
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease and the fourth leading cause of cancer deaths. It has one of the highest mortality rates for solid tumors and the overall five year survival rate is unacceptably low [1]. Invariably, the initial diagnosis occurs at a stage when the cancer is already advanced leading to poor prognosis [2]. Surgery is possible only in 25% of the patients and the gain in survival rises from a dismal 5% to a modest 25% [3, 4]. Tumor cell migration and invasion are the most critical steps in progression of pancreatic cancer and they both occur at an early stage. PDACs are surrounded by dense fibrous tissue due to the intense desmoplastic fibrotic response, one of the hall marks of pancreatic cancer [5]. Some of the associated risk factors PDAC are diabetes, chronic inflammation (pancreatitis), alcohol and cigarette smoking. Tobacco smokers are three times more prone to develop PDAC than nonsmokers[6]. In addition, hepatitis B or C infections can also increase the incidence of PDAC[7]. There are no effective PDAC screening tests available. Adjuvant therapies include fluorouracil-based chemo radiation (fluorouracil and gemcitabine) [8, 9]. However the benefits are marginal as the cancer rapidly acquires drug resistance [10, 11].
In PDAC, the pancreatic stellate cells (PSCs), responsible for the generation of fibrous tissue in the pancreas, play a vital role in tumorigenesis. During development of pancreatic cancer or during inflammation, PSCs undergo morphological and functional changes to become myofibroblast -like cells. They express α-smooth muscle actin (α-SMA) and have high capacity to produce ECM proteins like type-1 collagen and fibronectin. Key intracellular signaling pathways like mitogen activated protein kinases (MAPK), cytokines, growth factors and microRNAs activate PSCs and promote interaction between PSCs and cancer cells that further fuel pancreatic cancer development [12-14]. In vitro, PSC culture supernatant stimulated migration, invasion and colony formation of pancreatic cancer cells. In addition, injection of PSCs along with PDAC cells into orthotopic murine models increased tumorigenicity along with metastasis [15, 16]. PSCs are known to accompany cancer cells to metastatic sites and stimulate angiogenesis [17]. Recently Lu J. et al., using a modified Boyden chamber assay, showed that PSCs stimulate migration of pancreatic cells via haptotactic mechanisms, which are mediated through collagen-1, activated α2/β1 integrin-FAK signaling pathway [18]. Apart from their indispensable role in fibrogenesis, PSCs through their secretion of matrix metalloproteinases (MMP) and their inhibitors (tissue inhibitors of metalloproteinase, TIMPs) have the potential to promote metastasis [3, 19].
About 10% of the patients inherit PDAC, an aspect that was recently reviewed and will not be addressed here [20, 21]. The inheritance of familial pancreatic cancer (FPC) is mostly autosomal dominant with a heterogeneous phenotype. Germline mutations in BRCA2, PALB2 and ATM are known to trigger pancreatic cancer in some families [22]. Lipocalin-2 and tissue inhibitor of metalloproteinase 1 have recently been identified as potential serum markers for early detection of FPC [23].
Pancreatic cancer is characterized by several chromosomal abnormalities. There are frequent losses in multiple chromosome arms including 1p, 3p, 4q, 6q, 8p, 9p, 12q, 17p, 18q, and 21q and gains in 8q and 20q [24]. A seminal paper by Kinzler and coworkers [25] described detailed gene expression analysis of tumor transcripts amplified from 24 pancreatic cancers. The transcripts represented more than 23,000 genes. They identified 12 core cellular signaling pathways that favored pancreatic cancer tumor growth and metastasis which were genetically altered in 67-100% of the tumors. Here we highlight, in particular, those pathways involving FAK and paxillin as potential therapeutic targets in pancreatic cancer Figure 1 [26].
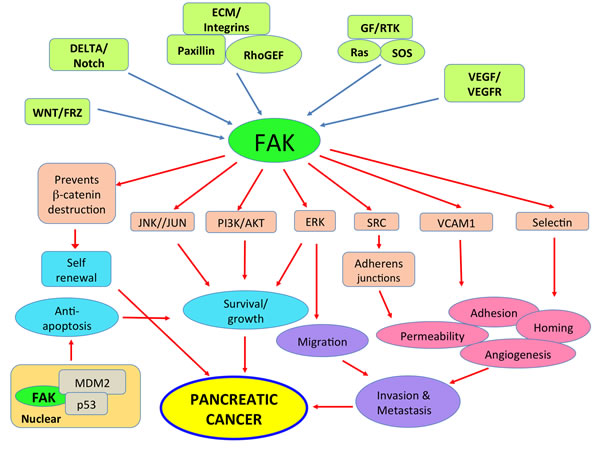
Figure 1: FAK plays a significant role in multiple signaling pathways that contribute to pancreatic cancer growth and metastasis. Several receptor systems induce FAK activation that then contributes to the unique function. For instance, RTK signaling through FAK contribute to pancreatic tumor growth and metastasis; however VEGFR mediated signaling through FAK triggers angiogenesis. In addition, K-RAS, which is frequently mutated in pancreatic cancer, is also linked to FAK. FAK also influences lamellipodia formation through activation of small GTPases and promotes homotypic cell adhesion indirectly through paxillin. Suppression of p53 expression by nuclear FAK may also indirectly contribute to tumor growth by inhibiting apoptosis. It is therefore very likely that there is subtle compartmentalization of FAK in the cell and the final effector function could be the result of a combination of FAK mediated and non-FAK mediated signals.
FOCAL ADHESION KINASE (PTK2)
FAK is an intracellular, highly conserved, non-receptor tyrosine kinase encoded by PTK2 located on human chromosome 8q24.3. It is ubiquitously expressed in all cells [27, 28] and was initially identified in v-Src transformed chicken embryo fibroblasts [29]. FAK is associated with many aspects of metastasis such as adhesion, migration and invasion. FAK is overexpressed and activated in a variety of cancers including colon, breast, lung, thyroid, head and neck, liver, pancreatic and esophageal and is correlated with poor survival rates [30, 31]. The underlying mechanism of FAK overexpression is unclear. FAK is upregulated in PDAC and this increased expression is correlated with the size of the tumor [32].
FAK serves as a scaffolding protein and an integral component of focal adhesions and is anchored via paxillin. It regulates paxillin function via phosphorylation and plays an important role in lamellipodia formation and cell motility. Figure 2 describes in brief, some of the key signaling molecules that FAK interacts with. The 125 kDa FAK protein is mainly composed of N-terminal FERM domain with an autophosphorylation site (Y397), followed by a proline rich region (PR1), central catalytic kinase domain, two additional proline rich regions (PR2 and PR3) and a C-terminal focal adhesion-targeting (FAT) domain (Figure 2). The FERM domain of FAK is structurally similar to cytoskeletal proteins such as talin and the ezrin-radixin-moesin (ERM) family of proteins and also signaling molecules such as the JAK family tyrosine kinases and tyrosine phosphatases [33, 34]. It mediates FAK interaction with integrins and growth factor receptors [27, 35, 36]. The N-terminal PR1 region serves as a docking site for SH3-containing proteins such as cellular Src, whereas the C-terminal PR2 and PR3 regions mediate interactions with other SH3-containing proteins such as p130Cas, endophilin A2, Graf, and ASAP1 [37-39]. The catalytic kinase domain of FAK is highly conserved and contains major phosphorylation sites Y576 and Y577and the ATP binding site K454 [40]. The crystal structure of FAK kinase domain shows an open confirmation, which is very similar to the fibroblast growth factor receptor-1 (FGFR-1) and vascular endothelial growth factor receptor (VEGFR) [41]. The binding of p130Cas with FAK plays an important role in promoting cell migration, which is mediated through RAC activation whereas the binding of FAK with GRAF and ASAP1 regulates cytoskeletal dynamics and focal contact assembly. The FAT domain is mainly required for interactions with other scaffold proteins such as paxillin or talin during focal adhesion formation [42-46]. The C-terminal domain of FAK is known as FRNK (FAK-Related Non-Kinase) acts as a negative regulator of FAK activity by inhibiting activation and signaling of endogenous FAK [47]. FRNK is expressed in significant quantities during neonatal development in vascular smooth muscle cells; however in adults its expression is highly down regulated. Overexpression of FRNK in cells is known to inhibit cell migration; cell spreading and growth factor induced signaling to MAP kinase. Suppression of FAK kinase activity by FRNK obviously can affect both kinase and scaffold mediated FAK functions; however the in vivo effects of FRNK in adult remain to be seen [48]. FAK activity is further regulated through its interaction with the FAK-inhibitory protein FIP200. The suppressor of cytokine signaling (SOCS) proteins also interacts with FAK and facilitates the poly-ubiquitination of FAK and its degradation [49].
