
INTRODUCTION
Merkel cell carcinoma is an extremely uncommon, biologically aggressive, cutaneous neuroendocrine cancer [1–4]. It typically presents on sun-exposed skin of elderly men as a rapidly enlarging asymptomatic flesh-colored or blue-red nodule. Local, regional, and distant recurrences are associated with a poor prognostic outcome.
Management for localized disease is surgery: a wide local excision and a sentinel lymph node biopsy. A complete lymph node dissection may follow for patients with a positive sentinel lymph node for cancer. In addition, adjuvant radiation therapy is usually given not only to patients with positive sentinel lymph nodes, but also to patients with Merkel cell carcinoma of the head and neck [1–4].
For patients with metastatic disease, chemotherapy is used. Unfortunately, after two to three cycles of treatment, resistance frequently develops. In addition to radiation therapy [5, 6], immunotherapy (such as systemic pembrolizumab [MK-3475] a humanized anti-PD1 antibody [7]) and targeted molecular therapy are investigational approaches that have been used for metastatic Merkel cell carcinoma [8–11].
Due to the rarity of the disease, data regarding response to therapy are often derived from case reports and retrospective series, rather than prospectively performed clinical trials. Thus, it has been challenging to define the role of chemotherapy in management of advanced Merkel cell carcinoma. Systemic chemotherapies currently used include platinum with or without etoposide, as well as cyclophosphamide, doxorubicin and vincristine [3–5]. Modest responses can be achieved with these cytotoxic agents (median progression-free survival of 3 months). Indeed, there are no drugs approved by the Food and Drug Administration (FDA) specifically for Merkel cell carcinoma.
Importantly, in Merkel cell carcinomas, several molecular abnormalities have been reported [12–30]. These include overexpression of Hedgehog (Hh) signal pathway proteins, telomerase activation (TERT), tumor suppressor anomalies (TP53, RB1 and SUFU), and tyrosine kinase signaling activation (AKT, KIT, PDGFRA, PIK3CA and PTEN). In addition, chromosomal abnormalities [29] and microRNA alterations [30] have been demonstrated in Merkel cell carcinomas.
Clinical trials using a variety of targeted tyrosine kinase inhibitors, either as monotherapy or in combination with chemotherapy or one or more additional tyrosine kinase inhibitors, have been initiated for Merkel cell carcinoma. Although a complete response with imatinib (targeting KIT and PDFGR) has been described [31], a low response rate to the agent was observed in a clinical trial [32]. Similarly, a complete response to pazopanib has been observed in Merkel cell carcinoma resistant to chemotherapy [33]; currently, a phase II trail (NCT01841736) is open to evaluate pazopanib in patients with neuroendocrine tumors including Merkel cell carcinoma. However, for several of the current trials, in which these therapies are being given to unselected patients rather than matched to individuals whose tumors harbored cognate aberrations, the results have yet to be reported. Indeed, we are unaware of any trials in which Merkel cell carcinoma patients Merkel cell carcinoma were selected for the presence of specific aberrations and were treated with appropriated targeting agents.
Given that additional effective treatment strategies are needed, the genomic profiles of Merkel cell carcinomas, as determined by comprehensive genomic profiling (targeted next-generation sequencing (NGS)), were examined and the data analyzed in the context of potential actionability.
