
INTRODUCTION
Glioma represents approximately 70% of primary brain tumors in adults, with an incidence of about 6 cases per 100,000 individuals worldwide [1]. The most advanced form of glioma, as well as the most invasive and resistant to therapy, is glioblastoma (GBM), which comprises 50%–60% of all gliomas. These tumors arise from three different types of glial cells that are normally found in the brain: astrocytes, oligodendrocytes, and ependymal cells. There are a number of histological subtypes, as stratified by the World Health Organization (WHO). Grade I and II low-grade diffuse gliomas are slow-growing, less aggressive tumors, while grade III and IV are more aggressive malignant tumors. The most common form, with the worst prognosis, is GBM (WHO grade IV). Patients with GBM usually survive less than 15 months following diagnosis. Currently, there are no effective long-term treatments for this disease [2, 3].
Recent research by The Cancer Genome Atlas (TCGA), together with intensive research over the last three decades, has revealed a large number of putative oncogenes that are amplified or overexpressed in glioma [4–6], including c-Myc and Cdc20. C-Myc is a well-known transcription factor with roles in cell cycle progression, apoptosis, and longevity during development [7] and oncogenesis [8] and in stem cell self-renewal, differentiation, and metabolic reprogramming [9, 10]. Elevated expression of c-Myc is associated with aggressive disease and a poor clinical outcome [11, 12]. It is also notable that both the p53 and PTEN/PI3K pathways can directly regulate c-Myc, with p53 repressing c-Myc transcription by directly binding to the c-Myc promoter, whereas downstream PI3K pathway arms can modulate c-Myc translation and protein degradation [13]. Cdc20 appears to act as a regulatory protein that interacts with several other proteins at multiple points in the cell cycle [14–16]. It is required for two microtubule-dependent processes, nuclear movement prior to anaphase and chromosome separation [15, 17]. Cdc20 is highly expressed in many types of cancer, including GBM [17, 18]. Recently, studies have shown that Cdc20 maintains tumor initiation cells and serves as a potential target for cancer treatment [18–21]. However, wether c-Myc and Cdc20 overexpression is a driver or passenger event in gliomagenesis remains unclear.
In this study, we used a powerful glia-specific mouse model (the RCAS/Ntv-a system) to characterize the oncogenic function of c-Myc and Cdc20 in gliomagenesis in vitro and in vivo. Then, because single c-Myc and Cdc20 genes are not sufficient to induce glioma in both CDKN2A+/+ and CDKN2A−/− Ntv-a mice, we explored the role of c-Myc and Cdc20 in glioma development in a RCAS/Ntv-a mouse model using the combination of kRas and Akt3.
RESULTS
C-Myc and Cdc20 promote glial progenitor cell proliferation and immortalization
We previously reported that some GBM signature genes, including c-Myc and Cdc20, distinguish GBM from other low-grade gliomas [22]. We analyzed the expression of these genes in patient tissue samples from the Rembrandt dataset and confirmed that they were both highly expressed in high-grade GBM samples compared to in normal control samples; Cdc20 expression was induced gradiently through low grade to high grade (Figure 1A). In addition, the expression of c-Myc and Cdc20 was significantly higher in the samples with GBM in the TCGA GBM dataset than in the normal control samples (data not shown). To understand the oncogenic function of c-Myc and Cdc20, we performed proliferation and transformation assays ex vivo using primary glial progenitor cells from Ntv-a mice. As shown in Figure 1B, overexpression of c-Myc and Cdc20 promoted glial progenitor cell proliferation (Figure 1B). Interestingly, the transduced glial progenitor cells with c-Myc or Cdc20 could be cultured over 6 months, whereas GFP control cells were arrested in growth within 1 month. Moreover, the glial progenitor cells immortalized by c-Myc overexpression not only showed higher proliferation rates than did those immortalized by Cdc20 overexpression (Figure 1C) but also gained the ability to form anchorage-independent colonies, unlike Cdc20-transduced cells (Figure 1D and 1E). C-Myc has stronger oncogenic activity than does Cdc20 in promoting glial progenitor cell transformation.
