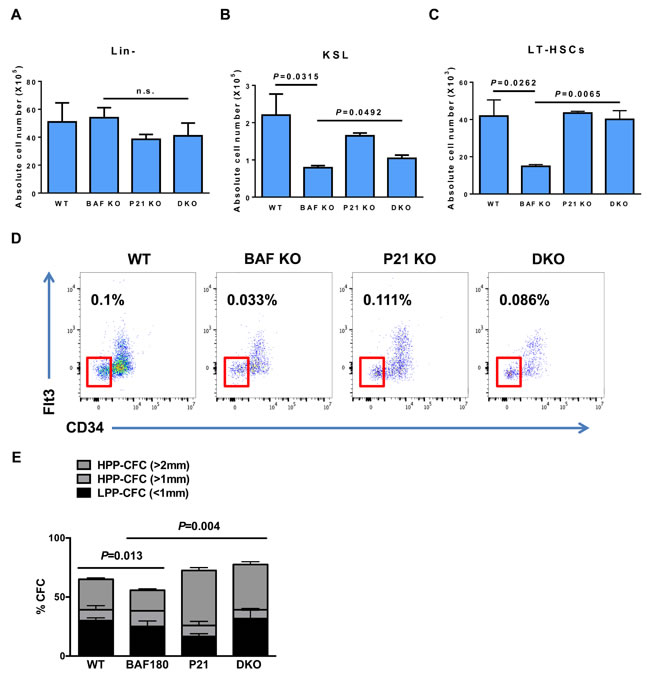
Figure 5: p21 deletion improves HSC defects in BAF180 deficient mice.A. Absolute number of Lineage negative cells (X105) in bone marrow (1 femur and 1 Tibia) of mice of the indicated genotypes. Cells were isolated at 8-10 months post tamoxifen injection (means±SD, n = 3, 4). B. Absolute number of KSL cells (X105) and C. absolute number of LT-HSCs (X103, CD34- Flt3- KSL) per 1 femur and 1 Tibia (means±SD, n = 2-4). D. Representative FACS profiles of KSL cells of indicated genotypes. FACS plots were gated on LT-HSCs. The numbers indicate the frequency of LT-HSCs in live bone marrow cells. E. Single cell CFC assay. Single CD34-CD150+ KSL cells were sorted into 96 well plates in the presence of mSCF, IL-3 and EPO. Colonies were evaluated at day 14. LPP-CFCs (colony diameter < 1mm), HPP-CFCs (colony diameter > 1mm). 60 single sorted cells per mouse were analyzed (means±SD, n = 2, 3).
Discussion
In this study, we show that inactivation of BAF180, a subunit of the SWI/SNF complex, led to premature senescence with G0/G1 cell cycle arrest in MEFs, and resulted in decreased hematopoietic stem/progenitor cells in aged mice with impaired repopulation capability. In addition, we showed that BAF180 deletion induced p21 expression in both MEFs and HSCs. We provided strong genetic evidence to support the model that BAF180 deletion-induced p21 is at least partially responsible for the phenotypic defects observed in BAF180 deficient cells or animals, as deletion of p21 in the backdrop of BAF180 deficiency could rescue the premature senescence phenotype in BAF180 KO MEFs, and significantly rescue the decreased number of HSCs in BAF180 KO mice. Together, our study suggest a model that, at least in the model system we have studied, BAF180 normally represses p21 expression, and BAF180 deficiency initiates a p21-dependent checkpoint to induce cell cycle arrest and premature senescence, which likely is responsible for the reduced HSC reserves observed in aged BAF180 deficient mice.
The aforementioned model is in line with the well-established function of p21 to promote cell cycle arrest and senescence. However, the exact roles of p21 in the regulation of stem cell homeostasis and aging are complex. On one hand, p21 controls HSC quiescence, which is critical in maintaining HSC self-renewal. p21 deletion resulted in increased HSC cycling and subsequent exhaustion, and defective repopulating potential [31]. On the other hand, it has been shown that activation of p21-dependent checkpoint limits HSC self-renewal in aging telomere dysfunctional mice. Correspondingly, deletion of p21 improved the repopulating potential and self-renewal of HSCs from mice with dysfunctional telomeres [23]. Similar to the above study conducted in the context of telomere dysfunction, our study suggests that BAF180 deficiency also likely induces the p21-dependent checkpoint to restrain HSC functionality in aged mice. It is possible that BAF180 is differentially regulated along aging. However, our current data showed that BAF180 expression does not significantly change during aging in different mouse tissues (Figure S3). The potential mechanism by which BAF180 is regulated under physiological aging process merits further investigation.