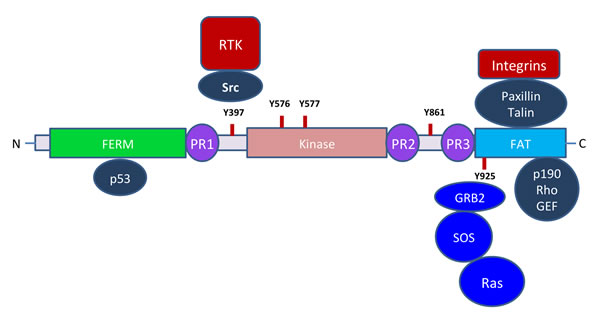
Figure 2: FAK and its potential interacting partners. In cancers, highly active receptor tyrosine kinases such as MET and EGFR phosphorylate Src that then interacts with FAK Y397 through SH2 domain. Juxtaposed Src phosphorylates FAK at Y576 and Y577 and possibly other sites, resulting in a highly active FAK both in terms of the kinase activity and as an adapter protein. In addition, it is also connected to RAS through GRB2 and SOS that feeds into MAPK pathway. The CT FAT domain interacts with paxillin that binds to the relatively short cytoplasmic tails of integrins. Although the FERM domain is capable of interacting with integrins and RTKs, the more favored interaction appears to be through paxillin anchored to focal adhesions.
ACTIVATION OF FAK BY INTEGRINS
Signaling pathways mediated by integrins play a critical role in cancers in general and pancreatic cancers in particular (Figure 3). Integrins belong to family of transmembrane receptors and they link extracellular matrix proteins (ECM) with the intracellular actin cytoskeleton to regulate cell shape and motility. Integrins are heterodimeric receptors composed of α and β subunits. There are 24 known α subunits and 9 β beta subunits and the particular combination of two subunits (α, β) generate a specific integrin receptor. Each integrin recognizes a specific ligand such as vitronectin, fibronectin, laminin and collagen, which are present in either interstitial spaces or in basement membrane. Integrins also serve as coreceptors to ICAM-1, VCAM-1. Apart from their role in cell adhesion, binding of integrins to ECM proteins activates integrin-associated tyrosine kinases, resulting in the tyrosine phosphorylation of downstream signaling molecules. Figure 3 describes some of the important mediators of these signaling pathways. Thus by controlling these signaling events, integrins play an important role in the regulation of cell migration and cell survival [50, 51]. The relatively short cytoplasmic tails of integrins interact with cytoskeletal adapter proteins, such as paxillin (described below in greater details), which then recruits FAK. Ligand mediated clustering of integrins promotes FAK autophosphorylation at Y397, which then interacts with the SH2 domain of c-Src that is activated by RTKs. This site can also potentially interact with the SH2 containing proteins such as Shc, PI3K, Grb2 and phospholipase C. Binding and subsequent activation of Src leads to further tyrosine phosphorylation of FAK at Y407, Y576 and Y577, in its kinase domain to facilitate maximum activation [35, 52, 53]. Src mediated phosphorylation of FAK at Y925 creates a docking site for GRB2 which activates the small GTP protein RAS and the downstream ERK2 (MAPK) that is known to play an important role in directed cell motility and invasion.
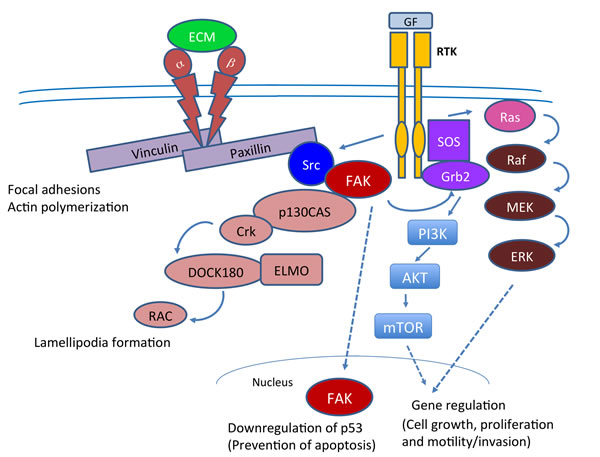
Figure 3: Cell signaling pathways in which FAK plays a key role in tumorigenesis. FAK plays a central role in both Integrin and RTK mediated signaling and plays a key role in cytoskeletal changes, lamellipodia formation and cell proliferation and motility. Interaction of integrins with ECM also triggers downstream activation of FAK resulting in its autophosphorylation at Y397. In parallel, the RTK mediated activation of c-Src and its related kinases such as Fyn and Lck, results in the generation of an open SH2 domain that docks on to Y397 and further propagation of the signals. Src mediated Y925 phosphorylation recruits GRB2 which then leads to the activation of RAS and subsequently ERK2. The Y925 site in FAK is a part of paxillin interaction site and its phosphorylation is likely to disrupt FAK localization to focal adhesions. Interestingly, ERK2 mediated FAK phosphorylation at S910 also destabilizes paxillin-FAK interaction. Moreover, ERK2 mediated phosphorylation of paxillin can positively influence FAK adherence to focal adhesions. FAK thus promote cell proliferation and motility that ultimately translates to cancer metastasis. In addition, FAK has the potential to interact with p53 promoter site and down regulate p53 transcription, thus promoting cell survival.
FAK AND RECEPTOR TYROSINE KINASES
Several of the well-known oncogenes are receptor tyrosine kinases (RTKs) such as EGFR, MET, EPH, VEGFR and KIT. They play an indispensable role in tumor growth and signaling through these receptors also contributes to cancer cell motility and metastasis. This is due in part to their ability to activate FAK and MAPK (Figure 2). The connection between RTKs and FAK appears to be indirect. The most likely scenario is through the ability of RTKs to activate Src family tyrosine kinases (STK) whose SH2 domain binds to FAK. This further feeds signals that promote cell motility.
FAK AND K-RAS
K-RAS protein is a GTPase, and an early player in the signal transduction pathways regulating cell proliferation and differentiation [54]. Pancreatic cancer has maximum K-RAS alterations as compared to any other tumor type. In 75 - 90% of pancreatic tumors, there are activating point mutations of the K-RAS oncogene at codons 12, 13 and 61 [55, 56]. The most common of which is substitution of the wild type glycine residue (codon 12) by cysteine, arginine, valine, or aspartic acid [55]. These mutations result in the generation of constitutively active K-RAS. The active K-RAS mutants in turn activate related downstream signaling pathways including RAF-MAPK, MEK1/2 and PI3K-AKT, resulting in uncontrolled cell growth and metastasis.