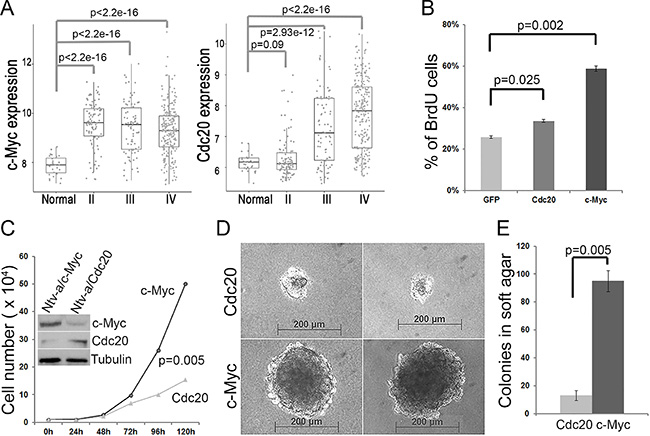
Figure 1: c-Myc and Cdc20 promote glial progenitor cell proliferation and transformation. (A) Expression of Cdc20 and c-Myc was induced in GBM. Expression of Cdc20 and c-Myc was analyzed in the patient tissue samples from the Rembrandt database, including normal tissues (28 cases), grade II glioma (99 cases), grade III glioma (85 cases), and grade IV GBM (228 cases). Expression of c-Myc and Cdc20 (signal intensity) was significantly different between normal tissues and grade IV GBM cases (P < 2.2e-16). (B) Glial progenitor cells were isolated from the brains of Ntv-a newborn mice and infected with RCAS-c-Myc, RCAS-Cdc20, or RCAS-GFP. The transduced cells were incubated with 10 μM BrdU for 1 hour and fixed with 70% ethanol. BrdU-positive cells were measured by flow cytometry after the cells were labeled with anti-BrdU antibody conjugated with FITC. The data were obtained from three replicates (p values, Student’s t-test). (C) The immortalized cells with c-Myc or Cdc20 were seeded in a 24-well plate at 1 × 104 cells/well. The cell growth curve was drawn according to the mean number of cells, in triplicate, at the indicated time points. (D) The immortalized cells with c-Myc or Cdc20 were seeded on a six-well plate with soft agar and incubated for 2 more weeks in cell culture conditions. A representative image of the colonies is shown. (E) The quantification of colonies was from three independent experiments.
Oncogenic signal of c-Myc or Cdc20 is insufficient to induce glioma, and the combination of kRas and Akt3 is sufficient to induce GBM in Ntv-a CDKN2A-null mice
To investigate the causal role of c-Myc and Cdc20 in glioma development, we injected Ntv-a transgenic mice with DF1 cells that produced RCAS-c-Myc or RCAS-Cdc20. A histopathologic examination of the brains from injected mice at the end of the 12-week study indicated that delivery of RCAS-c-Myc or RCAS-Cdc20 alone failed to induce glioma (Figure 2A). Therefore, we built a glioma mouse model by combining kRas and Akt family genes in the RCAS/Ntv-a glia-specific mouse model. An analysis of GBM samples from TCGA revealed that the Akt signaling pathway is one of the most altered pathways in this tumor. Akt3 is one of three closely related serine/threonine-protein kinases in the Akt family (Akt1, Akt2, and Akt3). In more recent years, it has become evident that Akt3 is more important than is Akt1 in glioma development and progression [23, 24]. Thus, we injected Ntv-a transgenic mice with DF1 cells that produced RCAS-kRas and RCAS-Akt3. As shown in Figure 2A, at the end of week 12, no gliomas had developed in 55 injected Ntv-a mice in two independent experiments. Because human high-grade glioma has additional deletions and mutations, such as loss of CDKN2A, which results in the disruption of cell cycle arrest pathways [25, 26], we further determined whether loss of CDKN2A is required for kRas- and Akt3-induced glioma by injecting CDKN2A-null Ntv-a transgenic mice with RCAS-kRas and RCAS-Akt3. Nine of 47 (19.1%) mice developed glioma within 12 weeks (Figure 2B). All of these tumors were GBM according to the histopathological criteria (microvascular proliferation and necrosis); other features included the appearance of mitotic cells, spindle cells, and giant cells (Figure 2C), as described in the WHO 2000 classification system. Therefore, loss of CDKN2A is required for GBM induction by kRas and Akt3.