RESULTS
Genetic aberrations in Merkel cell carcinomas (Tables 1 and 2, Figure 1)
Table 1: Genomic portfolio in each of 17 patients with Merkel cell carcinoma [a]
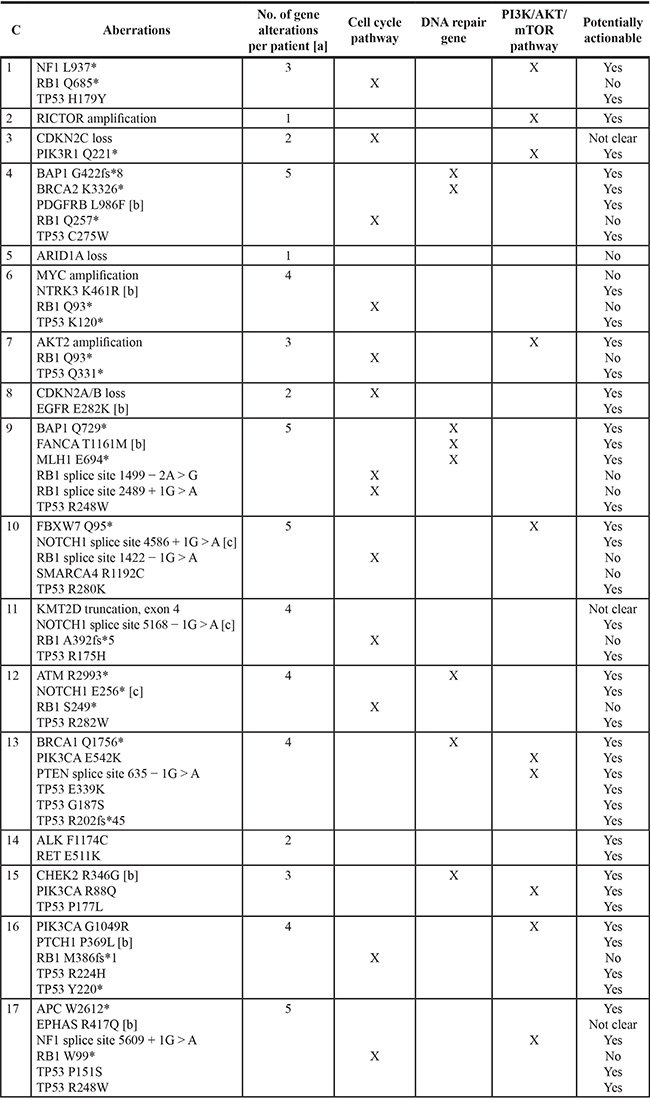
Abbreviations: C, case; No., number.
[a] 4 cases had more than one molecular aberration in the same gene: case 9 [RB1 = 2], case 13 [TP53 = 3], case 16 [TP53 = 2], and case 17 [TP53 = 2].
[b] Aberration is of uncertain clinical significance and relevance of therapeutic strategies is unknown.
[c] Aberration is an inactivating alteration and therapeutic strategies are not expected to be relevant.
Table 2: Summary of genomic alterations in patients with Merkel cell carcinoma
Aberration |
Number of patients |
Percent of patients |
Potentially actionable [a] |
|---|---|---|---|
TP53 |
12 |
71 |
Yes |
RB1 |
10 |
59 |
No |
NOTCH1 |
3 |
18 |
No [b] |
PIK3CA |
3 |
18 |
Yes |
BAP1 |
2 |
12 |
Yes |
BRCA1/2 |
2 |
12 |
Yes |
NF1 |
2 |
12 |
Yes |
AKT2 |
1 |
6 |
Yes |
ALK |
1 |
6 |
Yes |
APC |
1 |
6 |
Yes |
ARIDIA |
1 |
6 |
No |
ATM |
1 |
6 |
Yes |
CDKN2A/B |
1 |
6 |
Yes |
CDKN2C |
1 |
6 |
Not clear [c] |
CHEK2 |
1 |
6 |
Yes |
EGFR |
1 |
6 |
Yes |
EPHAS |
1 |
6 |
Not clear [c] |
FANCA |
1 |
6 |
Yes |
FBXW7 |
1 |
6 |
Yes |
KMT2D |
1 |
6 |
Not clear [c] |
MLH1 |
1 |
6 |
Yes |
MYC |
1 |
6 |
No |
NTRK3 |
1 |
6 |
Yes |
PDGFRB |
1 |
6 |
Yes |
PIK3R1 |
1 |
6 |
Yes |
PTCH1 |
1 |
6 |
Yes |
PTEN |
1 |
6 |
Yes |
RET |
1 |
6 |
Yes |
RICTOR |
1 |
6 |
Yes |
SMARCA4 |
1 |
6 |
No |
[a] Potentially actionable indicates some evidence in the literature that there are drugs that impact the target. This evidence may derive from clinical observations in other tumors or from preclinical evidence.
