
Introduction
The BCR-ABL1 oncogene results from the t(9;22)(q34;q11) reciprocal translocation generating the Philadelphia chromosome (Ph). BCR-ABL1+ CML is a stem cell-derived disease that progresses in three distinct phases: chronic phase (CP), which may last for several years, accelerated phase (AP), and finally blast crisis (BC) that is refractory to therapy [1]. The tyrosine kinase inhibitor (TKI) imatinib revolutionized the treatment of BCR-ABL1+ leukaemia being the first drug successfully targeting an oncogenic tyrosine kinase [2]. Imatinib paved the way for this group of signal interceptors and the TKIs imatinib, nilotinib and dasatinib are now used as first-line therapy in CP-CML patients [3-5]. Although these substances are highly effective in eradicating the vast majority of peripheral CML cells, residual BCR-ABL1+ progenitors persist in CML patients despite undetectable molecular disease and may only be eliminated after continuous treatment for several years [6-8]. In addition, some patients fail to respond to BCR-ABL1 TKIs (primary resistance) or relapse after an initial promising response (secondary resistance). Secondary resistance is most frequently caused by point mutations in the BCR-ABL1 kinase domain that render the kinase insensitive to TKI treatment [9, 10]. Imatinib resistance frequently represents the first signal that the disease progresses into a more advanced stage to accelerated phase and finally terminal blast crisis associated with short median survival times [11].
Genomic instability is one of the hallmarks of a transformed cell and provides the basis for the development of BCR-ABL1 mutations under the selection pressure of TKI therapy [12]. Genomic instability may result from increased DNA damage as well as an aberrant DNA repair machinery [13]. DNA damage may be caused by external triggers including UV-light, radiation, and certain chemicals. The production of endogenous reactive oxygen species (ROS) – a cell intrinsic process - is another well described inducer of DNA damage. Several studies have defined ROS - especially hydroxyl radicals (HO*) and lipid peroxidation products - as major sources for Amutations in various forms of cancer [14, 15]. The potential of ROS to induce spontaneous DNA double-strand breaks (DSBs) combined with impaired DNA repair responses, make ROS a potent trigger/inducer of chromosomal aberrations [16-19].
BCR-ABL1 activity has convincingly been linked to ROS production [20]. BCR-ABL1 induced ROS production combined with impaired DNA damage responses (mainly via RAD51 de-regulation) is thought to promote genomic instability and finally self-mutagenesis; a process which has the potential to provoke TKI resistance [11, 17, 18].
Aside from BCR-ABL1 expression, the pronounced activation of the transcription factor STAT5 is considered a signalling hallmark in CML cells [21-23]. STAT5 signalling is crucial for transformation by BCR-ABL1 and thus represents a bottle neck for disease induction. Importantly – as critical for therapeutic intervention and defining STAT5 as drug target - STAT5 is also essential for leukaemia maintenance [24-29]. The importance of STAT5 in CML pathogenesis is further underscored by the fact that STAT5 expression increases during disease progression and that high STAT5 levels significantly decrease imatinib sensitivity [30, 31].
In this study we investigated the potential connection between the transcription factor STAT5 and the occurrence of BCR-ABL1 mutations. We describe the highly significant correlation between STAT5 expression levels and the frequency of BCR-ABL1 mutations in human CML patients. We propose a model where STAT5 triggers ROS production and thereby induces DNA damage. The concomitant STAT5 dependent up-regulation of anti-apoptotic genes enables the cells to survive and allows for the development of BCR-ABL1 mutations thereby explaining the increased occurrence of inhibitor-resistance in advanced disease stages.
Results
Elevated STAT5A mRNA levels correlate with BCR-ABL1 mutation rates
We have recently shown that STAT5 expression levels increase during CML progression and that elevated activated STAT5 levels account for a reduced responsiveness to BCR-ABL1 tyrosine kinase inhibitor (TKI) therapy [30]. The STAT5 mediated TKI resistance was independent of BCR-ABL1 mutations or BCR-ABL1 expression levels but critically dependent on an intact STAT5 signalling. The initiative for our current study came from an experiment where p160v-ABL transformed murine cells infected with a retrovirus encoding for Stat5a-IRES-GFP (STAT5A) displayed enhanced colony formation over an empty vector (GFP) control when plated in methylcellulose containing 1 µM imatinib. Three weeks thereafter, clones were visible in the dishes containing cells ectopically expressing STAT5A (Supporting Information Fig S1A) whereas hardly any colony was visible in the control cultures. Similar observations were made with Ba/F3p210BCR-ABL1 cells that were seeded in 96 well plates in medium enriched with 2 µM imatinib. Ba/F3p210BCR-ABL1 cells ectopically expressing STAT5A gave rise to more imatinib-resistant clones compared to control cells (Supporting information Fig S1B). Hence, even under conditions where we continuously blocked BCR-ABL1 activity using high imatinib concentrations, STAT5 expression conferred an advantage and allowed some cells to escape BCR-ABL1 TKI therapy. One frequent cause for imatinib resistance is the occurrence of mutations within the BCR-ABL1 oncogene. To test a potential correlation between STAT5 expression levels and the frequency of BCR-ABL1 mutations we decided to analyse leukemic samples from a cohort of 50 CML patients including our initial patient sample collection [30] (For patient characteristics see Table 1).
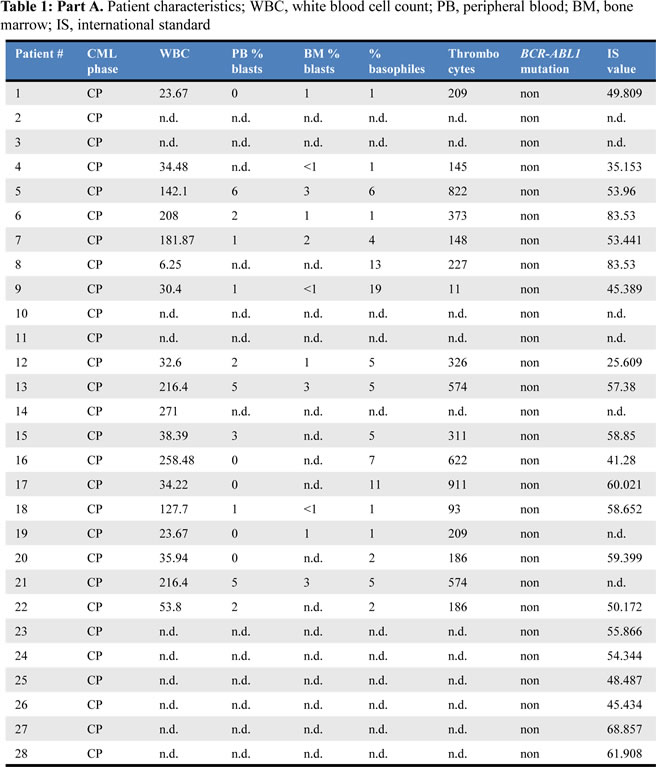
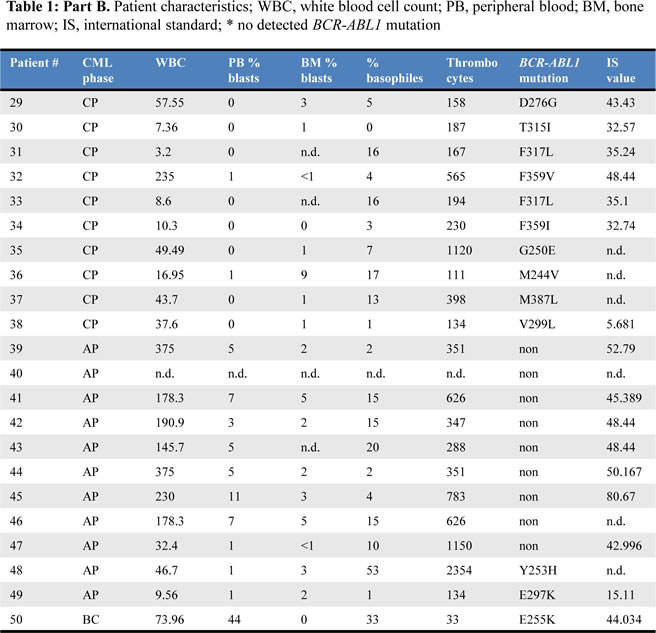
We confirmed our initial observation that STAT5A mRNA expression levels increase during disease progression from CP to AP (p < 0.0001, Fig 1A). Remarkably, the correlation between STAT5A expression and BCR-ABL1 mutations was of high significance (p < 0.0001 and p = 0.0054 for CP and AP, respectively). Patients harboring a mutated form of BCR-ABL1 displayed elevated levels of STAT5A compared to TKI sensitive patients. This correlation was observed in both groups analyzed, in CP- as well as AP-CML patients (Fig 1B and 1C). The distribution of the mutants appears random and not restricted to a distinct set of mutations (Fig 1D).