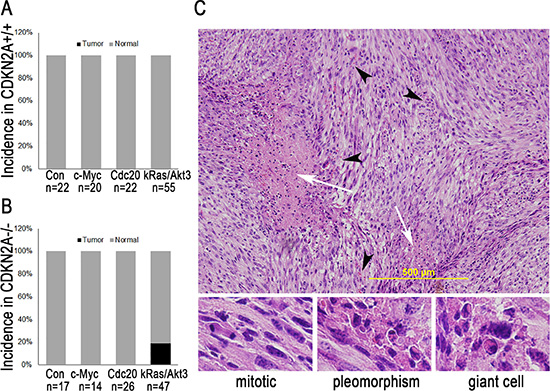
Figure 2: c-Myc or Cdc20 is insufficient to induce glioma, but kRas/Akt3 is sufficient to induce GBM in Ntv-a CDKN2A-null mice. (A) c-Myc or Cdc20 is insufficient to induce glioma in Ntv-a CDKN2A mice. (B) kRas/Akt3 is sufficient to induce GBM in Ntv-a CDKN2A-null mice. (C) H & E staining of one representative tumor from Ntv-a CDKN2A-null mice injected with kRas/Akt3 shows features of GBM, with palisading around necrotic foci (white arrow), glomeruloid vascular proliferation (black arrowhead) (up panel), and other features, such as mitotic cells, pleomorphism, and giant cells (low panel).
Combination of c-Myc and kRas/Akt3, but not Cdc20, enhances GBM development
To determine the role of c-Myc and Cdc20 in GBM development, we combined c-Myc or Cdc20 signaling with kRas/Akt3 oncogenic signals in the established mouse model. RCAS-c-Myc or RCAS-Cdc20, in combination with RCAS-kRas and RCAS-Akt3, were injected into CDKN2A-null Ntv-a mice. Nine of 19 mice (47.4%) with the c-Myc/kRas/Akt3 combination developed GBM, with microvascular proliferation and necrosis (Figure 3A). In contrast, only 2 of 21 mice with the Cdc20/kRas/Akt3 combination developed GBM (Figure 3A). Consistent with this, the overall survival duration was poor in mice injected with the c-Myc/kRas/Akt3 combination (Figure 3B). However, tumor-free survival did not significantly differ among the kRas/Akt3, kRas/Akt3/Cdc20, and kRas/Akt3/c-Myc mice (Figure 3C). A histopathologic examination of the brain tumors indicated that they were GBM with microvascular proliferation and necrosis (Figure 3D–3F).
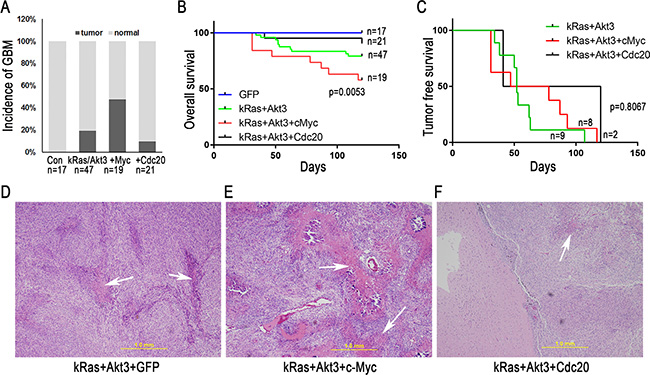
Figure 3: c-Myc promotes GBM development in CDKN2A-null mice with a combination of kRas and Akt3 signals. (A) GBM incidence was increased by c-Myc combined with kRas/Akt3 signals in Ntv-a CDNK2A-null mice. (B) Overall survival curve indicates the percentage of mice developing symptomatic tumors over time; the combination of c-Myc and kRas/Akt3 resulted in poor survival (p = 0.0053, log-rank test for trend). (C) Tumor-free survival curve indicates that mice with kRas/Akt3 and kRas/Akt3/c-Myc tumors had similar poor survival (p = 0.8067, log rank test for trend). (D–F) H & E staining of one representative tumor from each combination group with GBM features (microvascular proliferation and necrosis).