[b] Activating NOTCH mutations are potentially actionable but the ones in this series were inactivating.
[c] Not clear indicates mixed or inconclusive literature evidence for the potential of available drugs to impact the altered gene product.
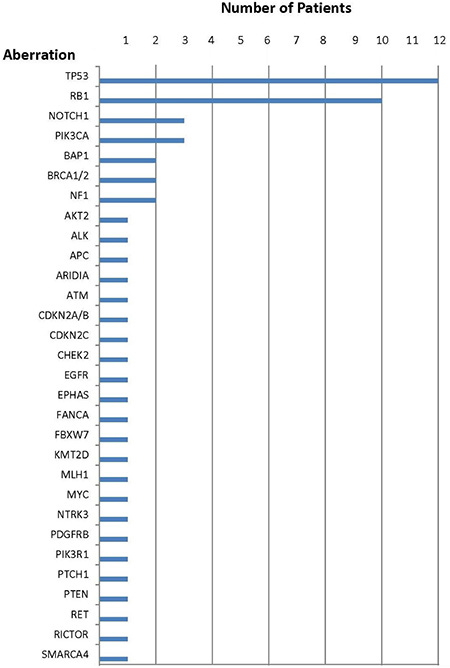
Figure 1: Number of patients with each aberration.
Specific genomic abnormalities were observed in all 17 Merkel cell carcinomas and ranged from one to five alterations per tumor; the median was four. Only two patients (cases 2 and 5) had one aberration and only three patients (cases 1, 7 and 15) had two aberrations. Indeed, more than half of the patients (9/17 [53%]) had four or more genetic anomalies.
The most common anomaly among all Merkel cell carcinomas was in the TP53 gene (12/17 patients [71%]). Abnormalities in the cell cycle pathway (CDKN2A/B, CDKN2C or RB1) were also observed in 71% of cases [12/17]. Aberrations in the PI3K/AKT/mTOR pathway (AKT2, FBXW7, NF1, PIK3CA, PIK3R1, PTEN or RICTOR) were the third most common set of aberrations (9/17 [53%]). Anomalies in DNA repair genes (ATM, BAP1, BRCA1/2, CHEK2, FANCA or MLH1) were seen in 29% (5/17) of patients. Aberrations in either ALK and RET (case 14) or ARIDIA (case 5) were each only noted in 6% (1/17) of patients.
Concurrent anomalies in both the cell cycle and PI3K/AKT/mTOR pathways were noted in 35% (6/17) of patients (cases 1, 3, 7, 10, 16 and 17). Abnormalities of both the cell cycle pathway and DNA repair genes occurred in 18% (3/17) of patients (cases 4, 9 and 12) and aberrations in PI3K/AKT/mTOR pathway and DNA repair genes were discerned in 12% (2/17) of patients (cases 13 and 15).
Number of genomic aberrations and the distinctness of the profiles (Tables 1 and 2)
There were 30 distinct genes involved with 60 distinct molecular alterations. Genomic twins refer to two or more patients that have alterations in the identical genes. Molecular twins refer to two or more patients that have alterations in the same genes and the specific alterations within the gene are also identical. There were no genomic or molecular twins in this study. Therefore, our analysis showed that each of the 17 Merkel cell carcinomas were distinct at the genomic and at the molecular level.
TP53 suppressor gene aberrations (Tables 1 and 2, Figure 1)
Genomic abnormalities in TP53 were found in 71% (12/17) of patients. However, amongst the 16 molecular aberrations, 15 were distinct; two patients (cases 9 and 17) had the same molecular abnormality: R248W. One tumor (case 13) harbored three distinct molecular TP53 abnormalities and two tumors (cases 16 and 17) harbored two distinct molecular aberrations.
Cyclin pathway aberrations (Tables 1 and 2, Figure 1)
Aberrations in cell cycle genes were observed in 71% (12/17) of patients. The most common aberration was in the RB1 tumor suppressor gene; RB1 was mutated in 10 patients. In one patient (case 9), there were two molecular aberrations in RB1. Genomic alterations in either CDKN2A/B (case 8) or CDKN2C (case 3) were each observed in one patient.