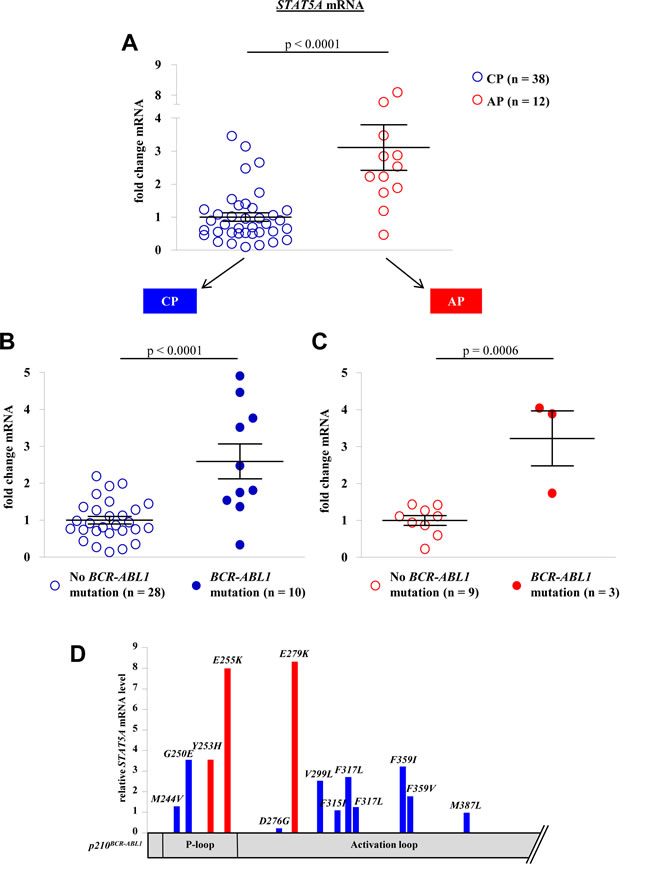
Figure 1: BCR-ABL1 mutations correlate with STAT5A expression levels in primary CML patient samples. (A) STAT5A mRNA levels of CML-CP patients (n = 38) versus CML-AP patients (n = 12). (B, C) STAT5A expression levels of CML-CP (B) and CML-AP (C) patients expressing wild type BCR-ABL1 versus mutated BCR-ABL1. (D) Scheme depicting BCR-ABL1 amino acid substitutions as well as the relative location on the BCR-ABL1 gene. The height of each bar represents the corresponding relative STAT5A mRNA level.
Enforced STAT5 expression leads to increased levels of ROS
The correlation between STAT5 expression levels and the frequency of BCR-ABL1 mutations suggests a causal relationship, which can be tested by exogenous retroviral add back of Stat5a. Several mechanisms may link STAT5 to DNA mutations including an increased expression of the PAX5 regulated B-cell mutator protein AID [32], an increased expression of RAD51 DNA-repair proteins [33, 34] or a STAT5 triggered production of ROS [35, 36].
As we failed to detect any changes in PAX5, AID and RAD51 expression in BCR-ABL1+ cells upon enforced STAT5 expression we focused our attention on ROS levels. When we used the ROS sensitive fluorescent dye DHE to stain p185BCR-ABL1+ cells we obtained a clear and consistent picture: the enforced expression of STAT5A - but not the control vector - provoked a significant up-regulation of endogenous ROS levels in 4/4 tested cell lines (Fig 2A and 2B). Comparable results were obtained in human K562 cells upon enforced STAT5A expression (Fig 2C). In line with our findings shRNA mediated down-regulation of STAT5A/B reduced ROS production in K562 cells (Fig 2D). In summary these experiments provided evidence, that STAT5 expression correlates with high ROS levels in BCR-ABL1+ cells.
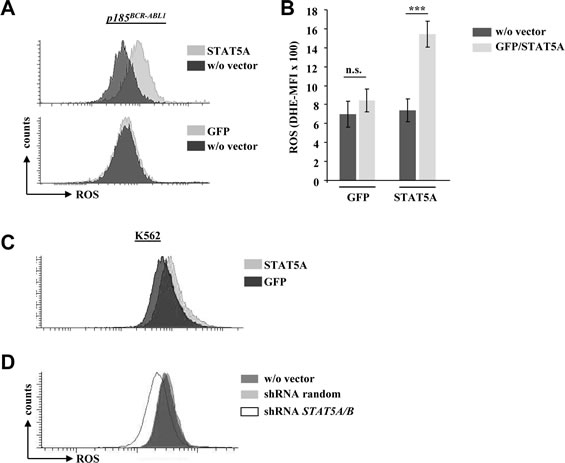
Figure 2: STAT5A mediates ROS production in murine and human BCR-ABL1 transformed cell lines. (A) Representative FACS histogram of p185BCR-ABL1 transformed murine cells infected with a retrovirus encoding for Stat5a-IRES-GFP (upper histogram) or a control vector (GFP, lower histogram). Cells have been stained with the ROS sensitive fluorescent dye DHE to measure differences in ROS levels upon enforced expression of STAT5A. (B) Statistical analysis showing DHE-MFI of individually derived p185BCR-ABL1+ cell lines (n = 4, data are mean ± SD., n.s. = not significant, *** p < 0.001). (C) FACS histogram depicting ROS level of the human CML cell line K562 upon enforced expression of STAT5A or GFP. (D) K562 cells expressing a random shRNA or a shRNA specific for STAT5A/B were stained with DHE to measure differences in ROS levels.
STAT5 induced ROS production accompanies enhanced DNA double-strand breaks
ROS is known to induce DNA damage including double-strand brakes (DSBs). Upon DSBs the kinases ATM and/or ATR phosphorylate serine 139 on histone H2A-X (γH2A-X) which quantifies DSBs. We performed intracellular anti-γH2Ax FACS-based staining to test the effects of enforced STAT5 signaling on DNA damage. Indeed, the increased STAT5A mediated ROS levels in p160v-ABL+ cells (Fig 3A) were accompanied by elevated levels of DSBs (Fig 3B). Comparable results were obtained in Ba/F3 cells transformed by p185BCR-ABL1. To investigate the potential causal link of STAT5 mediated ROS production and increased DNA-DSB frequency, we used the ROS scavenger N-acetyl-cystein (NAC). Ba/F3p185BCR-ABL1 cells ectopically expressing STAT5A showed an elevated level of ROS compared to control cells that was reduced upon NAC treatment (Fig 3C). In line with this, NAC-mediated decrease in ROS was accompanied by decreased γH2A-X level (Fig 3D).
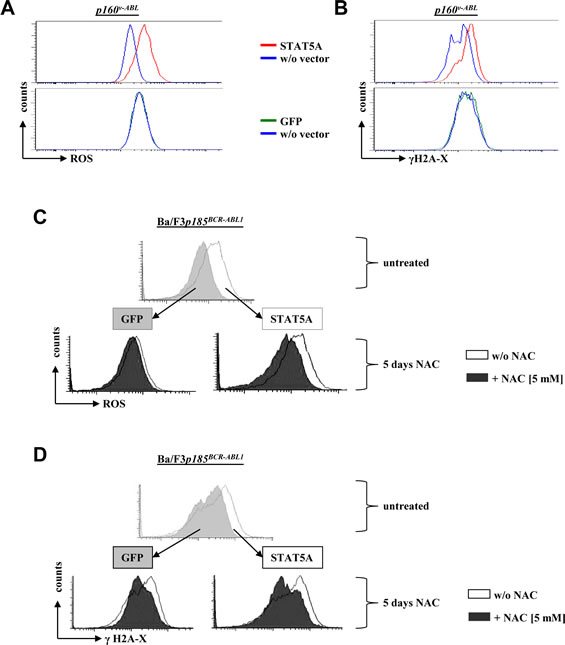
Figure 3: Enforced STAT5A expression induces DNA double-strand breaks. (A) FACS histogram depicting ROS levels of a p160v-ABL transformed murine cell ectopically expressing STAT5A or GFP. (B) Cells were stained with an intracellular FACS-antibody against γH2A-X to measure differences in DNA double-strand breaks. (C, D) Ba/F3p185BCR-ABL1 cells expressing STAT5A or a control vector (GFP) were treated for five days with the ROS scavenger NAC. Depicted are FACS histograms for (C) ROS and (D) γH2A-X of untreated cells (top of each panel, GFP versus STAT5A expressing cells) and cells treated with NAC compared to controls (bottom of each panel).