In contrast to our data showing that BAF180 deletion in MEFs led to premature senescence, cell cycle arrest, and increased p21 expression, previous studies showed that knockdown of BAF180 by shRNA promoted proliferation, or delayed senescence with decreased p21 expression [17, 18]. Since these studies were conducted in different cellular contexts (cancer cell lines in previous studies vs primary MEFs and HSCs in this study), it is possible that BAF180 plays context and cell-lineage specific functions in the regulation of cell cycle and p21 expression. For example, certain genetic mutations in cancer cells may turn BAF180 from a p21 repressor to a p21 activator. Alternatively, complete knockout by genetic deletion vs partial knockdown by shRNA may result in different outcomes, suggesting the gene dosage of BAF180 may be important for its function.
Recent cancer genomic studies have identified frequent inactivating mutations in BAF180 gene in various types of human cancers, most notably renal cell carcinoma and intrahepatic cholangiocarcinomas [10, 11]. For example, BAF180 is mutated in about 40% of clear cell renal cell carcinomas (ccRCCs), a predominant RCC subtype which accounts for up to 75% of all RCC cases, ranking BAF180 as the second most mutated gene in ccRCC only after VHL [11]. Most of these mutations are inactivating mutations. BAF180 is located on chromosome 3p, a loci frequently deleted in human cancers. These genomic data strongly suggest that BAF180 is a bona fide tumor suppressor in human cancers. However, our current study reveals that broad somatic deletion of BAF180 in adult mice is not sufficient to drive the development of renal cell carcinoma or other cancers (data not shown). In addition, in contrast to the expected enhanced proliferation or immortalization phenotype from MEFs with deficiency of typical tumor suppressors (such as p53), BAF180 KO MEFs exhibit premature senescence, cell cycle arrest, and are much more difficult to be immortalized (Figure 1). It should be noted that deletion of some tumor suppressors, such as PTEN or VHL, in primary MEFs also results in premature senescence [32, 33]. It has been proposed that senescence may serve as a failsafe program to restrain tumor suppressor deficiency-induced tumor development, which may explain the lack of tumor phenotypes in our BAF180 deficient mouse model. Whether combined deletion of BAF180 and p21 (or another senescence checkpoint regulator) would promote any tumor phenotype in vivo remains to be determined in the future studies. In the case of renal tumor development, since deletion of VHL in the mouse is not sufficient to drive renal tumor development [34], it will be interesting to determine whether combined deletion of BAF180 and VHL, the two most frequently mutated tumor suppressors in renal cancer, in the kidney would provoke renal tumor phenotype in vivo.
Materials and Methods
Generation and analysis of mice
The generation of BAF180L allele was described in the previous publication [19]. BAF180L/L mice were crossed with tamoxifen-inducible Cre deleter strain, Rosa26-CreERT2 to generate BAF180L/+ Rosa26-CreERT2+ mice. BAF180L/+ Rosa26-CreERT2+ mice were further backcrossed six generations with C57BL/6J mice and then F6 progeny were intercrossed to generate mice of the desired genotypes. Tamoxifen treatment of mice to induce Cre recombinase activation and target gene deletion was done as previously described [35-37]. Briefly, tamoxifen (120 ug/gram body weight; Sigma, T5648) was injected into 4 weeks old mice intraperitoneally for five consecutive days. Cre deleter mice (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J), p21 knockout mice (B6.129S6(Cg)-Cdkn1atm1Led/J) and Ly5.1 recipients for bone marrow transplantation assays (B6.SJL-Ptprca/BoyAiTac) were obtained from The Jackson Laboratory. Tissue samples were fixed in 10% neutral-buffered formalin (Fisher Scientific) overnight, washed with 1XPBS and stored in 70% ethanol at 4 °C. Tissues were processed for paraffin embedding and sections were prepared for H&E staining (5 um) and antibody immune staining (3 um). All animal manipulations were performed under MD Anderson IACUC (Institutional Animal Care and Use Committee)-approved protocols.