Effect of c-Myc and kRas/Akt3 on GBM development, with inhibition of glial progenitor cell differentiation
The experiments showed that c-Myc promotes GBM development when combined with kRas and Akt3 signals in CDKN2A-null Ntv-a transgenic mice. The mouse GBMs were marked by high cellularity, prominent vascularity, and necrosis that were identical to those seen in human GBM. To further characterize the mouse GBM, we performed immunohistochemical staining with antibodies specific for glial cell differentiation marker GFAP and cell proliferation marker Ki67 and found that c-Myc inhibited cell differentiation and promoted cell proliferation (Figure 4). To determine whether the inhibition of tumor cell differentiation is unique in c-Myc-induced GBM, we performed immunohistochemical staining with antibodies that were specific for glial cell differentiation markers containing GFAP and Nestin and found that the resultant tumor cells expressed both Nestin and GFAP in kRas/Akt3/c-Myc- and kRas/Akt3/Cdc20-induced GBM (Figure 5A). However, GFAP levels were dramatically decreased in the tumor cells from c-Myc/kRas/Akt3 combination mice. We also observed that GFAP expression was inhibited in c-Myc-transformed glial progenitor cells, while Cdc20 showed no effect in Cdc20-transformed cells (Figure 5B).
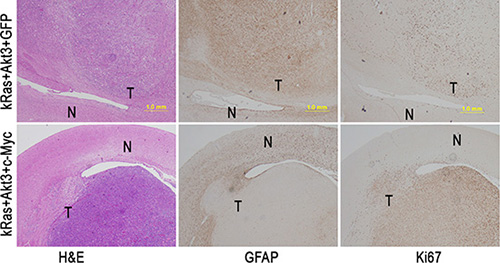
Figure 4: c-Myc promotes tumor cell proliferation with inhibition of cell differentiation. A representative immunohistochemical staining image was shown in the tumors from kRas/Akt3/c-Myc and kRas/Akt3/GFP control tumors with indicated markers for Ki67 and GFAP. N, normal cells; T, tumor cells; scale bar, 1.0 mm.
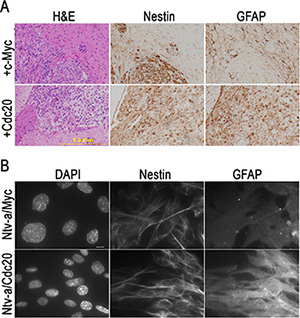
Figure 5: c-Myc inhibits glial progenitor cell differentiation in vivo and in vitro. (A) Immunohistochemical staining of two representative kRas/Akt3/c-Myc and Cdc20/kRas/Akt3 control tumors with indicated markers for Nestin and GFAP. (B) Immunofluorescence staining with GFAP and Nestin antibodies in c-Myc- or Cdc20-transformed glial progenitor cells. DAPI co-staining for genomic DNA, scale bar, 10 μm.