STAT5B is a transcriptional regulator of STAT5A
The Stat5 gene locus encodes for two distinct but closely related STAT5 isoforms; STAT5A and STAT5B have 97% amino acid homology and fulfill largely redundant and overlapping functions. Differences are present within the extreme C-terminus that account for distinct functions within some cell types. We asked whether the ability to induce ROS production is restricted to STAT5A. To answer this question we generated p160v-ABL transformed murine cell lines and infected them with a retrovirus encoding for STAT5B. RT-PCRs for Stat5a and Stat5b mRNA were performed to analyze the differences in the expression levels of the distinct Stat5 isoforms. To our surprise, the enforced expression of Stat5b also increased the levels of endogenous Stat5a in 2/3 analyzed cell lines (Fig 4A). The vice versa effect was not detected and enforced expression of Stat5a had no impact on expression levels of endogenous Stat5b mRNA (Fig 4B). Enhanced STAT5A protein levels were also detected in several cell lines transduced with a Stat5b retrovirus. However, the protein expression levels varied when cell lines were compared on different time points and culture conditions. Representative immunoblots of three distinct cell lines are shown in Fig 4C. Mouse embryonic fibroblasts that lack the Stat5a/b gene locus ectopically expressing STAT5A, STAT5B or GFP served as control. Based on these data we analysed a potential transcriptional regulation of Stat5a by STAT5B via ChIP. Three conserved candidate sites (Stat5_1 to Stat5_3) in the promoter region of STAT5A/B were chosen based on the consensus sequence of STAT5 (TTCN3GAA) (Supporting Information Fig S2A and S2B). Stat5_1 contains two adjacent conserved sites of which the second one (Stat5_1.2) showed prominent binding in ChIP experiments and this site was used for further analysis. A p160v-ABL+ cell line harboring low endogenous STAT5A/B levels ectopically expressing STAT5A or STAT5B was used for ChIP analysis. RT-PCR primers for Cis were used as positive control (Fig 4D). For primers amplifying region Stat5_1.2 we could detect a clear enrichment over Chr1 (negative control) for both cell lines analyzed (Fig 4E). These ChIP data support our initial findings and indicate a binding of STAT5 in its own promoter region.
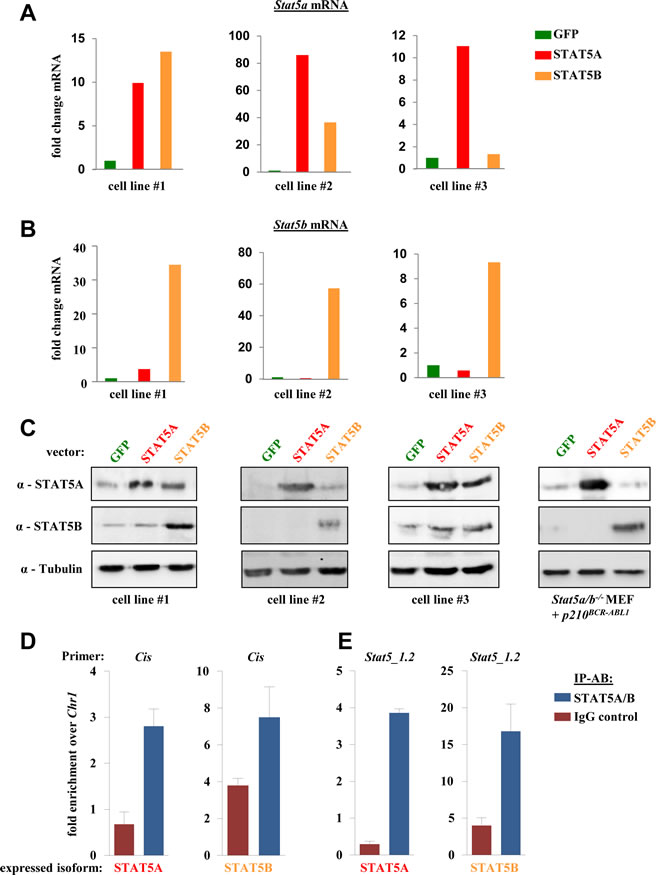
Figure 4: STAT5B acts as a transcriptional inducer of STAT5A. (A) Stat5a and (B) Stat5b mRNA levels of three individually derived p160v-ABL transformed murine cell lines ectopically expressing GFP, STAT5A or STAT5B. The fold change in mRNA levels upon enforced STAT5 expression compared to GFP expressing cells is depicted. (C) Immunoblot for STAT5A and STAT5B of three distinct p160v-ABL+ cell lines expressing indicated vectors. Stat5a/b-/- mouse embryonic fibroblasts ectopically expressing GFP, STAT5A or STAT5 were used as antibody control (right blot). (E, F) ChIP assays were performed using p160v-ABL transformed cell lines ectopically expressing STAT5A or STAT5B. Protein-DNA complexes were immunoprecipitated using antibodies directed against STAT5A/B or IgG (negative control) and analysed by RT-PCR for the presence of (E) Cis (positive control) and (F) the Stat5a promoter region Stat5_1.2. Bar graphs depict fold enrichment over Chr1. Experiments have been performed in triplicates.
STAT5A and STAT5B increase ROS levels in vivo
As depicted in Fig 5A and 5B both isoforms – STAT5A and STAT5B - were capable to enhance ROS production in Abelson transformed cells which was again accompanied by elevated levels of γH2A-X (Supporting Information Fig S3A). Based on our data presented above indicating a transcriptional regulation of Stat5a by STAT5B we cannot completely exclude that the mechanism of STAT5B to induce ROS production is partly through up-regulation of endogenous STAT5A.
One may argue that the enhanced ROS production lacks disease relevance as it may be antagonized in vivo by the microenvironment. To test this hypothesis we intravenously injected p160vABL transformed murine cells ectopically expressing STAT5A or STAT5B as well as control cells into recipient NSG mice. 12 days after injection the mice were sacrificed, spleens isolated, and single cell suspensions were immediately stained with DHE (Fig 5C). The spleens of diseased mice showed infiltration rates of GFP+ leukemic cells in the range of 50 to 60% indicating a successful engraftment (Fig 5D). Although this procedure is considerable more stressful for cells compared to an in vitro ROS analysis, a reproducible and significant increase in ROS levels upon expression of STAT5A or STAT5B was observed (Fig 5E, 5F and Supporting Information Fig S3B).
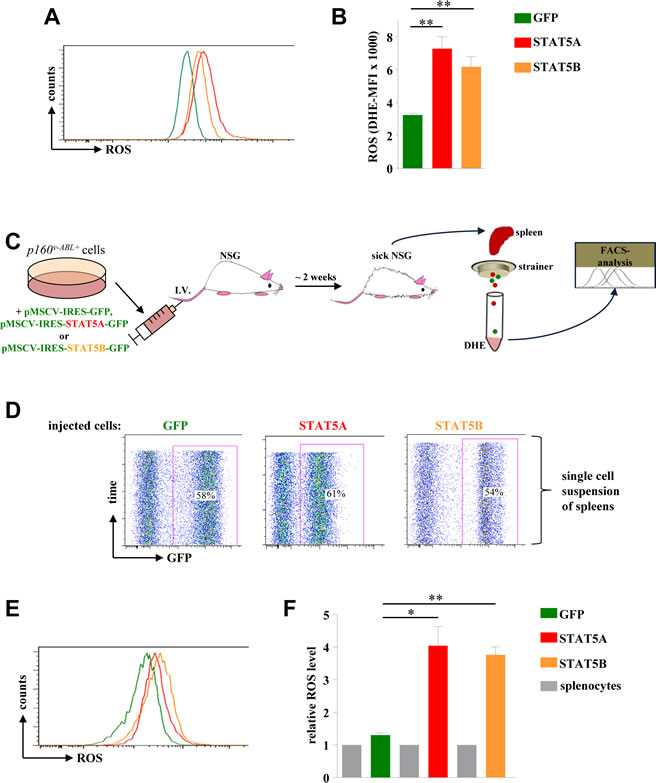
Figure 5: STAT5A and STAT5B expression mediates ROS production in a leukaemia mouse model. (A) p160v-ABL transformed murine cells were infected with a retrovirus encoding for GFP, STAT5A or STAT5B and subsequently used to measure ROS levels via DHE staining. (B) Statistical analysis showing DHE-MFI of individually derived p160v-ABL+ cell lines (n = 3). (C) Scheme of experimental procedure used to analyse ROS production of Abelson transformed cells in an in vivo mouse model for leukaemia. (D) FACS blots of single cell suspensions derived from the spleens of diseased mice. The percentage of leukemic cells infiltrating the spleen is shown within the blot. (E) FACS histogram of ex vivo derived leukemic cells expressing GFP, STAT5A or STAT5B stained with DHE to analyse differences in ROS levels. (F) Relative ROS level of ex vivo derived leukemic cells. The fold change in ROS (DHE-MFI) compared to GFP-negative splenocytes is depicted (n = 3). (B and F) Bar graphs represent mean ± SD. * p < 0.05, ** p < 0.01.