PI3K/AKT/mTOR pathway aberrations (Tables 1 and 2, Figure 1)
Genomic abnormalities in the PI3K/AKT/mTOR pathway were noted in 53% (9/17) of patients. There were 10 molecular abnormalities in these nine patients; one patient (case 13) had an aberration in both PIK3R1 and PTEN. The most common genomic aberration was in PIK3CA [found in three patients (cases 13, 15 and 16)]; two patients had a genomic aberration of the NF1 gene.
DNA repair gene aberrations (Tables 1 and 2, Figure 1)
DNA repair gene abnormalities were observed in 29% (5/17) of patients. They included eight molecular abnormalities. Two patients had genomic aberration of BAP1 (cases 4 and 9); two patients had BRCA1/2 alterations (case 13 had a BRCA1 abnormality and case 4 had a BRCA2 abnormality). In two of the five patients, there were abnormalities in multiple DNA repair genes; either BAP1 and BRCA2 (case 4) or BAP1, FANCA, and MLH1 (case 9). Genomic alterations in either ATM (case 12) or CHEK2 (case 15) were each observed in one patient.
Actionable aberrations (Tables 1, 2 and 3)
Table 3: Potential therapies for genomic aberrations in each of 17 patients with Merkel cell carcinoma [34–72] [a]
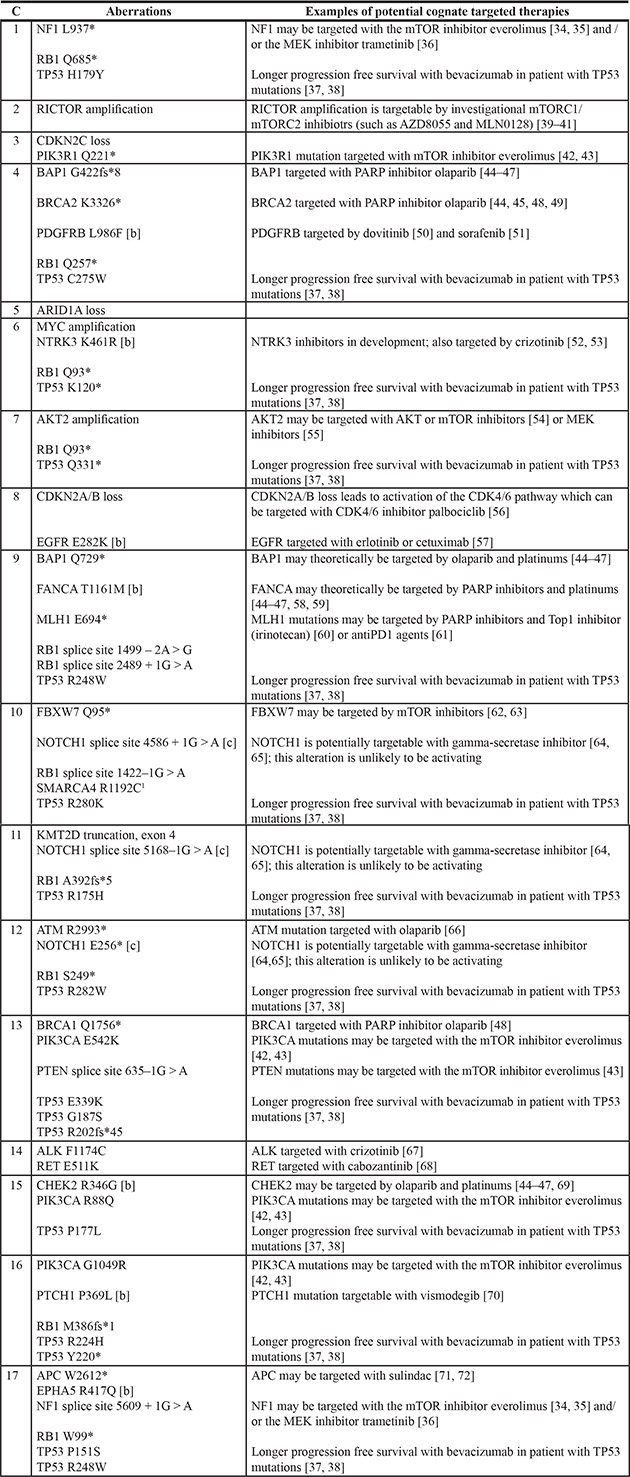
Abbreviations: C, case.