DISCUSSION
Glioma represents the most common type of primary brain tumor. GBM, the most advanced form of glioma, is the most invasive and resistant to therapy. Intensive research over the last three decades, especially recent research by TCGA, has revealed a large number of putative oncogenes and tumor suppressor genes that are altered in glioma, such as EGFR, PDGFRα, IGFBP2, c-Myc, Cdc20, and Akt3 oncogenes and CDKN2A, PTEN, and NF1 tumor suppressor genes. However, many of these signature genes are only markers in a subgroup of glioma cases; their causal role in gliomagenesis remains unexplored. In this study, we used the glia-specific RCAS/Ntv-a somatic gene transfer mouse model, which has been used extensively to test oncogenes, singly [27, 28] and in combination [29, 30], for their ability to induce gliomagenesis. We provided definitive evidence that c-Myc and Cdc20 alone are insufficient to induce glioma. Interestingly, glioma induction with the kRas and Akt3 combination required CDKN2A deletion. Moreover, the combination of the oncogenes c-Myc or Cdc20 in the context had distinct effects on the promotion and inhibition of GBM development.
We and others have shown that c-Myc and Cdc20 are both highly expressed in GBM samples compared to in normal control samples. To understand the oncogenic function of c-Myc and Cdc20, we first performed cell proliferation assays ex vivo using primary glial progenitor cells from Ntv-a mice. Overexpression of c-Myc and Cdc20 promoted glial progenitor cell proliferation. Moreover, the transduced glial progenitor cells with c-Myc or Cdc20 could be cultured over 6 months, whereas GFP control cells were arrested in growth within 1 month. However, the glial progenitor cells immortalized by c-Myc overexpression not only showed higher proliferation rates than did those immortalized by Cdc20 overexpression but also gained the ability to form anchorage-independent colonies, unlike Cdc20-transduced cells. This suggests that c-Myc has stronger oncogenic activity than does Cdc20 in promoting glial progenitor cell transformation.
To investigate the causal role of c-Myc and Cdc20 in glioma development, we induced c-Myc and Cdc20 expression in our RCAS/Ntv-a glia-specific mouse model. The data indicated that delivery of RCAS-c-Myc or RCAS-Cdc20 alone failed to induce glioma. Akt is among the most hyperactivated signaling pathways in human cancer and regulates key cellular functions, including cell growth, proliferation, angiogenesis, glucose metabolism, invasion, and survival. An analysis of human GBMs from TCGA revealed that the Akt signaling pathway is one of the top altered pathways in GBM [4, 5]. In addition, researchers have observed that Akt activation status is correlated with glioma grade [31]. The Akt family comprises three members (Akt1, Akt2, and Akt3) that share significant overall homological and kinase domain similarities but diverge in the linker region and the PH domain; they play distinct roles in development and tumor formation [23, 24, 32–35]. Akt1 is the most well characterized isoform of the three, but in recent years, it has become evident that Akt2 and Akt3 are more important than is Akt1 in glioma development and progression [23, 24, 36]. In this study, we used the well-characterized RCAS/Ntv-a glia-specific mouse model system to determine whether a combination of oncogenic signals induces GBM formation. First, we delivered RCAS-kRas and RCAS-Akt3 to Ntv-a mice; no glioma developed within the 12-week study period. CDNK2A deletion is common in human high-grade glioma, and we observed that additional CDKN2A loss in the mouse model was required for the induction of GBM by the combination of kRas and Akt3. However, the combination of kRas and Akt1 signals was sufficient to induce GBM in Ntv-a mice [29, 30]. These findings provide additional evidence that Akt1 and Akt3 play different roles in GBM development, dependent on their cellular location, interacting partners, and the context of other oncogenic signals.
Cancer development and progression involve the activation of multiple oncogenic signals. Identifying oncogenic drivers in glioma development and progression is a promising approach for targeted therapy. Oncogenic drivers can cause normal cells to become cancerous and promote the growth of tumors. They can be used to develop targeted therapies aimed at shutting down the signaling pathways. In this study, c-Myc not only induced primary glial progenitor cell proliferation, immortalization, and transformation but also promoted GBM formation (19.1% versus 47.7%) when combined with kRas/Akt3 signals in our glia-specific mouse model. Although Cdc20 induced primary glial progenitor cell proliferation and immortalization similar to c-Myc, it inhibited GBM formation (19.1% versus 9.1%) when combined with kRas/Akt3 signals. On the basis of these data, we conclude that c-Myc serves as an oncogenic driver and may be a promising target for GBM treatment; this is consistent with the results of a previous report [37]; however, Cdc20 is a passenger in GBM development and progression when combined with kRas/Akt3 oncogenic signals.