In pancreatic cancer, K-RAS mutations usually develop in the early phase of carcinogenesis and patients with mutated K-RAS have significantly less overall survival compared to patients with wild type K-RAS. This clearly suggests that mutations in K-RAS can initiate and advance pancreatic cancer. Apart from mutations, K-RAS is frequently amplified in pancreatic cancer. The role of FAK in K-RAS mediated cell migration and motility is controversial [57, 58]. K-RAS mediated activation of MEK1 results in phosphorylation of FAK at Ser910 resulting in the suppression of its kinase activity [59]. This most likely occurs through activated RAS that signals via Fgd1-Cdc42-PAK1- MEK-ERK signaling cascade. MEK1 phosphorylates FAK S910 that results in the recruitment of PIN1 and PTP-PEST. The co-localization with FAK occurs at the lamellipodia of migrating cells. PIN1 binding and prolyl isomerization of FAK can cause PTP-PEST to interact with and dephosphorylate FAK at Y397 resulting in suppression of FAK kinase activity. Despite the inhibition of FAK kinase activity, activated RAS can still promote cell migration, invasion, and metastasis through a FAK independent pathway. It is therefore possible that the use of FAK inhibitors in those pancreatic cancers wherein RAS is highly activated may not be of any use as FAK is already suppressed. This however remains to be seen as FAK is known to be highly active in majority of pancreatic cancers [32]. It is also noteworthy that despite the significant role played by mutated RAS in various cancers, to date no successful treatment has been developed that is based on targeting RAS.
FAK AND PAXILlIN
Paxillin is a major component of focal adhesions that form a structural link between extracellular matrix and actin cytoskeleton. It is a multidomain, 68 kDa protein, first identified as a phosphotyrosine protein in cells transformed by v-Src [60, 61]. In cancer cells, its function is regulated through Src and FAK mediated phosphorylation [62, 63]. Paxillin lacks any intrinsic enzymatic activity and functions mainly as an adaptor protein, by creating an array of docking sites for other proteins, thus promoting the assembly of multiprotein complexes.
Active FAK is located at the leading edge of the cell where it regulates the assembly and disassembly of focal adhesions that result in directional movement. Paxillin appears to be responsible for the disassembly of adhesions at the cell front. This is mediated by the interaction FAK/Src complex with paxillin. A recent study indicates that paxillin recruitment to the cell front protrusions occurs after the assembly of the focal adhesion [64]. Several studies have now shown that both FAK and paxillin are important for cell migration and participate in the dynamic assembly and disassembly of focal adhesions [65-68].
The interaction between paxillin and p130Cas with FAK is through the FAT domain in FAK and appears to be constitutive. The SH3 mediated binding of p130Cas to FAK results in the increased tyrosine phosphorylation of p130Cas at multiple sites. This in turn stimulates SH2 mediated binding of Crk adaptor protein to p130 Cas ultimately resulting in the activation of RAC mediated membrane ruffling or lamellipodia formation and the promotion of cell motility/invasion [69].
The significance of paxillin and FAK interaction can be appreciated by the recent report that a FAK mutant which fails to interact with paxillin resulted in a decrease in FAK in focal adhesions and a noticeable decrease in its phosphorylation. This ultimately led to a significant reduction in adhesion, migration and invasion. It is therefore clear that targeting FAK in cancer therapy is likely to also suppress paxillin-mediated functions [70]. In this fashion, paxillin could serve as a biomarker for FAK therapeutics.
Paxillin also interacts with various other structural proteins such as vinculin and actopaxin, regulators of actin organization (COOL/PIX and PKL/GIT) and the adaptor protein Crk. Through these protein-protein interactions paxillin plays a pivotal role in various physiological processes such as gene expression, matrix organization, tissue remodeling, cell proliferation, cell survival, cell motility and metastasis [44, 46, 71].
Hic-5 and Leupaxin are related to paxillin and are frequently found to coexist. While paxillin is ubiquitously expressed in most of the tissues (except for nervous system) [72-74], Hic-5 is mostly expressed in smooth muscle tissues, particularly the vasculature [75, 76] and Leupaxin is mainly expressed in leukocytes [77]. Gene knockout studies in mice resulted in distinct phenotypes for paxillin and Hic-5. Loss of paxillin resulted in early embryonic lethality whereas the Hic-5 knockouts were viable with minor vascular defects [78, 79] indicating that paxillin interactions are more vital.
The carboxy terminal of paxillin is composed of four LIM domains, which are zinc-binding structures, whereas the amino terminal has five LD motifs and multiple SH2- binding domains. LIM domains are double zinc finger motifs that target paxillin to focal adhesions. LD motifs mainly function as binding sites for other proteins. Moreover, the N terminal region of paxillin has a proline rich region that serves as a dock for SH3 containing proteins. There are several alternatively spliced isoforms of paxillin that, based on their expression levels, could influence the overall function, however this aspect needs further study [72, 80].
Paxillin is known to acquire gain of function mutations that are associated with alterations in the malignant progression of many tumors [81-83] including breast, lung [84-86], prostate [87], melanoma [88] and colorectal cancer [89]. The most common mutation, A128T as identified from our laboratory, is linked to invasive tumor growth [79, 84, 90-92].
In addition to integrin signaling, paxillin also plays a vital role in RTK mediated signaling which is especially important in tumor growth and metastasis. EGF, TGF-β, platelet derived growth factor (PDGF) and androgen receptor mediated signaling are all known to induce paxillin phosphorylation, an event linked to tumor growth and metastasis [93-97]. Paxillin expression levels are known to correlate with HER2 levels in breast cancer cells and patient samples and thus may be a predictor of therapeutic efficacy [83, 98].
Paxillin and its relative Hic-5 appear to play a key role in invadopodia, which are cell protrusions that aid tumor metastasis. They are rich in membrane-bound and soluble matrix metalloproteinases (MMPs) that dissolve the fibrotic and ECM envelope that surrounds the tumor. They have an actin core and several actin-binding and nucleating proteins including paxillin and Hic-5. In addition, paxillin tyrosine phosphorylation is necessary for promoting invadopodia dynamics. Hic-5 also appears to be an essential component of invadopodia formed in cells that have undergone a TGF-α-mediated EMT and forced expression of Hic-5 in epithelial cells can eliminate the need for TGF-α. Paxillin may be dispensed with in cases wherein Hic-5 expression levels are relatively high. A similar role for leupaxin in cancer cell invadopodia has been noted. The Rho GTPases along with the Rho proteins that are anchored through paxillin appear to act in concert and coordinate invadopodia movements.
Cancer cell motility can be described as mesenchymal or amoeboid and the ability of cancer cells to switch between the two is known as plasticity. Paxillin and Hic-5 play a critical role in breast cancer cell morphology and plasticity during invasion. Abrogation of Hic-5 expression promotes an amoeboid phenotype while suppression of paxillin levels results in favoring of a mesenchymal morphology [99].
Paxillin may have an important role in cervical cancer. It was shown to interact with the bovine form of the human papillomavirus (HPV) E6 protein and promote cell transformation [100-102]. Most importantly, paxillin also regulates anchorage independent growth and cell survival through its own tyrosine phosphorylation, which facilitates interaction with p210BCR/ABL, FAK and vinculin [73, 97, 103-105]. In addition, its interaction with the anti-apoptotic protein BCL-2 appears to promote cell survival in the absence of cell adhesion in both cancerous and normal cells. This is partly mediated through FAK signaling [106, 107]. It has been previously shown that Wnt5A, JNK and paxillin are overexpressed in pancreatic cancer and Wnt5A/JNK signaling stimulates cell migration in pancreatic cancer by activating paxillin [108, 109]. Our lab has studied the effect of activating mutations of paxillin on mitochondrial dynamics in lung cancer. Live cell imaging showed that compared to wild type, some mutant clones had enhanced focal adhesion and lamellipodia formation (A127, P233L and P487L). Paxillin mutants exhibited altered association with BCL-2, Dynamin-related Protein-1(DRP-1) and Mitofusion-2 (MFN-2) proteins resulting in dysregulated mitochondrial dynamics. Our results suggest that paxillin mutants through their interactions with BCL-2 and DRP-1 could regulate cisplatin drug resistance in human lung cancer cells [110].