[a] Many of these therapies have not been validated as effective in patients.
[b] Aberration is of uncertain clinical significance and relevance of therapeutic strategies is unknown.
[c] Aberration is an inactivating alteration and therapeutic strategies are not expected to be relevant.
Of the 30 distinct genomic aberrations, 73% (22/30) were theoretically targetable by either an off-label use of an FDA-approved drug (21/30) or an experimental drug in a clinical trial where an off-label use did not exist (1/30).
The vast majority of patients (94%, 16/17) had at least one aberration that was potentially targetable. There were between zero (case 5) and four (cases 4, 9 and 13) actionable genes affected per patient (median, two genes per patient). Potential therapies for the genomic aberrations in each of the 17 patients with Merkel cell carcinoma are summarized in Table 3 [34–72].
DISCUSSION
Merkel cell carcinoma is an ultra-rare neuroendocrine cancer of the skin that most commonly presents in elderly Caucasian men [5, 73]. The pathogenesis is related not only to ultraviolet light exposure, but also to immunosuppression [5, 73]. In addition, the presence of Merkel cell polyomavirus (MCPyV) has been demonstrated in about 45% [73] to 80% [74] of the cases. Gene mutations may have a role in the etiology of Merkel cell carcinoma, particularly in patients whose tumors are Merkel cell polymavirus-negative [75]. A recent study of nine virus-negative patients showed high mutational burden (as compared to that in virus-positive patients), and alterations in TP53, RB1, PIK3CA, HRAS, PRUNE2 and NOTCH (integrative sequencing that included data from whole-exome sequencing and whole-transcriptome sequencing) [13]. Another similar study (N = 619 genes analyzed; 21 virus-negative and 13 virus-positive patients) confirmed high mutation burden and a UV-induced DNA damage signature for virus-negative patients. All viral-negative tumors harbored mutations in RB1, TP53, and a high frequency of mutations in NOTCH1 and FAT1. Additional mutated or amplified cancer genes of potential clinical importance included those in the PI3K or MAPK pathway [14]. Of interest, a subset of virus-negative patients showed high PDL1, suggesting that they might respond to antiPD1 checkpoint inhibitors [15].
The prognosis for patients with Merkel cell carcinoma is poor; more than 33% of patients die from their disease and 50% of patients with advanced tumors live less than 9 months following diagnosis [76]. Of interest in this regard is that exome sequencing of Merkel cell revealed that TP53 was more common in the virus-negative group and predicted a poor survival (5-year survival in TP53 mutant versus wild-type stage I and II disease was 20% vs. 92%, respectively; P = 0.0036) [16]. In general, Merkel cell carcinoma has shown low response rates to chemotherapy [4-6] and to molecularly targeted therapies that are administered without molecular matching [32]. Thus, therapeutic options for Merkel cell carcinoma are limited. In addition, we are not aware of any reports that describe the response in Merkel cell carcinomas when genetic aberrations and therapies were matched. We therefore investigated the genomic landscape of Merkel cell carcinomas by comprehensive genomic profiling and analyzed potential pharmacologic tractability.
The most common genetic aberration among 17 patients with Merkel cell carcinoma was TP53 mutation (12/17 [71%]) (Tables 1 and 2, Figure 1). Our current study observed a markedly higher incidence of TP53 mutations than that noted in previous reports that demonstrated TP53 mutations ranging from 0% to 37% [16, 19, 20, 77, 78]. The TP53 gene is large and there are many areas that can be mutated [79]; our study used comprehensive genomic profiling that evaluated all areas of the gene; in contrast, some of the earlier reported results sequenced discrete regions of the TP53 gene and may not have identified all existing mutations. TP53 gene anomalies are generally seen in virus-negative Merkel cell cancers [16], but a limitation of our study is that viral status was not available. Finally, each of the reports of Merkel cell genomics have small numbers of patients, perhaps accounting in part for the variability in percent positive for TP53 mutations.