STAT5 mediated ROS production is independent of iron metabolism, Catalase, MnSOD and the mitochondrial respiratory chain
Intracellular ROS levels are maintained within a tight equilibrium and are subject to several layers of regulation including internal and external factors. Iron represents one prominent factor that interferes with ROS levels and ROS staining. Iron is present in culture media or it is an intracellular intermediate product of heme metabolism. As STAT5 has been reported to interfere with iron metabolism in erythroid cells through CD71 and IRP-2 gene regulation [37, 38] we used the iron chelators desferal (DFO) and diethylene triamine pentaacetic acid (DTPA) to test whether differences in iron metabolism or iron level account for STAT5 induced changes in ROS production. As depicted in Supporting Information Fig S4A we failed to detect any changes in ROS production upon iron chelator pre-treatment.
One way how cells control their ROS levels is by tightly regulating the expression of endogenous ROS detoxifying enzymes and scavengers. Two prominent ROS scavengers are Catalase and MnSOD, both regulated by FOXO3a [39, 40]. Expression of neither of these genes was significantly changed upon enforced expression of STAT5A or STAT5B (Supporting Information Fig S4B and S4C). The slight increase of Catalase and MnSOD mRNA may rather be indicative for an endogenous negative feedback loop caused by elevated ROS levels. It was also shown that inhibition of the mitochondrial respiratory chain (MRC) by rotenone significantly decreased intracellular ROS levels produced by BCR-ABL1 [20]. Thus, the MRC is an important source for ROS production in BCR-ABL1 transformed cells and prompted us to study the role of mitochondria as source of STAT5 mediated ROS production. To avoid off-target effects of inhibitors of the MRC we made use of electro spin resonance spectroscopy (ESR) analysis in combination with cyclic hydroxyl amines (CMH) and iron chelators. This method detects particularly superoxide radicals (O2•−) rather than other ROS members like hydroxyl radicals or hydrogen peroxide. No significant differences in the level of O2•− were measured upon retroviral expression of STAT5A or STAT5B (Supporting Information Fig S4D). However, if MRC was inhibited by antimycin A (maximizing O2•− from MRC by complex III inhibition) all cell lines displayed significantly increased levels of O2•−. This observation and the lacking differences under basal conditions suggest that the MRC is not a major source of O2•− upon overexpression of STAT5A or STAT5B. The exclusion of mitochondrial O2•− points to alternative sources for ROS production than the MRC. The discrepancy between the results obtained with the ESR/CMH method compared to the DHE-based analysis (total ROS) under basal conditions reflects different sensitivity and selectivity (ESR/CMH: O2•−, DHE: O2•−, H2O2, HO•) and provides evidence that STAT5A/B acts preferably on the downstream ROS species (H2O2, HO•) or their detoxification.
STAT5 mediated ROS production is JAK2 independent but depends on BCR-ABL1 signalling and the N-terminus of STAT5
Although STAT5 is a classical transcription factor recent evidence revealed a cytoplasmic function for STAT5 in transformed cells. Moreover, we have recently shown that particularly the C-terminal serine sites of STAT5A are needed for efficient myeloid transformation and that both C-terminal serine sites in STAT5A were prominently phosphorylated in CML cells [41]. To gain further insights how STAT5 regulates ROS production, we expressed distinct STAT5A variants in p160v-ABL transformed cells. Besides a GFP-control vector and wild type STAT5A, we expressed a STAT5A variant lacking the serine phosphorylation sites on position 725 and 779 (SASA)[41], an N-terminally truncated STAT5A devoid of the first 136 amino acids (ΔN)[42], a variant not efficiently capable to bind to DNA, but still able to translocate into the nucleus (EE/AA)[43], and a mutant lacking the essential tyrosine phosphorylation site on position 694 (Y/F) [43], devoid of dimerization, efficient nuclear translocation and incapable of DNA binding to STAT5 response elements (for scheme see Supporting Information Fig S5A). STAT5EE/AA and STAT5Y/F failed to increase the level of ROS indicating that the transcriptional activity, dimerization and DNA binding is required for elevating ROS levels. Remarkably, also the STAT5ΔN variant failed to increase ROS levels (Fig 6A). Oligomerization through the STAT5 N-terminus is essential for the transforming potential of constitutively active STAT5 and it was shown to be required for transcription of survival genes or cell cycle regulators [42, 44]. This led us to conclude that an N-terminally regulated target gene might be the trigger for enhanced ROS production.
In hematopoietic non-transformed cells JAK2 is the dominant upstream kinase of STAT5. Although JAK2 is not essential for disease maintenance in CML [45], it still has the potency to phosphorylate STAT5 and is part of a complex with the BCR-ABL1 network [46, 47]. To study the impact of JAK2 for STAT5 mediated ROS production we treated two individual p160v-ABL transformed cell lines expressing GFP, STAT5A or STAT5B with 1 µM of the highly potent and specific JAK1/2 inhibitor INCB-018424 [48] for 24 hours. DHE FACS analysis revealed no impact of JAK2 on cellular ROS levels irrespective whether STAT5 was ectopically expressed or not (Fig 6B).
We asked whether the STAT5 mediated ROS production depends on the presence of the BCR-ABL1 oncogene. To address this question we made use of parental Ba/F3 and 32D cells growing in IL-3 enriched medium. IL-3 is a potent activator of the JAK2-STAT5 pathway in these cells (Supporting Information Fig S5B-S5D and [45]) thereby giving us the opportunity to compare BCR-ABL1-dependent versus –independent STAT5 mediated ROS production side by side. As expected, Ba/F3p210BCR-ABL1 and 32Dp185BCR-ABL1 cells reacted with elevated ROS levels to the enforced expression of STAT5A or STAT5B (Fig 6C, upper panel). Remarkably, the parental Ba/F3 and 32D cells devoid of the BCR-ABL1 oncogene showed no signs of elevated ROS levels despite enforced STAT5 expression and activation by JAK2 in IL-3 supplemented growth medium (Fig 6C, lower panel). This led us to conclude that oncogenic BCR-ABL1 signalling acts in concerted action with STAT5 to trigger ROS production and to turn STAT5 into a harmful ROS-triggering DNA mutator.
As BCR-ABL1 and STAT5 are both required to increase ROS we analysed the relation between BCR-ABL1 levels (IS value) and BCR-ABL1 mutations. To our surprise, we failed to observe a positive correlation. We even detected a slightly negative correlation between IS values and mutation rate (Fig 6D-F) underscoring the importance of STAT5 as mediator of ROS production.