FAK AND PI3 KINASE
PI3K is a cellular oncogene and an essential intracellular lipid kinase that plays an important role in cell survival. FAK through phosphorylated Y397 is known to directly interact with the SH2 domain of p85, the regulatory subunit of PI3K[111, 112]. Both SH2 and SH3 domains of p85 appear to play a role in the binding of PI3K to FAK [113]. FAK has recently been shown to activate the PI3K pathway to suppress doxorubicin-induced apoptosis[114]. PI3K-AKT pathway is essential for cell survival and shown to be constitutively active in most PDACs. Targeting this pathway with small molecule inhibitors or by knock down strategies results in growth inhibition, both in vitro and in vivo [115]. In another study, dual targeting of PI3K-AKT2 with RNA interference resulted in significantly increased apoptosis and reduced proliferation and colony formation in vitro and in vivo. This clearly indicates that simultaneous targeting of key molecules in PDAC is an effective treatment strategy [116, 117].
FAK AND TUMOR SUPPRESSOR GENES
TP53
TP53 is an important tumor suppressor gene. According to cosmic database 49% of pancreatic cancers have TP53 mutations [118]. MK 1775 a Wee 1 inhibitor targets aberrant p53 by blocking cell cycle checkpoint regulation [119]. The interaction between p53 and FAK appears to serve two purposes. The fact that FAK has been discovered in nucleus and that it can bind to p53 and inhibit apoptosis supports a role for FAK activity in tumorigenesis. It was also found that p53 can bind to the FAK promoter site in the nucleus and suppress its transcription thereby aiding p53-mediated cell cycle arrest and apoptosis [120].
SMAD4
In normal cells, the cytokine TGF-β acts, as a tumor suppressor; however in cancer cells it is known to promote metastasis. It signals through SMAD4, a transcription cofactor, that stimulates gene transcription. SMAD4 is known interact with FAK. There appears to be a cross talk between TGF-β and RTKs. TGF- signaling is also known to induce clustering of some RTKs through FAK. Moreover 20% of pancreatic cancers are known to harbor SMAD4 mutations that correlate with poorer prognosis and increased metastasis [121, 122].
MERLIN
Merlin is a tumor suppressor protein, coded by the NF2 gene (Neurofibromatosis type II). It is a membrane cytoskeleton protein, which links actin filaments to the cell membrane, and mediates tumor suppression through contact-mediated growth inhibition [123, 124]. It has been shown that NF2 function or expression is lost in various cancers through mutation or chromosome deletion [125-127]. This is particularly relevant in malignant mesothelioma where 50% of patients have loss or inactivation of Merlin. Merlin deficient cancer cells, including mesothelioma cells are very sensitive to FAK inhibition. A study by Shapiro and coworkers reported that mesothelioma cells showed increased sensitivity to the small molecule FAK inhibitor VS-4718 in vitro as well as in vivo tumor xenograft models. This study clearly demonstrated a synthetic lethal relationship between Merlin loss and FAK inhibitor sensitivity in MPM [128].
FAK INHIBITORS
Several approaches are used to target and inhibit FAK activity in cancer cells. Initial trials included, knockdown of FAK using siRNA, antisense oligonucleotides and adenoviral dominant-negative FAK-CD. These methods induced significant FAK down regulation, inhibited cancer cell proliferation, increased apoptosis and thus decreased tumorigenicity [129-131]. However these approaches have limitations for clinical research because of their toxicity in vivo. This led to development of small molecule inhibitors of FAK. These are divided mainly into two groups. The first group is comprised of inhibitors that target enzymatic or catalytic kinase dependent functions of FAK. The other group consists of the compounds that inhibit kinase independent functions of FAK, such as its protein-protein interactions with other binding partners [53, 132, 133].
FAK Kinase inhibitors
These are ATP analogs that effectively suppress the kinase activity of FAK [134-137]. These inhibitors bind to the residues surrounding ATP- binding pocket of kinases and since this pocket is similar in majority of the kinases, they unfortunately tend to have far off target effects. The most well-known and specific FAK inhibitors are either pyrimidine (NVP-TAE-226, PF-573228, PF-562271 and GSK2256098) or pyridine based (VS-6063, VS-4718 and VS-5095).
FAK Scaffold inhibitors
FAK is known to interact with many proteins such as Paxillin, Src, EGFR, Her-2, MET, PI3K, VEGFR-3 and the scaffolding function of FAK plays an important role in cancer cell signaling. Disrupting the formation of these complexes can inhibit downstream FAK signaling in cancer cells.
All the recent inhibitors are listed in the Table 1.
Table 1: Inhibitors of FAK kinase and scaffold function
Name |
Target |
Specificity |
Cancers targeted |
Clinical Trial |
References |
TAE-226 Novartis |
Kinase inhibitor ATP competitive |
FAK & PYK2 |
Glioma & ovarian |
Preclinical |
136, 143,144 |
PF-573,228 Pfizer |
Kinase inhibitor ATP competitive |
FAK |
Prostate & breast |
Preclinical |
137 |
GSK2256098 GlaxoSmithKline |
Kinase inhibitor ATP competitive |
FAK |
Ovarian & pancreatic |
Phase I |
26 |
NVP-TAC544 |
Kinase inhibitor ATP competitive |
FAK |
N/A |
Preclinical |
26 |
VS-4718 (PND-1186) Verastem |
Kinase inhibitor ATP competitive |
FAK & PYK2 |
Breast & ovarian |
Phase I |
26 |
VS-6062 (PF562271 and PF271) Verastem |
Kinase inhibitor ATP competitive |
FAK & PYK2 |
Breast, prostate, pancreatic, head & neck |
Phase I |
26, 135 |
VS-6063 Verastem |
Kinase inhibitor ATP competitive |
N/A |
Ovarian |
I/Ib and II |
26, 151 |
1H-Pyrrolo(2,3-b) Merk Serono |
Kinase inhibitor Non-ATP competitive |
Hinge region of FAK |
N/A |
Preclinical |
138 |
Compound 1 and 2 Takeda |
Kinase inhibitor Non-ATP competitive |
FAK Y397 site |
N/A |
Preclinical |
138 |
Y15 (Compound 14) Cure FAKtor Pharmaceuticals |
Kinase inhibitor Non-ATP competitive |
FAK Y397 site |
Colon |
Preclinical |
133, 146, 147 |
C4 Cure FAKtor Pharmaceuticals |
Scaffold inhibitor |
FAK /VEGFR pathway |
Pancreatic & breast |
Preclinical |
148 |
R2 (Roslins) Cure FAKtor Pharmaceuticals |
Scaffold inhibitor |
FAK & p53 |
Colon |
Preclinical |
149 |
Y11 Cure FAKtor Pharmaceuticals |
Scaffold inhibitor |
FAK Y397 site |
Colon & Breast |
Preclinical |
26 |
FAK and combinatorial therapy
Multiple signaling pathways such as those mediated through RTKs, FAK, Src, AKT, MAPK and PI3K/mTOR are known to play important roles in tumorigenesis and also contribute to drug resistance. A strategy specifically to overcome resistance of cancer cells to chemotherapy and also to increase the efficacy of drugs is to use a combinatorial approach. A combination of FAK inhibitors along with inhibitors of other signaling molecules has proven more effective than single drug alone [138].
The dual inhibition of FAK with a dominant negative form FAK-CD and EGFR inhibitor AG-1478 or Src inhibitor PP2 was much more effective as compared to FAK inhibition alone. This combination treatment resulted in increased cell detachment, inhibition of AKT/ERK1/2 and Src, and increased apoptosis, as evidenced by increased cleaved caspase 3 and 8 in breast and colon cancer cells [139, 140].