TP53 has proven difficult to target. MDM2 inhibitors can theoretically be used in patients with wild-type TP53. Recent data suggest that TP53 mutations result in increased levels of VEGFA, which is the target of bevacizumab [80]. Said et al. showed that bevacizumab-containing regimens were associated with longer progression-free survival when compared to non-bevacizumab-containing regimens in patients with TP53-mutated advanced solid tumors (median 11.0 versus 4.0 months (p < 0.01) [37]. Wee-1 inhibitors, which are in experimental trials, may also target TP53 [81] (Table 3 [34–72]).
The cell cycle pathway (CDKN2A/B, CDKN2C or RB1 genes) was also abnormal in 71% of patients (12/17) with Merkel cell carcinomas (Tables 1 and 2, Figure 1). Aberrations in the cyclin D-cyclin-dependent kinase pathway that regulates the cell cycle restriction point is a common feature of human cancer, contributing to tumor proliferation, genomic instability and chromosomal instability [12, 82, 83]. This pathway can be altered through multiple mechanisms including increased signaling through CDK4 and CDK6 amplification, overexpression of cyclin D1, and loss of inhibitors including CDKN2A and/or CDKN2B [84–87]. Regarding therapeutic implications, the cell cycle pathway is possibly targetable with CDK4/6 inhibitors such as palbociclib [56], and further investigation is warranted (Table 3 [34–72]).
Mutation or loss of RB1, a tumor suppressor gene, also alters the cell cycle pathway. RB1 gene alterations in Merkel cell cancers are associated with virus-negative disease [88]. Merkel cell polyomavirus large-T antigen binds RB1 with high affinity, suppressing its anti-neoplastic function [89]. Aberration of RB1 renders tumors resistant to CDK4/6 inhibitors such as palbociclib [38]. Ten patients in our series had RB1 mutations (Tables 1 and 2, Figure 1).
Importantly, aberrations in the PI3K/AKT/mTOR pathway (AKT2, FBXW7, NF1, PIK3CA, PIK3R1, PTEN or RICTOR) were also commonly seen in Merkel cell carcinomas (9/17 [53%]) (Tables 1 and 2, Figure 1). PIK3CA is a key regulator of cell motility and chemotaxis. Aberrations in PI3KCA usually occur in tumors that do not have Merkel cell polyomavirus [24, 25].
The PI3K/AKT/mTOR pathway can be targeted by PI3K/AKT/mTOR inhibitors such as everolimus and temsirolimus, both of which are FDA-approved mTOR inhibitors [42, 43]. Since Merkel cell carcinomas—regardless of whether they are positive or negative for Merkel cell polyomavirus—show activated PI3K/AKT signaling, PI3K and dual PI3K/mTOR inhibitors may be used as potential targeted therapies, though the literature suggests that for many tumors with pathway activation, they are not effective as single agents [24, 25]. With regards to RICTOR amplification, recent studies have shown that this aberration may be targetable by investigational mTORC1/mTORC2 inhibitors such as AZD8055 and MLN0128 [39–41] (Table 3 [34–72]).
Several investigators have also previously shown that MAP (mitogen-activated protein) kinase-related genes—such as KRAS and BRAF—are more frequently aberrant in the presence of mutant PIK3CA, as compared with wild-type PIK3CA [90]. These genes may confer resistance to PI3K/AKT/mTOR inhibitors. Interestingly, none of our patients had KRAS or BRAF alterations.
Abnormalities in the DNA repair gene pathway (ATM, BAP1, BRCA1/2, CHEK2, FANCA or MLH1) were also observed in 29% of patients (5/17) (Tables 1 and 2, Figure 1). Drugs such as platinums, PARP inhibitors, and possibly immunotherapeutic agents can target DNA repair gene abnormalities (Table 3 [34–72]). Some of these abnormalities (such as BRCA1/2 or ATM) can be germline; germline testing was not conducted in the patients included in this analysis.