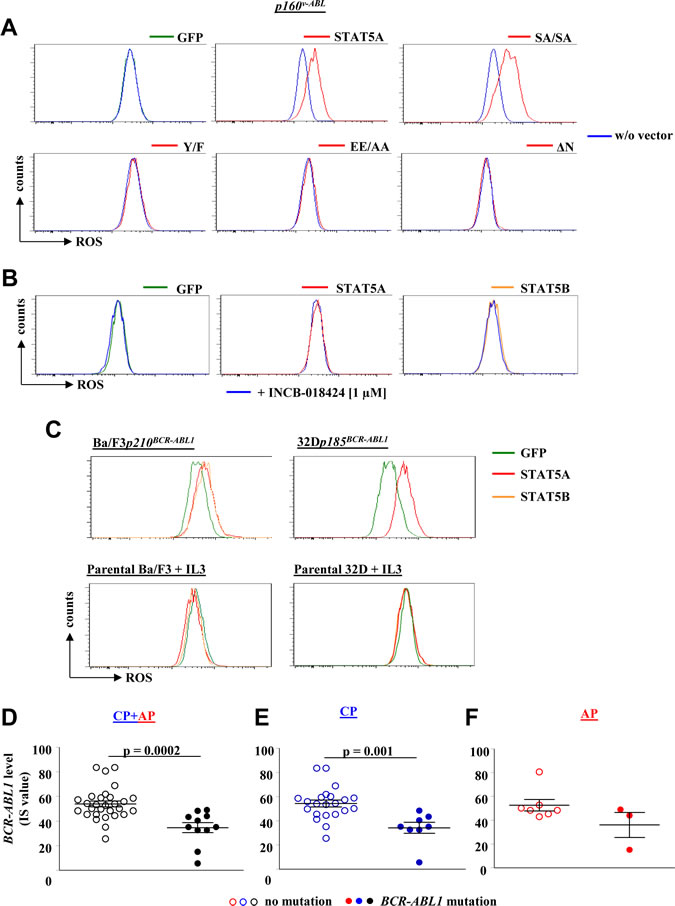
Figure 6: STAT5 mediated ROS production depends on its N-terminal domain and on BCR-ABL signalling but is independent of JAK2. (A) A p160v-ABL+ cell line ectopically expressing GFP, wild type STAT5A or one of the indicated STAT5A mutants was used to detect differences in ROS level. The experiment has been performed in triplicates. A representative FACS histogram of each cell line is depicted. (B) Two individually derived Abelson transformed cell lines ectopically expressing GFP, STAT5A or STAT5B were treated with the JAK1/2 inhibitor INCB-018424 for 24 hours. Representative histograms of one cell line for each expressed vector treated with INCB-018424 compared to untreated counterparts are depicted. (C) FACS histograms of DHE stained Ba/F3 and 32D cells transformed with p210BCR-ABL1 and p185BCR-ABL1, respectively (upper panel). The lower panel depicts the ROS level of untransformed parental Ba/F3 and 32D cells growing in IL-3 supplemented medium and ectopically expressing GFP, STAT5A or STAT5B. (D – F) Relative BCR-ABL1 levels (IS value) of CML patients harbouring wild type BCR-ABL1 or mutated BCR-ABL1. Shown are patients in (D) CP and AP (n = 39), (E) only CP (n = 29) and (F) only AP (n = 10).
In summary we propose the following model for CML disease progression: In the presence of BCR-ABL1 the increase in STAT5 expression leads to an increased probability to acquire a ROS mediated mutation rendering the BCR-ABL1 kinase less responsive to TKIs, a well-documented phenomenon of CML progression [49]. The increased mutation rate may provoke apoptosis induction which is effectively counteracted by STAT5 itself as the anti-apoptotic BCL2 family members BCL2, BCLXL, or MCL1 are STAT5 target genes as documented by many studies [38, 50-53]. Furthermore STAT5 represses miRNA15/16 that counteracts BCL-2 and BCLXL [44]. Therefore, we propose a dual role for STAT5; while increasing the rate of DSBs and therefore BCR-ABL1 mutation frequency it provides the molecular link to survival gene upregulation to accept elevated DNA damage rate in a subset of leukemic cells with high STAT5 activity (Supporting Information Fig S6A and S6B).
Discussion
The introduction of imatinib has revolutionized the treatment of CML which has once been a deadly disease before the laboratory of Brian Druker pioneered TKI development. However, the occurrence of BCR-ABL1 TKI resistance limits this success story and additional therapeutic intervention strategies are required. It is noteworthy, that the probability to acquire a BCR-ABL1 mutation rendering the cells unresponsive to TKIs increases during disease progression from the CP to the more advanced disease stages AP and BCs [49, 54, 55]. In the present study we propose a model how elevated STAT5 expression levels contribute to the enhanced BCR-ABL1 mutation rates observed in advanced disease phases. The expansion of our cohort of CML patients confirmed our initial finding that STAT5A expression increases during disease progression. It furthermore revealed a highly significant correlation between STAT5A mRNA levels and the occurrence of BCR-ABL1 mutations. As high STAT5 levels in BCR-ABL1+ cells are accompanied by elevated endogenous ROS levels and ROS is a well-known factor responsible for DNA damage and subsequent mutation, we propose a causal link between STAT5 expression, ROS levels and BCR-ABL1 mutations. The continuous selection pressure subsequently exerted by TKI therapy might select for mutations conferring TKI resistance under a situation where enhanced STAT5 levels and activity promotes survival in a subset of CML clones.
ROS are generated by different sources including NADPH oxidases [56],[57], mitochondria [58], xanthine oxidase [59], as well as endothelial nitric oxide synthase under specific conditions [60]. ROS primarily serves as mediator of important processes including differentiation, host defence [61], oxygen sensing [62], proliferation, apoptosis, and response to mechanical strain [63]. ROS is considered an important signalling mediator [64] and may interfere with gene regulatory pathways such as mitogen-activated protein kinase [65, 66] or hypoxia-responsive-element via hypoxia-inducible factor [67, 68], also known to be regulated by STAT5 transcription factors [69]. Deregulation of ROS metabolism has been implicated in several patho-physiological conditions and has been associated with inflammation, vascular atherosclerosis [70], diabetes [71], hypertension [72], and tumourigenesis including CML [73].
Recent evidence pinpointed at a key role for the Rac2 GTPase in BCR-ABL1 driven ROS production. Rac GTPase alter mitochondrial membrane potentials and electron flow through the mitochondrial respiratory chain complex III (MRC-cIII) in CML cells, thereby generating significant ROS levels [74]. There is at least one additional mechanism how BCR-ABL1 enhances ROS as STAT5 clearly acts via an independent mechanism; electro spin resonance analysis excluded mitochondria as a source of STAT5 triggered ROS production. Of all alternative sources for ROS, NADPH oxidases are of particular interest as NOX4 NADPH has recently been identified as direct transcriptional target of STAT5 in liver and fibroblast cells [75]. All our attempts to detect NOX4 in BCR-ABL1 transformed cells failed. Tissue specific differences in NOX4 expression are the most likely reason underlying this phenomenon. Although we currently cannot identify one or more distinct target genes, we could show that STAT5 induced ROS production require the transcriptional activity of STAT5. Using several STAT5 variants we predict the involvement of the N-terminus which is essential for tetramer formation and the regulation of survival genes (Cite again Li et al., Blood, 2010). Experiments employing STAT5A versus STAT5B revealed that STAT5 is also capable to regulate its own transcription which may at least partially account for the up-regulation of STAT5 protein and mRNA levels observed in CML-AP. Despite high homology between STAT5A and STAT5B pronounced differences are present within the C-terminal transactivation domain of STAT5 isoforms, which are also known to undergo differential post-translational modifications (such as serine phosphorylation, methylation, acetylation), or splicing. Differential STAT5 activities may account for different binding partners and thereby may lead to different sets of target gene regulation.
STAT5 alone does not suffice to regulate ROS but requires the concomitant presence of the BCR-ABL1 oncogene. This phenomenon is evident from experiments showing that STAT5 fails to increase ROS in Ba/F3 and 32D cells lacking BCR-ABL. Despite culture of these parental cells in IL-3 enriched medium leading to constitutive JAK2-STAT5 signalling, the enforced expression of STAT5A or STAT5B did not impact on ROS levels. Several reasons may account for this discrepancy; STAT5 may have a different set of target genes due to distinct co-factors in BCR-ABL1+ cells compared to non-transformed hematopoietic cells. Alternatively, one may envision that decreases in ROS scavenger pathways occur downstream of BCR-ABL1 expression that act in concert with STAT5 to enhance intracellular ROS. In the absence of BCR-ABL1, STAT5 triggered ROS levels in untransformed cells may be “buffered” by scavenger pathways. Along these lines STAT5 may depend on a second signalling pathway activated by BCR-ABL1 to synergistically turn on the ROS-producing machinery. Our results show that STAT5 – albeit harmless to untransformed cells – becomes a potential threat once an oncogene induced signal re-wiring had occurred.
Although the evidence for STAT5 as proto-oncogene is overwhelming one should consider that STAT5 has also been assigned tumour suppressor, differentiation or senescence functions, all counteracting transformation [36, 76]. Constitutively STAT5 (cSTAT5) – induced senescence involves increased production of ROS which accounts for an elevated DNA damage rate linking cSTAT5A expression to p53 induced senescence. These findings again underscore the tight connection between STAT5, ROS and DNA damage [35]. As BCR-ABL1+ cells are largely resistant to senescence induction, CML cells can survive DNA-damage that predisposes them to the development of mutations.