Primary and LT-immortalized MEFs
All MEFs were prepared from E13.5 embryos as we previously described [38]. The generation of BAF180 WT and KO MEFs from BAF180 L/L, Rosa26-CreERT2 MEFs was conducted as previous described [39, 40]. Briefly, MEFs (passage 0 or 1) were treated with either 200 nM 4-hydroxytamoxifen (4-OHT; Sigma, H7904) or vehicle (ethanol control) for 4 days, resulting in BAF180 WT and KO MEFs. For immortalized BAF180 WT and KO MEFs, primary BAF180 L/L, Rosa26-CreERT2 MEFs were first immortalized by infection of SV40 large T antigen, followed by 4-OHT treatment.
BrdU incorporation assay
BrdU staining was performed using FITC BrdU Flow kit BD (BD Pharmingen, 559619) according to manufacturer’s protocol. Briefly, MEFs were pulsed with 10 uM BrdU for 90 min and trypsinized, fixed and permeabilized. After treatment of DNase for 1 h at 37 °C, cells were stained with FITC-labeled anti-BrdU antibody for 20 min at RT, incubated with RNase and PI for 30 min and analyzed with LSRII flow cytometer.
In situ senescence-associated β-gal assay
MEFs (passage 4-6 as indicated) were cultured for 4 days and cellular senescence was determined using a Senescence β-galactosidase staining kit (Cell signaling, #9860). Cells were fixed and β-galactosidase activity was detected at pH6.0. Cells were incubated with X-gal staining solution for 8-12h at 37 °C in a dry incubator without CO2.
FACS
Single-cell suspensions were prepared from spleen and thymus by passing cells through a 70 um cell strainer (Falcon). Bone marrow cells from tibias, femurs and spine were crushed with PBS +2% FBS and filtered. After cell counting, cells were stained with following antibody (BD Biosciences, eBiosciences) combinations: Biotin-conjugated lineage marker cocktail (Ter-119, Gr-1, Mac-1, B220, CD4, CD8), HSC (Sca1, c-Kit, CD34, Flt3), T lymphocytes (CD4, CD8, CD25, CD44, Ter119), B lymphocytes (B220, CD4, CD8, CD19, CD43). For KSL cells sorting, cKit positively selected cells were used for staining. Briefly, cells were stained with c-Kit-APC antibody for 30 min on ice, washed with PBS and incubated with anti-APC microbeads for 15 min at room temperature. c-Kit positive cells were collected using a MACS separation column and used for HSC staining. FACS analysis was carried out on LSRII (BD Biosciences) and Gallios (Beckman coulter) at the MDACC Flow Cytometry and Cellular Imaging Core Facility. FACS data was analyzed with Flowjo software V10.0.8.
Western blot
Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25 % deoxycholic acid, 1 % NP-40, 1 mM EDTA) containing protease inhibitors (Roche, 4693159001) and phosphatase inhibitors (EMD Millipore, 524625). Protein concentrations were determined by Bradford assay (Bio-Rad, 500-0006) and western blots were performed using 20-40 ug of protein. The following antibodies were used in this study: BAF180 (Bethyl Laboratories, A301-591A, 1:500), p16 (Santa Cruz, sc-1207, 1:1,000), p53 (Santa Cruz, sc-6243, 1:1,000), BRG1 (Santa Cruz, sc-7796, 1:1,000), BRM (Santa Cruz, sc-6450, 1:1,000), p19 (Abcam, Ab80, 1:2,000), p21 (Santa Cruz, sc-6246, 1:1,000), vinculin (Sigma, V4505, 1:50,000).