TRAIL is known to induce apoptosis in cancer cells; however its clinical use has been limited due to rapid development of resistance [141]. Dao et al., using PANC-1 cells (TRAIL resistant pancreatic cancer cells), showed that PH11, a novel Focal Adhesion Kinase (FAK) inhibitor in combination with TRAIL rapidly induced apoptosis. PH11 appeared to downregulate c-FLIP via inhibition of FAK and the PI3K/AKT pathways, thereby rendering PANC-1 cells susceptible to TRAIL induced apoptosis [142].
The combination of FAK inhibitor TAE-226 and docetaxel, an antimitotic drug, demonstrated a significant decrease in ovarian tumor growth, increased apoptosis in endothelial cells, reduced microvessel density and prolonged survival [143]. In another study it was shown that TAE-226 also increased radio-sensitivity of head and neck cancer cells [144]. The combination of FAK inhibitor PF-562271 with SU11248, an angiogenesis inhibitor, decreased tumor growth and inhibited angiogenesis of human hepatocellular carcinoma in rat xenograft model [145]. The combination of FAK inhibitor Y15 with Src inhibitor PP2 significantly decreased viability of colon cancer cells along with decrease in Y397 FAK and Y418 Src phosphorylation [146]. Also recently it was shown that the combination of FAK inhibitor Y15 with gemcitabine was more effective in suppressing tumor growth in a pancreatic cancer mouse xenograft model than the use of a single drug alone [147]. The combination of another FAK inhibitor, C4 with doxorubicin was more effective in inhibiting breast cancer xenograft tumor growth and angiogenesis in mice [148]. In another study Y15 was used in combination with temozolomide and was shown to be very effective in blocking U87 glioblastoma xenograft tumor growth model [149]. Roslin (R2), a small molecule that disrupts the interaction of FAK with P53 significantly decreased tumor growth in colon cancer. It also sensitized HCT16 cells to doxorubicin and 5-fluorocil [150].
A recent development in cancer therapeutics further strengthens FAK as a viable cancer therapeutic target. In ovarian cancers resistant to taxane treatment, it was observed using reverse-protein arrays, that levels of YB-1, a RNA binding protein, which regulates transcription, are elevated. AKT mediated phosphorylation of YB-1 promotes resistance; however FAK inhibition prevents this, thereby making these cancers susceptible to taxane treatment [151]. The above studies further reinforce the importance of combination therapy, where FAK inhibitor sensitizes cancer cells to chemotherapy.
CONCLUSIONS AND FUTURE PERSPECTIVES
PDAC is a devastating disease with poor prognosis. Of the known signaling pathways that contribute to PDAC, FAK plays a vital role in signaling pathways mediated through integrins, RTKs, RAS, and TGF β Moreover, it is also likely to suppress p53 expression. We therefore speculate that therapeutic targeting of FAK in PDAC is therefore likely to succeed, particularly when this strategy is combined with other chemotherapeutic treatments. Signaling pathways downstream of activated FAK including paxillin will be important to study in the context of FAK inhibition and other therapeutics to identify novel biomarkers. In the future, emerging technologies may allow for direct therapeutic targeting of paxillin.
Acknowledgments
In part supported by National Cancer Institute of the National Institutes of Health under award number P30CA33572 (City of Hope).
conflicts of interest
None.
REFERENCES
1. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2015. CA. 2015; 65(1):5-29.
2. Hackert T and Buchler MW. Pancreatic cancer: advances in treatment, results and limitations. Digestive diseases. 2013; 31(1):51-56.
3. Phillips PA, Wu MJ, Kumar RK, Doherty E, McCarroll JA, Park S, Pirola RC, Wilson JS and Apte MV. Cell migration: a novel aspect of pancreatic stellate cell biology. Gut. 2003; 52(5):677-682.
4. Bilimoria KY, Bentrem DJ, Ko CY, Ritchey J, Stewart AK, Winchester DP and Talamonti MS. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: report from the National Cancer Database. Cancer. 2007; 110(4):738-744.
5. Mahadevan D and Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Molecular cancer therapeutics. 2007; 6(4):1186-1197.
6. Herreros-Villanueva M, Hijona E, Banales JM, Cosme A and Bujanda L. Alcohol consumption on pancreatic diseases. World journal of gastroenterology. 2013; 19(5):638-647.
7. Fiorino S. HBV and HCV infection and pancreatic ductal adenocarcinoma. Hepatobiliary & pancreatic diseases international. 2013; 12(6):570-574.
8. Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC and Grindey GB. Evaluation of the antitumor activity of gemcitabine (2’,2’-difluoro-2’-deoxycytidine). Cancer Res. 1990; 50(14):4417-4422.
9. Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD and Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997; 15(6):2403-2413.
10. O’Reilly EM and Abou-Alfa GK. Cytotoxic therapy for advanced pancreatic adenocarcinoma. Seminars in oncology. 2007; 34(4):347-353.
11. Strobel O, Hartwig W, Hackert T, Hinz U, Berens V, Grenacher L, Bergmann F, Debus J, Jager D, Buchler M and Werner J. Re-resection for isolated local recurrence of pancreatic cancer is feasible, safe, and associated with encouraging survival. Annals of surgical oncology. 2013; 20(3):964-972.
12. Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A and Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005; 128(4):907-921.
13. Bachem MG, Zhou S, Buck K, Schneiderhan W and Siech M. Pancreatic stellate cells—role in pancreas cancer. Langenbeck’s archives of surgery. 2008; 393(6):891-900.
14. Schneiderhan W, Diaz F, Fundel M, Zhou S, Siech M, Hasel C, Moller P, Gschwend JE, Seufferlein T, Gress T, Adler G and Bachem MG. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. Journal of cell science. 2007; 120(Pt 3):512-519.
15. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB and Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008; 68(3):918-926.
16. Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D and Apte MV. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008; 68(7):2085-2093.
17. Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, Biankin AV, Goldstein D, Pirola RC, Wilson JS and Apte MV. Role of pancreatic stellate cells in pancreatic cancer metastasis. The American journal of pathology. 2010; 177(5):2585-2596.
18. Lu J, Zhou S, Siech M, Habisch H, Seufferlein T and Bachem MG. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. British journal of cancer. 2014; 110(2):409-420.
19. Lugea A, Nan L, French SW, Bezerra JA, Gukovskaya AS and Pandol SJ. Pancreas recovery following cerulein-induced pancreatitis is impaired in plasminogen-deficient mice. Gastroenterology. 2006; 131(3):885-899.
20. Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nature reviews Cancer. 2013; 13(1):66-74.
21. Lerch MM and Mayerle J. [50 years of progress in pathophysiology, diagnosis and treatment of chronic pancreatitis]. Zeitschrift fur Gastroenterologie. 2013; 51(4):358-362.
22. Bartsch DK, Gress TM and Langer P. Familial pancreatic cancer—current knowledge. Nature reviews Gastroenterology & hepatology. 2012; 9(8):445-453.
23. Slater EP, Fendrich V, Strauch K, Rospleszcz S, Ramaswamy A, Matthai E, Chaloupka B, Gress TM, Langer P and Bartsch DK. LCN2 and TIMP1 as Potential Serum Markers for the Early Detection of Familial Pancreatic Cancer. Translational oncology. 2013; 6(2):99-103.
24. Fukushige S, Waldman FM, Kimura M, Abe T, Furukawa T, Sunamura M, Kobari M and Horii A. Frequent gain of copy number on the long arm of chromosome 20 in human pancreatic adenocarcinoma. Genes, chromosomes & cancer. 1997; 19(3):161-169.
25. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321(5897):1801-1806.
26. Sulzmaier FJ, Jean C and Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer. 2014; 14(9):598-610.
27. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR and Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Molecular and cellular biology. 1994; 14(3):1680-1688.
28. Zachary I. Focal adhesion kinase. The international journal of biochemistry & cell biology. 1997; 29(7):929-934.
29. Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB and Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proceedings of the National Academy of Sciences of the United States of America. 1992; 89(11):5192-5196.