Further protection of BCR-ABL1+ cells stems from the fact that BCR-ABL1-STAT5 not only triggers ROS production but also promotes survival. This protective effect is pronounced in cells expressing high levels of STAT5 as BCL-2, BCLXL and MCL-1 protein levels are under the control of STAT5. This allows the cells to tolerate elevated ROS levels; STAT5 acts in a dual way – while enhancing ROS levels it also provides the cells with the molecular machinery to handle the elevated ROS levels by up-regulating the molecular tools to accept the elevated DNA-damage frequency.
Materials and Methods
Mice
NOD/Shi-scid/IL-2Rɣnull (NOG) mice were maintained at the Veterinary University of Vienna. NOG mice were used for leukaemia engraftment and ROS ex vivo studies. In detail, 5 x 105 cells were injected intravenously and engraftment was monitoring by analysing the percentage of GFP+ cells in the peripheral blood of mice. All animal experiments were carried out in accordance with protocols approved by Austrian law.
Tissue culture conditions and infections
Tissue culture conditions, virus preparation, infection of bone marrow cells with viral supernatant from A010 cells or gp + E86 producer cell lines and establishment of cell lines was described previously [27].
Immunoblotting
Immunoblots were carried out as described previously.[27, 77] Membranes were probed with antibodies directed against α-tubulin (T9026 Sigma-Aldrich), STAT5A and STAT5B (both described in [78].
Plasmids
Wild type Stat5a, wild type Stat5b, the Stat5a mutants, Stat5a∆N [42], Stat5aEE/AA, Stat5aY/F [43], and the double serine mutant Stat5aSS/AA [41] were expressed in the retroviral vector pMSCV-IRES-eGFP. p185BCR-ABL1 and p210BCR-ABL1 was expressed via a pMSCV. Ecotropic, replication incompetent gp + E86 producers were generated and selected for high virus titer production by FACS as described previously [42, 79].
Primary patient samples
Primary cells were obtained from patients treated at the General Hospital, Vienna, Austria. Cells were obtained from patients with CML at routine blood and bone marrow examinations after informed consent was given. Peripheral blood and bone marrow mononuclear cells were isolated using Ficoll. Samples were analysed for BCR-ABL1 mutations. Use of human samples was approved by the Ethical Committee of the Medical University of Vienna and is in compliance with Austrian legislation.
Colony formation and 96-well imatinib resistance assay
For colony formation assays, p160v-ABL transformed cells were plated in cytokine-free methylcellulose (StemCell Technologies) at a density of 2 x 102 cells/ml or 3 x 106 cells/ml without or in the presence of 1 µM imatinib. For the 96-well imatinib resistance assay, 1 x 106 cells/ml were plated in 96-well plates containing RPMI supplemented with 2 µM imatinib. Medium with fresh supplemented imatinib was added every other day. Number of imatinib-resistant clones was evaluated 28 days after the initial imatinib exposure.
Intracellular pSTAT5 and γH2A-X staining
Cells were analysed by a BD FACS-Canto II FACS device and BD FACS Diva software (Beckton Dickinson). For the intracellular staining 5 x 106 cells were fixed by 2% paraformaldehyde (Aldrich)/PBS at room temperature (RT) for 10 minutes. All washing steps were performed for 15 minutes with 10 ml PBS/2% FCS/0.2% Tween-20 per sample. Cells were washed twice and permeabilized with 99% ice-cold methanol for 20 minutes at -20°C. Cells were washed twice and incubated with αCD16/CD32 (FCγIII/II receptor; BD Bioscience Pharmingen) and 5 µl γH2A-X (phospho histone H2AX S139, Alexa Fluor 647; Cell Signalling) or pSTAT5 (PY-STAT5-Alexa Fluor 647; BD) at RT for one hour. Cells were washed two times before analysing via flow cytometry.
Real-time PCR
Total RNA was isolated using Tri Reagent (Sigma) according to the manufacturer’s instructions. One µg of total RNA was used for cDNA synthesis using the GeneAmp RNA PCR Kit (Roche) and used for the RT-PCR reaction performed on a Biorad MyiQ2 (Biorad) using Sso Fast Eva Green Supermix (Biorad). All experiments were performed in triplicates and repeated at least twice. Sequences of primer pairs used during the course of the study (5’-3’): human STAT5A for: GGCTCCCTATAACATGTACCC, rev: AAGACTGTCCATTGGTCGGCG; human GAPDH for: TCTCCTCTGACTTCAACAGCG; rev: ACCACCCTGTTGCTGTAGCC; mouse Bcl2 for: ACTGAGTACCTGAACCGGCATC, rev: GGAGAAATCAAACAGAGGTCGC; mouse GAPDH for: AGAAGGTGGTGAAGCAGGCATC, rev: CGGCATCGAAGGTGGAAGAGTG; mouse MnSOD for: TTAACGCGCAGATCATGCA, rev: GGTGGCGTTGAGATTGTTCA; mouse Catalase for: TGAGAAGCCTAAGAACGCAATTC, rev: CCCTTCGCAGCCATGTG; mouse Stat5a for: ACATGGACCAGGCTCCTTCCC, rev: CTCATCCAGGTCAAACTCGCC; mouse Stat5b for: GGCAGGGTCAGTAACGGAAG, rev: GGCTCTGCAAAGGCGTTGTC.
Small interfering RNA mediated knockdown of STAT5A/B
For knockdown of STAT5, an oligonucleotide targeting human and murine STAT5A and STAT5B mRNA [80] (5’-GCAGCAGACCATCATCCTG-3’) was cloned into a modified pLKO.1 lentiviral vector expressing mCherry as a marker. Recombinant VSV-G pseudotyped lentiviruses were produced as described previousely [81]. K562 cells were transduced in the presence of polybrene (7 µg/ml) and knockdown of STAT5 was confirmed by immunoblotting.
ROS staining
Two days before the staining procedure growth medium was changed and cells were splitted. Only cell lines with less than 10% of dead or apoptotic cells on the day of staining were used. 5 x 105 cells were washed and stained in 4 µM dihydroethidium (DHE; MGT Inc.)/PBS solution for 20 minutes at 37°C. Cells were washed once and immediately used for FACS analysis.
ChIP analysis
ChIP analyses were performed as described in [82]. For 20 x 106 cells, 5 µg of the respective antibodies were used. For STAT5, a human/mouse STAT5A/B pan specific antibody from R&D systems was used. For IgG isotype control an antibody from Santa Cruz (sc-2027) was used. Conserved STAT5 binding sites were obtained by analyzing sequence conservation between the human and murine Stat5 locus using the ECR browser (http://ecrbrowser.dcode.org). Putative STAT5 binding sites were identified employing MULTITF (http://rvista.dcode.org). The precipitated DNA was quantified by real-time PCR with a MyiQ instrument (Bio-Rad) [83]. RT-PCR primers: Chr.1 for: CATAGATGAAGCTGCCACATAGGT, rev: GTGGGCAAGGACAAAGCATTA;
Stat5_1.2 for: TCCCTCCCATCCCTCTATTC, rev: AAGCCCCCTTTCCATCTCT; Cis for: GTCCAAAGCACTAGACGCCTG, rev: TTCCCGGAAGCCTCATCTT [84].
Electron spin resonance spectroscopy (ESR)
Cells (6 - 32 Mio. cells/ml) were centrifuged and pellets were resuspended in PBS buffer containing 100 µM DFO (desferal) and 25 µM DTPA (diethylene triamine pentaacetic acid). Cell suspensions were aliquoted and incubated at 30 or 37°C until measurement. Prior to ESR measurements CMH (1-Hydroxy-2,2,5,5-tetramethyl-pyrrolidine-3-carboxylic acid methyl ester) was added. For each data point 17µl of cell suspension were aspirated into a Teflon tube (0.9 mm ID) and transferred to a flexline dielectric resonator ER4118X-MD5 (Bruker, Rheinstetten) for measurement. ESR measurements were performed using a Bruker EMX spectrometer with the following parameters: microwave frequency 9.686 GHz, modulation frequency 100 kHz, modulation amplitude 1 G, time constant 0.082 sec, center field 3448 G, scan rate 71 G/min, sweep 80 G, sweep time 84 s, receiver gain 2 ×104. Always two consecutive scans were recorded and from the peak-to-peak intensity of the middle line the absolute CM• (3-(Carboxy-methyl)-2,2,5,5-tetramethyl-1-pyrrolidinyloxy) concentrations were obtained by comparison with a calibration curve constructed from different CP• (3-(Carboxy)-2,2,5,5-tetramethyl-1-pyrrolidinyloxy) concentrations.