MATERIALS AND METHODS
RCAS constructs and RCAS propagation in DF1 cells
RCAS-c-Myc and RCAS-Cdc20 were constructed in our laboratory and have been validated by DNA sequencing and protein expression in DF1 cells. RCAS-kRas encodes full-length human mutant G12D-activated kRas. RCAS-Akt3 carries the full length of human Akt3 genes with the HA tag, kindly provided by Dr. Peter Vogt (The Scripps Research Institute La Jolla, CA 92037, USA). RCAS-GFP, containing full-length GFP, was used as an infection marker and was kindly provided by Yi Li (Baylor College of Medicine, Houston, TX). DF1 immortalized chicken fibroblasts were grown in DMEM with 10% FCS in a humidified incubator containing 10% CO2 at 39°C.
Primary glial progenitor cell culture
All animal experiments were performed in accordance with The University of Texas MD Anderson Cancer Center (Houston, TX) Institutional Animal Care and Use Committee guidelines. Six newborn Ntv-a transgenic mice were killed on day one; their whole brains were mechanically cut into small pieces, washed with 1 × PBS (pH 7.4), and digested with 3 ml of 0.25% trypsin/1 mM EDTA in HBSS in sterile tubes that were then incubated in a 37°C water bath for 15 min, with gentle shaking every 5 min. After incubation, fresh DMEM/F12 with 10% FCS was added to terminate digestion. The supernatant containing primary cells was pelleted, washed once with fresh medium, and plated into 10-cm dishes for primary culture.
Ex vivo infection of glial progenitor cells, cell proliferation, and soft agar assays
The supernatants containing various RCAS virus particles from DF-1 cell cultures were collected and filtered through 0.22-μm filters before being transferred into 60% confluent primary glial progenitor cells from Ntv-a newborn mice. The cells were infected once per day for 2 days. They were then seeded into three six-well plates overnight and incubated with 20 μM BrdU for 1 hour before being harvested, fixed with ethanol, stained with FITC-conjugated anti-BrdU antibody, and incubated with 7-AAD. Cell proliferation was measured by flow cytometry on the basis of BrdU-positive cells. For the immortalization assay, the transduced cells continued to pass for up to 6 months; 1 × 104 cells were then seeded into 24-well plates, and cell growth was measured by counting the cell number. To determine whether c-Myc or Cdc20 promotes colony formation in soft agar, the log phase cells were plated into soft agar plates, as described previously [38]. The plates were incubated for 2 weeks, and the colonies were counted under a microscope. The morphological features of the colony were captured using AxioVision3.1 microscopy.
Injection of Ntv-a transgenic mice with RCAS viral particles
Both Ntv-a CDNK2A+/+ and CDNK2A−/− mice express tv-a, a receptor of RCAS virus that is modulated by glia-specific Nestin promoter; this virus has been previously described [27]. The mice were of mixed genetic background, including C57BL, 129, Balb/C, and FVB/N. RCAS viral particles were injected into Ntv-a transgenic mice, as described by Marucci et al. [17]. In brief, newborn mice (postnatal days 1–3) were intracranially injected (in the right frontal region) with 1 × 104 DF1 cells producing RCAS virus alone or a combination using a 10-μl gas-tight Hamilton syringe. All mice were monitored daily and killed at the end of week 12, or earlier if they showed signs or symptoms of severe sickness or lethargy. The whole brains were collected and fixed in 4% formaldehyde in PBS for at least 24 h. Sections were analyzed by standard hematoxylin and eosin (H & E) staining.