30. Miyazaki T, Kato H, Nakajima M, Sohda M, Fukai Y, Masuda N, Manda R, Fukuchi M, Tsukada K and Kuwano H. FAK overexpression is correlated with tumour invasiveness and lymph node metastasis in oesophageal squamous cell carcinoma. British journal of cancer. 2003; 89(1):140-145.
31. Itoh S, Maeda T, Shimada M, Aishima S, Shirabe K, Tanaka S and Maehara Y. Role of expression of focal adhesion kinase in progression of hepatocellular carcinoma. Clin Cancer Res. 2004; 10(8):2812-2817.
32. Furuyama K, Doi R, Mori T, Toyoda E, Ito D, Kami K, Koizumi M, Kida A, Kawaguchi Y and Fujimoto K. Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World journal of surgery. 2006; 30(2):219-226.
33. Chishti AH, Kim AC, Marfatia SM, Lutchman M, Hanspal M, Jindal H, Liu SC, Low PS, Rouleau GA, Mohandas N, Chasis JA, Conboy JG, Gascard P, Takakuwa Y, Huang SC, Benz EJ, Jr., et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends in biochemical sciences. 1998; 23(8):281-282.
34. Girault JA, Labesse G, Mornon JP and Callebaut I. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends in biochemical sciences. 1999; 24(2):54-57.
35. Hanks SK and Polte TR. Signaling through focal adhesion kinase. BioEssays. 1997; 19(2):137-145.
36. Schaller MD and Parsons JT. Focal adhesion kinase and associated proteins. Current opinion in cell biology. 1994; 6(5):705-710.
37. Harte MT, Hildebrand JD, Burnham MR, Bouton AH and Parsons JT. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996; 271(23):13649-13655.
38. Liu Y, Loijens JC, Martin KH, Karginov AV and Parsons JT. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Molecular biology of the cell. 2002; 13(6):2147-2156.
39. Taylor JM, Hildebrand JD, Mack CP, Cox ME and Parsons JT. Characterization of graf, the GTPase-activating protein for rho associated with focal adhesion kinase. Phosphorylation and possible regulation by mitogen-activated protein kinase. J Biol Chem. 1998; 273(14):8063-8070.
40. Corsi JM, Rouer E, Girault JA and Enslen H. Organization and post-transcriptional processing of focal adhesion kinase gene. BMC genomics. 2006; 7:198.
41. Nowakowski J, Cronin CN, McRee DE, Knuth MW, Nelson CG, Pavletich NP, Rogers J, Sang BC, Scheibe DN, Swanson RV and Thompson DA. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure. 2002; 10(12):1659-1667.
42. Salgia R, Avraham S, Pisick E, Li JL, Raja S, Greenfield EA, Sattler M, Avraham H and Griffin JD. The related adhesion focal tyrosine kinase forms a complex with paxillin in hematopoietic cells. J Biol Chem. 1996; 271(49):31222-31226.
43. Brown MC, Perrotta JA and Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. The Journal of cell biology. 1996; 135(4):1109-1123.
44. Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001; 20(44):6459-6472.
45. Schaller MD, Otey CA, Hildebrand JD and Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. The Journal of cell biology. 1995; 130(5):1181-1187.
46. Turner CE. Paxillin and focal adhesion signalling. Nature cell biology. 2000; 2(12):E231-236.
47. Nolan K, Lacoste J and Parsons JT. Regulated expression of focal adhesion kinase-related nonkinase, the autonomously expressed C-terminal domain of focal adhesion kinase. Molecular and cellular biology. 1999; 19(9):6120-6129.
48. Richardson A, Malik RK, Hildebrand JD and Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Molecular and cellular biology. 1997; 17(12):6906-6914.
49. Liu E, Cote JF and Vuori K. Negative regulation of FAK signaling by SOCS proteins. The EMBO journal. 2003; 22(19):5036-5046.
50. Hood JD and Cheresh DA. Role of integrins in cell invasion and migration. Nature reviews Cancer. 2002; 2(2):91-100.
51. Giancotti FG and Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annual review of cell and developmental biology. 2003; 19:173-206.
52. Calalb MB, Polte TR and Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Molecular and cellular biology. 1995; 15(2):954-963.
53. McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG and Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nature reviews Cancer. 2005; 5(7):505-515.
54. Fry LC, Monkemuller K and Malfertheiner P. Molecular markers of pancreatic cancer: development and clinical relevance. Langenbeck’s archives of surgery. 2008; 393(6):883-890.
55. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N and Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988; 53(4):549-554.
56. Malats N, Porta M, Corominas JM, Pinol JL, Rifa J and Real FX. Ki-ras mutations in exocrine pancreatic cancer: association with clinico-pathological characteristics and with tobacco and alcohol consumption. PANK-ras I Project Investigators. International journal of cancer. 1997; 70(6):661-667.
57. Antonyak MA and Cerione RA. Ras and the FAK paradox. Molecular cell. 2009; 35(2):141-142.
58. Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X, Zhou XZ, Aldape K, Cobb MH, Xie K, He J and Lu Z. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Molecular cell. 2009; 35(1):11-25.
59. Zheng D, Kurenova E, Ucar D, Golubovskaya V, Magis A, Ostrov D, Cance WG and Hochwald SN. Targeting of the protein interaction site between FAK and IGF-1R. Biochemical and biophysical research communications. 2009; 388(2):301-305.
60. Glenney JR, Jr. Tyrosine phosphorylation of a 22-kDa protein is correlated with transformation by Rous sarcoma virus. J Biol Chem. 1989; 264(34):20163-20166.
61. Turner CE, Glenney JR, Jr. and Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. The Journal of cell biology. 1990; 111(3):1059-1068.
62. Tachibana K, Sato T, D’Avirro N and Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. The Journal of experimental medicine. 1995; 182(4):1089-1099.
63. Burridge K, Turner CE and Romer LH. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. The Journal of cell biology. 1992; 119(4):893-903.
64. Hu YL, Lu S, Szeto KW, Sun J, Wang Y, Lasheras JC and Chien S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Scientific reports. 2014; 4:6024.
65. Hanks SK, Ryzhova L, Shin NY and Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 2003; 8:d982-996.
66. Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochimica et biophysica acta. 2001; 1540(1):1-21.
67. Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT and Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nature cell biology. 2004; 6(2):154-161.
68. Brown MC, Cary LA, Jamieson JS, Cooper JA and Turner CE. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Molecular biology of the cell. 2005; 16(9):4316-4328.
69. Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA and Schlaepfer DD. Differential regulation of cell motility and invasion by FAK. The Journal of cell biology. 2003; 160(5):753-767.
70. Deramaudt TB, Dujardin D, Noulet F, Martin S, Vauchelles R, Takeda K and Ronde P. Altering FAK-paxillin interactions reduces adhesion, migration and invasion processes. PloS one. 2014; 9(3):e92059.
71. Brown MC and Turner CE. Paxillin: adapting to change. Physiological reviews. 2004; 84(4):1315-1339.
72. Mazaki Y, Hashimoto S and Sabe H. Monocyte cells and cancer cells express novel paxillin isoforms with different binding properties to focal adhesion proteins. J Biol Chem. 1997; 272(11):7437-7444.
73. Salgia R, Li JL, Lo SH, Brunkhorst B, Kansas GS, Sobhany ES, Sun Y, Pisick E, Hallek M, Ernst T and et al. Molecular cloning of human paxillin, a focal adhesion protein phosphorylated by P210BCR/ABL. J Biol Chem. 1995; 270(10):5039-5047.
74. Yuminamochi T, Yatomi Y, Osada M, Ohmori T, Ishii Y, Nakazawa K, Hosogaya S and Ozaki Y. Expression of the LIM proteins paxillin and Hic-5 in human tissues. The journal of histochemistry and cytochemistry. 2003; 51(4):513-521.