Chemical compound treatment
To analyse the impact of JAK2 signalling, cells were treated with 1 µM of the JAK1/2 inhibitor INCB-01842 24 hours before DHE staining. To decrease the level of ROS the ROS scavenger N-acetyl-cystein (NAC) was added to the growth medium at a concentration of 5 mM for five days. To analyse differences in pSTAT5 level, cells were treated with 1 µM INCB-0184224 or 1 µM nilotinib 4 hours prior intracellular FACS staining. To decrease levels of endogenous iron, the iron chelators DFO (desferal, 100 µM) and DTPA (diethylene triamine pentaacetic acid, 25 µM) were added for 4 hours prior to measurements.
Statistics
Two-tailed Student’s t tests were used for statistical analysis if not stated otherwise. Difference was considered statistically significant when P < 0.05. The data are represented as mean ± SD of the number of the determinations and were analysed by Graph Pad® software (San Diego, CA).
Acknowledgements
The authors thank Thomas Decker, Graham Tebb and Mathias Müller for continuous discussion and scientific input. This work was supported by the Austrian Science Foundation (FWF-SFB 28 to V.S. and R.M., FWF P-24295-B23; A.H.).
Author Contributions
Experiments were designed, analysed and interpreted by W.W. and V.S; Experiments were performed and analysed by W.W., E. G., A.B., A.T., and L.G.; P.V. and S.C. provided patient material and advice; P.V. and R.M provided vital reagents. W.W. and V.S. wrote the paper with valuable input and research reagents from A. H., P.V. and R.M.
Conflict of Interest
The authors declare no conflict of interests
Reference
1. Kantarjian HM, Keating MJ, Talpaz M, Walters RS, Smith TL, Cork A, McCredie KB and Freireich EJ. Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. The American journal of medicine. 1987; 83(3):445-454.
2. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J and Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996; 2(5):561-566.
3. Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. The New England journal of medicine. 2006; 355(23):2408-2417.
4. Sawyers CL. Even better kinase inhibitors for chronic myeloid leukemia. The New England journal of medicine. 2010; 362(24):2314-2315.
5. Lenaerts T, Castagnetti F, Traulsen A, Pacheco JM, Rosti G and Dingli D. Explaining the in vitro and in vivo differences in leukemia therapy. Cell cycle. 2011; 10(10):1540-1544.
6. Chomel JC, Bonnet ML, Sorel N, Bertrand A, Meunier MC, Fichelson S, Melkus M, Bennaceur-Griscelli A, Guilhot F and Turhan AG. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood. 2011; 118(13):3657-3660.
7. Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS and Bhatia R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011; 118(20):5565-5572.
8. Chomel JC and Turhan AG. Chronic myeloid leukemia stem cells in the era of targeted therapies: resistance, persistence and long-term dormancy. Oncotarget. 2011; 2(9):713-727.
9. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008; 112(13):4808-4817.
10. Shah NP and Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003; 22(47):7389-7395.
11. Nicolini FE, Mauro MJ, Martinelli G, Kim DW, Soverini S, Muller MC, Hochhaus A, Cortes J, Chuah C, Dufva IH, Apperley JF, Yagasaki F, Pearson JD, Peter S, Sanz Rodriguez C, Preudhomme C, et al. Epidemiologic study on survival of chronic myeloid leukemia and Ph(+) acute lymphoblastic leukemia patients with BCR-ABL T315I mutation. Blood. 2009; 114(26):5271-5278.
12. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5):646-674.
13. Slupphaug G, Kavli B and Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutation research. 2003; 531(1-2):231-251.
14. Ziech D, Franco R, Pappa A and Panayiotidis MI. Reactive oxygen species (ROS)--induced genetic and epigenetic alterations in human carcinogenesis. Mutation research. 2011; 711(1-2):167-173.
15. Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes D, Daumer KM, Lin Z, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Knudsen ES, Sotgia F and Lisanti MP. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell cycle. 2010; 9(16):3256-3276.
16. Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, Small D and Rassool F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008; 111(6):3173-3182.
17. Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, Blasiak J and Skorski T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006; 108(1):319-327.
18. Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, Nieborowska-Skorska M, Blasiak J and Skorski T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004; 104(12):3746-3753.
19. Walz C, Crowley BJ, Hudon HE, Gramlich JL, Neuberg DS, Podar K, Griffin JD and Sattler M. Activated Jak2 with the V617F point mutation promotes G1/S phase transition. The Journal of biological chemistry. 2006; 281(26):18177-18183.
20. Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T, Greenfield EA, Salgia R and Griffin JD. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. The Journal of biological chemistry. 2000; 275(32):24273-24278.
21. Carlesso N, Frank DA and Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. The Journal of experimental medicine. 1996; 183(3):811-820.
22. Ilaria RL, Jr. and Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. The Journal of biological chemistry. 1996; 271(49):31704-31710.
23. Shuai K, Halpern J, ten Hoeve J, Rao X and Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996; 13(2):247-254.
24. Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T, Calabretta B and Skorski T. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. The Journal of experimental medicine. 1999; 189(8):1229-1242.
25. Scherr M, Chaturvedi A, Battmer K, Dallmann I, Schultheis B, Ganser A and Eder M. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML). Blood. 2006; 107(8):3279-3287.
26. Ye D, Wolff N, Li L, Zhang S and Ilaria RL, Jr. STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood. 2006; 107(12):4917-4925.
27. Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R and Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006; 107(12):4898-4906.
28. Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, Fajmann S, Grebien F, Warsch W, Stengl G, Hennighausen L, Poli V, Beug H, Moriggl R and Sexl V. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO molecular medicine. 2010; 2(3):98-110.
29. Kovacic B, Hoelbl A, Litos G, Alacakaptan M, Schuster C, Fischhuber KM, Kerenyi MA, Stengl G, Moriggl R, Sexl V and Beug H. Diverging fates of cells of origin in acute and chronic leukaemia. EMBO molecular medicine. 2012; 4(4):283-297.
30. Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Holbl A, Gleixner KV, Dworzak M, Mayerhofer M, Hoermann G, Herrmann H, Sillaber C, Egger G, Valent P, Moriggl R and Sexl V. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011; 117(12):3409-3420.
31. Jalkanen SE, Lahesmaa-Korpinen AM, Heckman CA, Rantanen V, Porkka K, Hautaniemi S and Mustjoki S. Phosphoprotein profiling predicts response to tyrosine kinase inhibitor therapy in chronic myeloid leukemia patients. Experimental hematology. 2012; 40(9):705-714 e703.
32. Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, Henke N, Li Z, Hoffmann TK, Kim YM, Hofmann WK, Jumaa H, Groffen J, Heisterkamp N, Martinelli G, Lieber MR, et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer cell. 2009; 16(3):232-245.
33. Slupianek A, Dasgupta Y, Ren SY, Gurdek E, Donlin M, Nieborowska-Skorska M, Fleury F and Skorski T. Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia. Blood. 2011; 118(4):1062-1068.
34. Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R and Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Molecular cell. 2001; 8(4):795-806.
35. Mallette FA, Moiseeva O, Calabrese V, Mao B, Gaumont-Leclerc MF and Ferbeyre G. Transcriptome analysis and tumor suppressor requirements of STAT5-induced senescence. Annals of the New York Academy of Sciences. 2010; 1197:142-151.
36. Mallette FA and Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle. 2007; 6(15):1831-1836.
37. Starzynski RR, Goncalves AS, Muzeau F, Tyrolczyk Z, Smuda E, Drapier JC, Beaumont C and Lipinski P. STAT5 proteins are involved in down-regulation of iron regulatory protein 1 gene expression by nitric oxide. The Biochemical journal. 2006; 400(2):367-375.
38. Kerenyi MA, Grebien F, Gehart H, Schifrer M, Artaker M, Kovacic B, Beug H, Moriggl R and Mullner EW. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood. 2008; 112(9):3878-3888.
39. Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T and Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101(27):10042-10047.
40. Tan WQ, Wang K, Lv DY and Li PF. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. The Journal of biological chemistry. 2008; 283(44):29730-29739.