Brain sectioning and immunohistochemistry
Each brain was coronally sectioned into five 5-μm-thick slices with a Leica microtome (Bannockburn, IL). Immunostaining was performed with LSAB2 kits (DAKO Cytomation, Carpinteria, CA). In brief, deparaffinized slides were treated with antigen-unmasking reagent (DAKO) by being heated in a microwave steamer for 16 min, and sections were blocked with 0.03% peroxidase for 10 min at ambient temperature. Antibodies against Nestin and GFAP were purchased from Abcom, Inc. (Cambridge, MA) and Millipore (Billerica, MA). Primary antibodies were diluted (1:500–1000) in TBST and incubated with sections at 4°C overnight. After being washed with TBST, sections were incubated with secondary antibodies (DAKO) at ambient temperature for 30 min. Sections were again washed with TBST and developed after the addition of DAB mixture (DAKO). After the reaction was terminated, sections were counterstained with freshly filtered hematoxylin and mounted.
ACKNOWLEDGMENTS AND FUNDING
We thank Ms. Ann M. Sutton in the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for editing this manuscript. This work was partially supported by NIH grants CA141432, CA09850305, and U24CA143835 to WZ, an MD Anderson core grant from the National Cancer Institute (CA16672), and grants for the Center for Cancer Systems Informatics from the National Foundation for Cancer Research to WZ. QL was supported by a grant from Tianjin Medical University Cancer Institute and Hospital.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
REFERENCES
1. Hofer S, Rushing E, Preusser M, Marosi C. Molecular biology of high-grade gliomas: what should the clinician know? Chin J Cancer. 2014; 33:4–7.
2. Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam do H, Mazurkiewicz M, Salacz M, Ashby L, Zagonel V, Depenni R, Perry JR, Hicking C, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015; 17:708–17.
3. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro Oncol. 2013; 15:4–27.
4. Cancer Genome Atlas Research N, Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–8.
5. Brennan C.W, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, et al. The somatic genomic landscape of glioblastoma. Cell. 2013; 155:462–77.
6. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006; 9:157–73.
7. Hofmann JW. Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, Hubbard GB, Ikeno Y, Zhang Y, Feng B, Li X, Serre T, Qi W, et al. Reduced Expression of MYC Increases Longevity and Enhances Healthspan. Cell. 2015; 160:477–88.
8. Dang CV. MYC on the path to cancer. Cell. 2012; 149:22–35.
9. Carroll PA, Diolaiti D, McFerrin L, Gu H, Djukovic D, Du J, Cheng PF, Anderson S, Ulrich M, Hurley JB, Raftery D, Ayer DE, Eisenman RN. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell. 2015; 27:271–85.
10. Conacci-Sorrell M, McFerrin L, Eisenman R.N, An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014; 4:a014357.
11. Wasylishen AR, Penn LZ. Myc: the beauty and the beast. Genes Cancer. 2010; 1:532–41. doi: 10.1177/1947601910378024.
12. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, Perez-Atayde A, Wong KK, Yuan GC, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014; 159:1126–39.
13. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, Stommel JM, Dunn KL, Wiedemeyer R, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008; 455:1129–33.
14. Yu H, Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell. 2007; 27:3–16.
15. Amador V, Ge S, Santamaría PG, Guardavaccaro D, Pagano M. APC/C (Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol Cell. 2007; 27:462–73.
16. Ji P, Smith SM, Wang Y, Jiang R, Song SW, Li B, Sawaya R, Bruner JM, Kuang J, Yu H, Fuller GN, Zhang W. Inhibition of gliomagenesis and attenuation of mitotic transition by MIIP. Oncogene. 2010; 29:3501–8.
17. Marucci G, Morandi L, Magrini E, Farnedi A, Franceschi E, Miglio R, Calò D, Pession A, Foschini MP, Eusebi V. Gene expression profiling in glioblastoma and immunohistochemical evaluation of IGFBP-2 and CDC20. Virchows Arch. 2008; 453:599–609.