75. Kim-Kaneyama JR, Suzuki W, Ichikawa K, Ohki T, Kohno Y, Sata M, Nose K and Shibanuma M. Uni-axial stretching regulates intracellular localization of Hic-5 expressed in smooth-muscle cells in vivo. Journal of cell science. 2005; 118(Pt 5):937-949.
76. Shibanuma M, Mashimo J, Kuroki T and Nose K. Characterization of the TGF beta 1-inducible hic-5 gene that encodes a putative novel zinc finger protein and its possible involvement in cellular senescence. J Biol Chem. 1994; 269(43):26767-26774.
77. Lipsky BP, Beals CR and Staunton DE. Leupaxin is a novel LIM domain protein that forms a complex with PYK2. J Biol Chem. 1998; 273(19):11709-11713.
78. Hagel M, George EL, Kim A, Tamimi R, Opitz SL, Turner CE, Imamoto A and Thomas SM. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Molecular and cellular biology. 2002; 22(3):901-915.
79. Kim-Kaneyama JR, Takeda N, Sasai A, Miyazaki A, Sata M, Hirabayashi T, Shibanuma M, Yamada G and Nose K. Hic-5 deficiency enhances mechanosensitive apoptosis and modulates vascular remodeling. Journal of molecular and cellular cardiology. 2011; 50(1):77-86.
80. Mazaki Y, Uchida H, Hino O, Hashimoto S and Sabe H. Paxillin isoforms in mouse. Lack of the gamma isoform and developmentally specific beta isoform expression. J Biol Chem. 1998; 273(35):22435-22441.
81. Deakin NO, Pignatelli J and Turner CE. Diverse roles for the paxillin family of proteins in cancer. Genes cancer. 2012; 3:362-70. doi: 10.1177/1947601912458582.
82. Madan R, Smolkin MB, Cocker R, Fayyad R and Oktay MH. Focal adhesion proteins as markers of malignant transformation and prognostic indicators in breast carcinoma. Human pathology. 2006; 37(1):9-15.
83. Short SM, Yoder BJ, Tarr SM, Prescott NL, Laniauskas S, Coleman KA, Downs-Kelly E, Pettay JD, Choueiri TK, Crowe JP, Tubbs RR, Budd TG and Hicks DG. The expression of the cytoskeletal focal adhesion protein paxillin in breast cancer correlates with HER2 overexpression and may help predict response to chemotherapy: a retrospective immunohistochemical study. The breast journal. 2007; 13(2):130-139.
84. Jagadeeswaran R, Surawska H, Krishnaswamy S, Janamanchi V, Mackinnon AC, Seiwert TY, Loganathan S, Kanteti R, Reichman T, Nallasura V, Schwartz S, Faoro L, Wang YC, Girard L, Tretiakova MS, Ahmed S, et al. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res. 2008; 68(1):132-142.
85. Mackinnon AC, Tretiakova M, Henderson L, Mehta RG, Yan BC, Joseph L, Krausz T, Husain AN, Reid ME and Salgia R. Paxillin expression and amplification in early lung lesions of high-risk patients, lung adenocarcinoma and metastatic disease. Journal of clinical pathology. 2011; 64(1):16-24.
86. Salgia R, Li JL, Ewaniuk DS, Wang YB, Sattler M, Chen WC, Richards W, Pisick E, Shapiro GI, Rollins BJ, Chen LB, Griffin JD and Sugarbaker DJ. Expression of the focal adhesion protein paxillin in lung cancer and its relation to cell motility. Oncogene. 1999; 18(1):67-77.
87. Sen A, De Castro I, Defranco DB, Deng FM, Melamed J, Kapur P, Raj GV, Rossi R and Hammes SR. Paxillin mediates extranuclear and intranuclear signaling in prostate cancer proliferation. The Journal of clinical investigation. 2012; 122(7):2469-2481.
88. Velasco-Velazquez MA, Salinas-Jazmin N, Mendoza-Patino N and Mandoki JJ. Reduced paxillin expression contributes to the antimetastatic effect of 4-hydroxycoumarin on B16-F10 melanoma cells. Cancer cell international. 2008; 8:8.
89. Yang HJ, Chen JZ, Zhang WL and Ding YQ. Focal adhesion plaque associated cytoskeletons are involved in the invasion and metastasis of human colorectal carcinoma. Cancer investigation. 2010; 28(2):127-134.
90. Radvanyi L, Singh-Sandhu D, Gallichan S, Lovitt C, Pedyczak A, Mallo G, Gish K, Kwok K, Hanna W, Zubovits J, Armes J, Venter D, Hakimi J, Shortreed J, Donovan M, Parrington M, et al. The gene associated with trichorhinophalangeal syndrome in humans is overexpressed in breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(31):11005-11010.
91. Kim MS, Yoo NJ and Lee SH. Absence of paxillin gene mutation in lung cancer and other common solid cancers. Tumori. 2011; 97(2):211-213.
92. Pallier K, Houllier AM, Le Corre D, Cazes A, Laurent-Puig P and Blons H. No somatic genetic change in the paxillin gene in nonsmall-cell lung cancer. Molecular carcinogenesis. 2009; 48(7):581-585.
93. Yu JA, Deakin NO and Turner CE. Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Molecular biology of the cell. 2009; 20(22):4706-4719.
94. Ren Y, Meng S, Mei L, Zhao ZJ, Jove R and Wu J. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem. 2004; 279(9):8497-8505.
95. Tatsumi Y, Cho YY, He Z, Mizuno H, Seok Choi H, Bode AM and Dong Z. Involvement of the paxillin pathway in JB6 Cl41 cell transformation. Cancer Res. 2006; 66(11):5968-5974.
96. Tumbarello DA and Turner CE. Hic-5 contributes to epithelial-mesenchymal transformation through a RhoA/ROCK-dependent pathway. Journal of cellular physiology. 2007; 211(3):736-747.
97. Zhao Y, Zhang X, Guda K, Lawrence E, Sun Q, Watanabe T, Iwakura Y, Asano M, Wei L, Yang Z, Zheng W, Dawson D, Willis J, Markowitz SD, Satake M and Wang Z. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(6):2592-2597.
98. Vadlamudi R, Adam L, Talukder A, Mendelsohn J and Kumar R. Serine phosphorylation of paxillin by heregulin-beta1: role of p38 mitogen activated protein kinase. Oncogene. 1999; 18(51):7253-7264.
99. Deakin NO and Turner CE. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Molecular biology of the cell. 2011; 22(3):327-341.
100. Tong X and Howley PM. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proceedings of the National Academy of Sciences of the United States of America. 1997; 94(9):4412-4417.
101. Tong X, Salgia R, Li JL, Griffin JD and Howley PM. The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J Biol Chem. 1997; 272(52):33373-33376.
102. Vande Pol SB, Brown MC and Turner CE. Association of Bovine Papillomavirus Type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene. 1998; 16(1):43-52.
103. Sachdev S, Bu Y and Gelman IH. Paxillin-Y118 phosphorylation contributes to the control of Src-induced anchorage-independent growth by FAK and adhesion. BMC cancer. 2009; 9:12.
104. Subauste MC, Pertz O, Adamson ED, Turner CE, Junger S and Hahn KM. Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. The Journal of cell biology. 2004; 165(3):371-381.
105. Zouq NK, Keeble JA, Lindsay J, Valentijn AJ, Zhang L, Mills D, Turner CE, Streuli CH and Gilmore AP. FAK engages multiple pathways to maintain survival of fibroblasts and epithelia: differential roles for paxillin and p130Cas. Journal of cell science. 2009; 122(Pt 3):357-367.
106. Frisch SM, Vuori K, Ruoslahti E and Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. The Journal of cell biology. 1996; 134(3):793-799.
107. Lin HM, Lee YJ, Li G, Pestell RG and Kim HR. Bcl-2 induces cyclin D1 promoter activity in human breast epithelial cells independent of cell anchorage. Cell death and differentiation. 2001; 8(1):44-50.