41. Friedbichler K, Kerenyi MA, Kovacic B, Li G, Hoelbl A, Yahiaoui S, Sexl V, Mullner EW, Fajmann S, Cerny-Reiterer S, Valent P, Beug H, Gouilleux F, Bunting KD and Moriggl R. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood. 2010; 116(9):1548-1558.
42. Moriggl R, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, Bunting KD, Wagner EF, Sonneck K, Valent P, Ihle JN and Beug H. Stat5 tetramer formation is associated with leukemogenesis. Cancer cell. 2005; 7(1):87-99.
43. Wang D, Moriggl R, Stravopodis D, Carpino N, Marine JC, Teglund S, Feng J and Ihle JN. A small amphipathic alpha-helical region is required for transcriptional activities and proteasome-dependent turnover of the tyrosine-phosphorylated Stat5. The EMBO journal. 2000; 19(3):392-399.
44. Li G, Miskimen KL, Wang Z, Xie XY, Brenzovich J, Ryan JJ, Tse W, Moriggl R and Bunting KD. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood. 2010; 115(7):1416-1424.
45. Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, Superti-Furga G and Sexl V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012; 8(3):285-293.
46. Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, Perrotti D and Arlinghaus RB. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009; 28(14):1669-1681.
47. Samanta A, Perazzona B, Chakraborty S, Sun X, Modi H, Bhatia R, Priebe W and Arlinghaus R. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2011; 25(3):463-472.
48. Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010; 115(15):3109-3117.
49. Deininger M, Buchdunger E and Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005; 105(7):2640-2653.
50. de Groot RP, Raaijmakers JA, Lammers JW and Koenderman L. STAT5-Dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Molecular cell biology research communications : MCBRC. 2000; 3(5):299-305.
51. Horita M, Andreu EJ, Benito A, Arbona C, Sanz C, Benet I, Prosper F and Fernandez-Luna JL. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. The Journal of experimental medicine. 2000; 191(6):977-984.
52. Gesbert F and Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000; 96(6):2269-2276.
53. Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, Akgul C, Derdak S, Pickl WF, Wacheck V, Selzer E, Monia BP, Moriggl R, Valent P and Sillaber C. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005; 105(8):3303-3311.
54. Hochhaus A, La Rosee P, Muller MC, Ernst T and Cross NC. Impact of BCR-ABL mutations on patients with chronic myeloid leukemia. Cell cycle. 2011; 10(2):250-260.
55. Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, Pane F, Muller MC, Ernst T, Rosti G, Porkka K, Baccarani M, Cross NC and Martinelli G. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011; 118(5):1208-1215.
56. Goyal P, Weissmann N, Rose F, Grimminger F, Schafers HJ, Seeger W and Hanze J. Identification of novel Nox4 splice variants with impact on ROS levels in A549 cells. Biochemical and biophysical research communications. 2005; 329(1):32-39.
57. Gabig TG, Kipnes RS and Babior BM. Solubilization of the O2(-)-forming activity responsible for the respiratory burst in human neutrophils. The Journal of biological chemistry. 1978; 253(19):6663-6665.
58. Dawson TL, Gores GJ, Nieminen AL, Herman B and Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. The American journal of physiology. 1993; 264(4 Pt 1):C961-967.
59. Sanders SA and Massey V. The thermodynamics of xanthine oxidoreductase catalysis. Antioxidants & redox signaling. 1999; 1(3):371-379.
60. Wang W, Wang S, Yan L, Madara P, Del Pilar Cintron A, Wesley RA and Danner RL. Superoxide production and reactive oxygen species signaling by endothelial nitric-oxide synthase. The Journal of biological chemistry. 2000; 275(22):16899-16903.
61. Babior BM. Oxygen-dependent microbial killing by phagocytes (second of two parts). The New England journal of medicine. 1978; 298(13):721-725.
62. Porwol T, Ehleben W, Brand V and Acker H. Tissue oxygen sensor function of NADPH oxidase isoforms, an unusual cytochrome aa3 and reactive oxygen species. Respiration physiology. 2001; 128(3):331-348.
63. Wolin MS, Burke-Wolin TM and Mohazzab HK. Roles for NAD(P)H oxidases and reactive oxygen species in vascular oxygen sensing mechanisms. Respiration physiology. 1999; 115(2):229-238.
64. Thannickal VJ and Fanburg BL. Reactive oxygen species in cell signaling. American journal of physiology Lung cellular and molecular physiology. 2000; 279(6):L1005-1028.
65. Garban HJ and Bonavida B. Nitric oxide disrupts H2O2-dependent activation of nuclear factor kappa B. Role in sensitization of human tumor cells to tumor necrosis factor-alpha -induced cytotoxicity. The Journal of biological chemistry. 2001; 276(12):8918-8923.
66. Papaconstantinou J and Hsieh CC. Activation of senescence and aging characteristics by mitochondrially generated ROS: how are they linked? Cell cycle. 2010; 9(19):3831-3833.
67. Goyal P, Weissmann N, Grimminger F, Hegel C, Bader L, Rose F, Fink L, Ghofrani HA, Schermuly RT, Schmidt HH, Seeger W and Hanze J. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free radical biology & medicine. 2004; 36(10):1279-1288.
68. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM and Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. The Journal of biological chemistry. 2000; 275(33):25130-25138.
69. Fatrai S, Wierenga AT, Daenen SM, Vellenga E and Schuringa JJ. Identification of HIF2alpha as an important STAT5 target gene in human hematopoietic stem cells. Blood. 2011; 117(12):3320-3330.
70. Ohara Y, Peterson TE and Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. The Journal of clinical investigation. 1993; 91(6):2546-2551.
71. Tesfamariam B and Cohen RA. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. The American journal of physiology. 1992; 263(2 Pt 2):H321-326.
72. Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA and Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997; 95(3):588-593.
73. Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK and Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999; 401(6748):79-82.
74. Nieborowska-Skorska M, Kopinski PK, Ray R, Hoser G, Ngaba D, Flis S, Cramer K, Reddy MM, Koptyra M, Penserga T, Glodkowska-Mrowka E, Bolton E, Holyoake TL, Eaves CJ, Cerny-Reiterer S, Valent P, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012; 119(18):4253-4263.
75. Yu JH, Zhu BM, Riedlinger G, Kang K and Hennighausen L. The liver-specific tumor suppressor STAT5 controls expression of the reactive oxygen species-generating enzyme NOX4 and the proapoptotic proteins PUMA and BIM in mice. Hepatology. 2012; 56(6):2375-2386.
76. Ferbeyre G and Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochimica et biophysica acta. 2011; 1815(1):104-114.
77. Moriggl R, Gouilleux-Gruart V, Jahne R, Berchtold S, Gartmann C, Liu X, Hennighausen L, Sotiropoulos A, Groner B and Gouilleux F. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996; 16(10):5691-5700.
78. Quelle FW, Wang D, Nosaka T, Thierfelder WE, Stravopodis D, Weinstein Y and Ihle JN. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Molecular and cellular biology. 1996; 16(4):1622-1631.
79. Grebien F, Kerenyi MA, Kovacic B, Kolbe T, Becker V, Dolznig H, Pfeffer K, Klingmuller U, Muller M, Beug H, Mullner EW and Moriggl R. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood. 2008; 111(9):4511-4522.
80. Scheeren FA, Naspetti M, Diehl S, Schotte R, Nagasawa M, Wijnands E, Gimeno R, Vyth-Dreese FA, Blom B and Spits H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nat Immunol. 2005; 6(3):303-313.
81. Mayerhofer M, Gleixner KV, Mayerhofer J, Hoermann G, Jaeger E, Aichberger KJ, Ott RG, Greish K, Nakamura H, Derdak S, Samorapoompichit P, Pickl WF, Sexl V, Esterbauer H, Schwarzinger I, Sillaber C, et al. Targeting of heat shock protein 32 (Hsp32)/heme oxygenase-1 (HO-1) in leukemic cells in chronic myeloid leukemia: a novel approach to overcome resistance against imatinib. Blood. 2008; 111(4):2200-2210.
82. Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA and Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007; 27(1):49-63.
83. Decker T, Pasca di Magliano M, McManus S, Sun Q, Bonifer C, Tagoh H and Busslinger M. Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity. 2009; 30(4):508-520.
84. Basham B, Sathe M, Grein J, McClanahan T, D’Andrea A, Lees E and Rascle A. In vivo identification of novel STAT5 target genes. Nucleic acids research. 2008; 36(11):3802-3818.