18. Wang L, Zhang J, Wan L, Zhou X, Wang Z, Wei W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol Ther. 2015; 151:141–51.
19. Xie Q, Wu Q, Mack SC, Yang K, Kim L, Hubert CG, Flavahan WA, Chu C, Bao S, Rich JN. CDC20 maintains tumor initiating cells. Oncotarget. 2015; 6:13241–54. doi: 10.18632/oncotarget.3676.
20. Zhang L, Niu T, Huang Y, Zhu H, Zhong W, Lin J, Zhang Y. Compound 331 selectively induces glioma cell death by upregulating miR-494 and downregulating CDC20. Sci Rep. 2015; 5:12003.
21. Kidokoro T, Tanikawa C, Furukawa Y, Katagiri T, Nakamura Y, Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene. 2008; 27:1562–71.
22. Kim S, Dougherty ER, Shmulevich I, Hess KR, Hamilton SR, Trent JM, Fuller GN, Zhang W. Identification of combination gene sets for glioma classification. Mol Cancer Ther. 2002; 1:1229–36.
23. Mure H, Matsuzaki K, Kitazato KT, Mizobuchi Y, Kuwayama K, Kageji T, Nagahiro S. Akt2 and Akt3 play a pivotal role in malignant gliomas. Neuro Oncol. 2010; 12:221–32.
24. Turner KM, Sun Y, Ji P, Granberg KJ, Bernard B, Hu L, Cogdell DE, Zhou X, Yli-Harja O, Nykter M, Shmulevich I, Yung WK, Fuller GN, et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc Natl Acad Sci U S A. 2015; 112:3421–6.
25. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998; 90:1473–9.
26. Acquaviva J, Jun HJ, Lessard J, Ruiz R, Zhu H, Donovan M, Woolfenden S, Boskovitz A, Raval A, Bronson RT, Pfannl R, Whittaker CA, Housman DE. Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation. Cancer Res. 2011; 71:7198–206.
27. Dunlap SM, Celestino J, Wang H, Jiang R, Holland EC, Fuller GN, Zhang W. Insulin-like growth factor binding protein 2 promotes glioma development and progression. Proc Natl Acad Sci U S A. 2007; 104:11736–41.
28. Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014; 26:288–300.
29. Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009; 15:110–6.
30. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000; 25:55–7.
31. Testa JR, Bellacosa A, AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001; 98:10983–5.
32. Peres J, Mowla S, Prince S. The T-box transcription factor, TBX3, is a key substrate of AKT3 in melanomagenesis. Oncotarget. 2015; 6:1821–33. doi: 10.18632/oncotarget.2782.
33. Ji P, Turner KM, Zhang W, OverAKT3: tumor progression and chemoresistance. Cell Cycle. 2015; 14:1993–4.
34. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007; 25:917–31.
35. Hu B, Emdad L, Bacolod MD, Kegelman TP, Shen XN, Alzubi MA, Das SK, Sarkar D, Fisher PB. Astrocyte elevated gene-1 (AEG-1) interacts with Akt isoform 2 to control glioma growth, survival and pathogenesis. Cancer Res. 2014; 74:7321–32.
36. Phung TL, Du W, Xue Q, Ayyaswamy S, Gerald D, Antonello Z, Nhek S, Perruzzi CA, Acevedo I, Ramanna-Valmiki R, Rodriguez-Waitkus P, Enayati L, Hochman ML, et al. Akt1 and akt3 exert opposing roles in the regulation of vascular tumor growth. Cancer Res. 2015; 75:40–50.
37. Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I, Redondo-Campos S, Folch G, Gonzàlez-Juncà A, Sodir NM, Massó-Vallés D, Beaulieu ME, Swigart LB, et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun. 2014; 5:4632.
38. Parke BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, Li X, Gumin J, Zheng H, Hu L, Yli-Harja O, Haapasalo H, Visakorpi T, et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Invest. 2013l; 123:855–65.