108. Lu W, Wei W, de Bock GH, Zhou H, Li Q and Shen X. The roles of Wnt5a, JNK and paxillin in the occurrence of metastasis of pancreatic adenocarcinoma. International journal of clinical oncology. 2014; 19(6):1011-1019.
109. Wei W, Li H, Li N, Sun H, Li Q and Shen X. WNT5A/JNK signaling regulates pancreatic cancer cells migration by Phosphorylating Paxillin. Pancreatology. 2013; 13(4):384-392.
110. Kawada I, Hasina R, Lennon FE, Bindokas VP, Usatyuk P, Tan YH, Krishnaswamy S, Arif Q, Carey G, Hseu RD, Robinson M, Tretiakova M, Brand TM, Iida M, Ferguson MK, Wheeler DL, et al. Paxillin mutations affect focal adhesions and lead to altered mitochondrial dynamics: relevance to lung cancer. Cancer biology & therapy. 2013; 14(7):679-691.
111. Chen HC and Guan JL. Stimulation of phosphatidylinositol 3’-kinase association with foca adhesion kinase by platelet-derived growth factor. J Biol Chem. 1994; 269(49):31229-31233.
112. Guan JL. Role of focal adhesion kinase in integrin signaling. The international journal of biochemistry & cell biology. 1997; 29(8-9):1085-1096.
113. Bachelot C, Rameh L, Parsons T and Cantley LC. Association of phosphatidylinositol 3-kinase, via the SH2 domains of p85, with focal adhesion kinase in polyoma middle t-transformed fibroblasts. Biochimica et biophysica acta. 1996; 1311(1):45-52.
114. van Nimwegen MJ, Huigsloot M, Camier A, Tijdens IB and van de Water B. Focal adhesion kinase and protein kinase B cooperate to suppress doxorubicin-induced apoptosis of breast tumor cells. Mol Pharmacol. 2006; 70(4):1330-1339.
115. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, Michalski CW, Schlitter AM, Esposito I, Kind AJ, Rad L, Schnieke AE, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer cell. 2013; 23(3):406-420.
116. Pawaskar DK, Straubinger RM, Fetterly GJ, Ma WW and Jusko WJ. Interactions of everolimus and sorafenib in pancreatic cancer cells. The AAPS journal. 2013; 15(1):78-84.
117. Pawaskar DK, Straubinger RM, Fetterly GJ, Hylander BH, Repasky EA, Ma WW and Jusko WJ. Synergistic interactions between sorafenib and everolimus in pancreatic cancer xenografts in mice. Cancer chemotherapy and pharmacology. 2013; 71(5):1231-1240.
118. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR and Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011; 39(Database issue):D945-950.
119. Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, Itadani H, Takahashi-Suzuki I, Fukasawa K, Oki H, Nambu T, Jiang J, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Molecular cancer therapeutics. 2009; 8(11):2992-3000.
120. Golubovskaya VM and Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. International review of cytology. 2007; 263:103-153.
121. Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, Goggins M, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Cameron JL, Olino K, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009; 15(14):4674-4679.
122. Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P, Herman JM, Cameron JL, Yeo CJ, Halushka MK, Eshleman JR, Raben M, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009; 27(11):1806-1813.
123. Bretscher A, Edwards K and Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002; 3(8):586-599.
124. Li W, Cooper J, Karajannis MA and Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 2012; 13(3):204-215.
125. Bianchi AB, Mitsunaga SI, Cheng JQ, Klein WM, Jhanwar SC, Seizinger B, Kley N, Klein-Szanto AJ and Testa JR. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92(24):10854-10858.
126. Cheng JQ, Lee WC, Klein MA, Cheng GZ, Jhanwar SC and Testa JR. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: evidence for a two-hit mechanism of NF2 inactivation. Genes, chromosomes & cancer. 1999; 24(3):238-242.
127. Morrow KA, Das S, Metge BJ, Ye K, Mulekar MS, Tucker JA, Samant RS and Shevde LA. Loss of tumor suppressor Merlin in advanced breast cancer is due to post-translational regulation. J Biol Chem. 2011; 286(46):40376-40385.
128. Shapiro IM, Kolev VN, Vidal CM, Kadariya Y, Ring JE, Wright Q, Weaver DT, Menges C, Padval M, McClatchey AI, Xu Q, Testa JR and Pachter JA. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Science translational medicine. 2014; 6(237):237ra268.
129. Xu LH, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ and Cance WG. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996; 7(4):413-418.
130. Xu LH, Yang X, Bradham CA, Brenner DA, Baldwin AS, Jr., Craven RJ and Cance WG. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol Chem. 2000; 275(39):30597-30604.
131. Golubovskaya VM, Zheng M, Zhang L, Li JL and Cance WG. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC cancer. 2009; 9:280.
132. Cance WG, Kurenova E, Marlowe T and Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Science signaling. 2013; 6(268):pe10.
133. Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D and Cance WG. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. Journal of medicinal chemistry. 2008; 51(23):7405-7416.
134. Parsons JT, Slack-Davis J, Tilghman R and Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008; 14(3):627-632.
135. Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L, Martinez-Alsina L, Lorenzen M, Berliner M, Luzzio M, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008; 68(6):1935-1944.
136. Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS and Rich JN. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Molecular carcinogenesis. 2007; 46(6):488-496.
137. Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG and Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007; 282(20):14845-14852.
138. Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci (Landmark Ed). 2014; 19:687-706.
139. Golubovskaya V, Beviglia L, Xu LH, Earp HS, 3rd, Craven R and Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002; 277(41):38978-38987.
140. Golubovskaya VM, Gross S, Kaur AS, Wilson RI, Xu LH, Yang XH and Cance WG. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Molecular cancer research. 2003; 1(10):755-764.
141. Trivedi R and Mishra DP. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front Oncol. 2015; 5:69.
142. Dao P, Smith N, Scott-Algara D, Garbay C, Herbeuval JP and Chen H. Restoration of TRAIL-induced apoptosis in resistant human pancreatic cancer cells by a novel FAK inhibitor, PH11. Cancer Lett. 2015; 360(1):48-59.
143. Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, Tari AM, Bornmann W, Fernandez A, Lopez-Berestein G and Sood AK. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007; 67(22):10976-10983.
144. Hehlgans S, Lange I, Eke I and Cordes N. 3D cell cultures of human head and neck squamous cell carcinoma cells are radiosensitized by the focal adhesion kinase inhibitor TAE226. Radiotherapy and oncology. 2009; 92(3):371-378.
145. Bagi CM, Christensen J, Cohen DP, Roberts WG, Wilkie D, Swanson T, Tuthill T and Andresen CJ. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer biology & therapy. 2009; 8(9):856-865.
146. Heffler M, Golubovskaya VM, Dunn KM and Cance W. Focal adhesion kinase autophosphorylation inhibition decreases colon cancer cell growth and enhances the efficacy of chemotherapy. Cancer biology & therapy. 2013; 14(8):761-772.
147. Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, Magis A, Ostrov D, Cance WG and Golubovskaya VM. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell cycle. 2009; 8(15):2435-2443.
148. Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA and Cance WG. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. Journal of medicinal chemistry. 2009; 52(15):4716-4724.
149. Golubovskaya VM, Ho B, Zheng M, Magis A, Ostrov D, Morrison C and Cance WG. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC cancer. 2013; 13:342.
150. Golubovskaya VM, Huang G, Ho B, Yemma M, Morrison CD, Lee J, Eliceiri BP and Cance WG. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Molecular cancer therapeutics. 2013; 12(2):162-172.
151. Kang Y, Hu W, Ivan C, Dalton HJ, Miyake T, Pecot CV, Zand B, Liu T, Huang J, Jennings NB, Rupaimoole R, Taylor M, Pradeep S, Wu SY, Lu C, Wen Y, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. Journal of the National Cancer Institute. 2013; 105(19):1485-1495.