Quantitative real-time PCR
RNA from tissue samples was extracted using Trizol (Invitrogen) and RNA from sorted LT-HSC was extracted using PicoPure RNA isolation kit (Life technologies, KIT0204). RNA was treated with RQ1 RNase-free DNase (Promega, M6101) and cDNA was prepared using Superscript III reverse transcriptase (Invitrogen, 18080-051). qRT-PCR was carried out in triplicate for each cDNA sample with TaqMan probe for p21 (CDKN1A, Mm00432448) and following primers : BAF180 (forward 5’- GCCCATATCCTGGAGAAGTTA-3’, reverse 5’-GGCGACCCCTCTTATCAGTA-3’) Ribosomal protein R15 (internal control, forward 5’- CTTCCGCAAGTTCACCTACC-3’, reverse 5’-TACTTGAGGGGGATGAATCG-3’). RT-PCR was run on stratagene Mx3000P (Agilent Technologies).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using a commercial kit SimpleChIP® Plus Enzymatic Chromatin IP Kit (Cell signalling) according to manufacture’s protocol. Briefly, MEFs were cross-linked at passage 4 and digested chromatin was immunoprecipitated with 2ug of BAF180 antibody (Bethyl Laboratories, A301-591A), 1 ug of H3K9Ac (Abcam, 4441), 1ug of H3K27Ac (Abcam, 4729), 1 ug of H3K4me1 (Abcam, 8895) and 1 ug of IgG (Cell signaling, 2729). The resulting DNA was used for quantitative real-time PCR using primers located on p21 promoter regions. PM1 (F: 5’-GGGCAGTTTTGACATCCTGT-3’, R: 5’-ACAGGCCTCCTCTTCTGTGA-3’), PM2 (F: 5’-AGTGTGGTCCCAGTCAGGTC-3’, R: 5’-AGACGAGGAAAGCAGTTCCA-3’), -4159/-3958 (F: 5’-GCTTCTGGTTGCCAATGTTT-3’, R: 5’-AAACAGGGCACTTGGTTCAC-3’), Exon1 (F: 5’-CGGTCCCGTGGACAGTGAGCAG-3’, R: 5’-GTCAGGCTGGTCTGCCTCCG-3’).
Competitive transplantation assay
Bone marrow cells from 2 month-old BAF180+/+, CreERT2+/- or BAF180L/L, CreERT2+/- mice (both CD45.2) were isolated and mixed with bone marrow cells from a CD45.1 mouse at the 1:1 ratio. Cells were then injected into lateral tail vein of lethally irradiated (12 Gy) CD45.1 recipients (1X106 cells/recipient). Six weeks after transplantation, peripheral blood was collected from the recipients to confirm successful reconstitution and recipients were injected with tamoxifen (120 ug/gram body weight) intraperitoneally for five consecutive days. Peripheral blood was collected and analyzed at 4, 8, 12 and 16 weeks post transplantation. For secondary transplantation, total bone marrow cells were isolated from three primary recipients, mixed together in equal proportion and injected into lethally irradiated CD45.1 recipients. Donor chimerism was analyzed from blood every 4 weeks. Three donor mice and eight-ten recipients were used for each transplant.
HSC single cell colony assay
The assay was conducted as previously reported [41]. Briefly, bone marrow cells were isolated from wild-type, BAF180 KO, p21 KO and BAF180/p21 double KO mice six weeks after tamoxifen injection. Single CD34-CD150+ KSL cells were sorted into 96-well plates and cultured in serum-free expansion media in the presence of 10 ng ml−1 of mouse stem cell factor (mSCF), 10 ng ml−1 interleukin-3 (IL-3) and 10 ng ml−1 erythropoietin (EPO), 10% fetal bovine serum and penicillin/streptomycin. The number of colony-forming cells was evaluated at day 14.
Statistics
Statistical analyses were performed using GraphPad Prism 6. The unpaired two-tailed Student’s t-test was used to calculate P values.
Acknowledgments
We thank all members of the Gan laboratory for their advice and technical assistance.
Conflicts of Interest
The authors declare competing interest in the work presented.
Funding
This research has been supported by grants from UT MD Anderson Cancer Center, Cancer Prevention & Research Institute of Texas (RP130020), National Institutes of Health (CA181196, CA190370), Ellison Medical Foundation (AG-NS-0973-13), and Gabrielle’s Angel Foundation for Cancer Research (to B. G.). M. J. Y is supported in part by NIH/NCI R01 CA164346, CPRIT RP140402, and Center for Genetics and Genomics, Center for Inflammation and Cancer, Institutional Research Grant (IRG), and Sister Institution Network fund of UT MD Anderson Cancer Center. B. G. is a Kimmel Scholar and an Ellison Medical Foundation New Scholar. B. G., L. M, and M. J. Y. are members of the UT M.D. Anderson Cancer Center, and are supported by the National Institutes of Health Core Grant CA016672.
References
1. Wilson BG and Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews Cancer. 2011; 11:481-492.
2. Kwon H, Imbalzano AN, Khavari PA, Kingston RE and Green MR. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. 1994; 370:477-481.
3. Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B and Kornberg RD. RSC, an essential, abundant chromatin-remodeling complex. Cell. 1996; 87:1249-1260.
4. Roberts CW and Orkin SH. The SWI/SNF complex--chromatin and cancer. Nature reviews Cancer. 2004; 4:133-142.
5. Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL and Crabtree GR. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. The EMBO journal. 1996; 15:5370-5382.
6. Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A and Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998; 394:203-206.
7. Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L, Perin JC, Xie H, Shaikh TH and Biegel JA. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clinical cancer research. 2009; 15:1923-1930.
8. Reisman DN, Sciarrotta J, Wang W, Funkhouser WK and Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer research. 2003; 63:560-566.
9. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363:1532-1543.
10. Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, Offerhaus GJ, Roa JC, Roberts LR, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nature genetics. 2013; 45:1470-1473.
11. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, Bignell G, Butler A, Cho J, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011; 469:539-542.
12. Ho L and Crabtree GR. Chromatin remodelling during development. Nature. 2010; 463:474-484.
13. Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G and Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000; 6:1287-1295.
14. Gao X, Tate P, Hu P, Tjian R, Skarnes WC and Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U S A. 2008; 105:6656-6661.
15. Thompson M. Polybromo-1: the chromatin targeting subunit of the PBAF complex. Biochimie. 2009; 91:309-319.
16. Chandrasekaran R and Thompson M. Polybromo-1-bromodomains bind histone H3 at specific acetyl-lysine positions. Biochem Biophys Res Commun. 2007; 355:661-666.
17. Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T, Memeo L, Hibshoosh H and Parsons R. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer research. 2008; 68:1667-1674.
18. Burrows AE, Smogorzewska A and Elledge SJ. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci U S A. 2010; 107:14280-14285.
19. Wurster AL, Precht P, Becker KG, Wood WH, 3rd, Zhang Y, Wang Z and Pazin MJ. IL-10 transcription is negatively regulated by BAF180, a component of the SWI/SNF chromatin remodeling enzyme. BMC immunology. 2012; 13:9.
20. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
21. Collado M and Serrano M. Senescence in tumours: evidence from mice and humans. Nature reviews Cancer. 2010; 10:51-57.
22. Kuilman T, Michaloglou C, Mooi WJ and Peeper DS. The essence of senescence. Genes Dev. 2010; 24:2463-2479.
23. Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, Lee HW, von Zglinicki T, Ganser A, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature genetics. 2007; 39:99-105.
24. Campisi J and d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8:729-740.
25. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75:685-705.
26. Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD et al.. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005; 8:19-30.
27. Wang Z, Zhai W, Richardson JA, Olson EN, Meneses JJ, Firpo MT, Kang C, Skarnes WC and Tjian R. Polybromo protein BAF180 functions in mammalian cardiac chamber maturation. Genes Dev. 2004; 18:3106-3116.
28. Adams GB and Scadden DT. The hematopoietic stem cell in its place. Nat Immunol. 2006; 7:333-337.
29. Moore KA and Lemischka IR. Stem cells and their niches. Science. 2006; 311:1880-1885.
30. Orkin SH and Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008; 132:631-644.
31. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M and Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000; 287:1804-1808.
32. Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, Signoretti S and Kaelin WG, Jr. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008; 10:361-369.
33. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C and Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005; 436:725-730.
34. Rankin EB, Tomaszewski JE and Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer research. 2006; 66:2576-2583.
35. Gan B, Lim C, Chu G, Hua S, Ding Z, Collins M, Hu J, Jiang S, Fletcher-Sananikone E, Zhuang L, Chang M, Zheng H, Wang YA, Kwiatkowski DJ, Kaelin WG, Jr., Signoretti S, et al. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell. 2010; 18:472-484.
36. Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L and Depinho RA. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010; 468:701-704.
37. Gan B, Sahin E, Jiang S, Sanchez-Aguilera A, Scott KL, Chin L, Williams DA, Kwiatkowski DJ and DePinho RA. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008; 105:19384-19389.
38. Gan B, Peng X, Nagy T, Alcaraz A, Gu H and Guan JL. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol. 2006; 175:121-133.
39. Lin A, Yao J, Zhuang L, Wang D, Han J, Lam EW, Network TR and Gan B. The FoxO-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene. 2014; 33:3183-3194.
40. Lin A, Piao HL, Zhuang L, Sarbassov DD, Ma L and Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacological inhibition of the PI3K-AKT pathway. Cancer Res. 2014; 74:1682-93.
41. Ema H, Morita Y, Yamazaki S, Matsubara A, Seita J, Tadokoro Y, Kondo H, Takano H and Nakauchi H. Adult mouse hematopoietic stem cells: purification and single-cell assays. Nature protocols. 2006; 1:2979-2987.