Interestingly, 16/17 patients (94%) had potentially actionable aberrations (Table 1). The number of actionable genes affected per patient ranged between zero (case 5) and four (cases 4, 9 and 13), with a median of two per patient. Indeed, the majority of the genomic alterations were theoretically druggable (Tables 1 and 2). Of the 22 (73%) actionable aberrations, 21 were targetable by an FDA-approved drug (off-label) (representing 70% [21/30] of all distinct alterations). An additional one (3% [1/30]) distinct alteration (RICTOR) was targetable by an experimental drug in a clinical trial. As there are no FDA-approved targeted therapies for Merkel cell carcinoma and most conventional chemotherapy has been shown to be associated with poor clinical outcomes: therefore, matched targeted therapies based on molecular profiling merits investigation [91].
Our current study has some limitations. First, it was performed retrospectively with a relatively limited number of patients. Second, molecular analysis was done on archival tumor tissue, which was obtained at different time points in relationship to the clinical history; there was no information regarding the status of the patients, whether the tumors were primary or metastatic, the location of the tumor and the presence or absence of Merkel cell polyomavirus, or cytokeratin-20 positivity (found in most, but not all, Merkel cell cancers) [95]. However, despite these limitations, the genomic characterization of Merkel cell carcinomas has uncovered interesting and possibly clinically relevant results.
In summary, our 17 patients with Merkel cell carcinomas harbored 30 genomic alterations (median = 4 per patient) of which 60 were distinct molecular aberrations. The most common genomic aberrations in patients with Merkel cell carcinoma were in the TP53 gene and the cell cycle pathway (CDKN2A/B, CDKN2C or RB1), followed by the PI3K/AKT/mTOR pathway (AKT2, FBXW7, NF1, PIK3CA, PIK3R1, PTEN or RICTOR) and DNA repair genes (ATM, BAP1, BRCA1/2, CHEK2, FANCA or MLH1). The vast majority of patients (94%) had at least one aberration that was potentially pharmacologically tractable by an FDA-approved drug or an investigational agent in a clinical trial. Indeed, of the 30 distinct genomic aberrations, 22 (73%) were potentially actionable. These observations suggest that matching patients with appropriately targeted agents is feasible and warrants study. Finally, no two patients had an identical molecular portfolio. This result is similar to that reported in metastatic breast cancer, where 131 distinct aberrations in 57 patients with no two patients having the same molecular portfolio were recently described [92–94]. Taken together, these observations suggest that customized targeted combination therapy merits investigation in patients with Merkel cell carcinoma.
MATERIALS AND METHODS
Patients
We investigated the genomic alterations of patients with Merkel cell carcinoma referred to Foundation Medicine (Cambridge, MA) for next-generation sequencing (December 2011 to April 2014 (N = 17)). Here, we report the prevalence and frequencies of these aberrations. This study was performed in accordance with University of California San Diego IRB guidelines for a de-identified database.
Tissue samples and mutational analysis
Available tissues from diagnostic and therapeutic procedures were used to assess molecular aberrations. Samples from formalin-fixed paraffin-embedded tissue were sent for targeted next-generation sequencing at Foundation Medicine (Cambridge, MA). The test sequences the entire coding sequence of 182, or more recently 236, cancer-related genes plus 47 introns from 19 genes often rearranged or altered in cancer to an average depth-of-coverage of greater than 250X (http://foundationone.com/docs/FoundationOne_tech-info-and-overview.pdf).
This method of sequencing allows for detection of copy number alterations, gene rearrangements, and somatic mutations with 99% specificity and > 99% sensitivity for base substitutions at > five mutant allele frequency and > 95% sensitivity for copy number alterations. Foundation Medicine uses a threshold of > eight copies for gene amplification. The submitting physicians provided a diagnosis of the tumor. Next-generation sequencing data were collected and interpreted by N-of-One, Inc. (Lexington, MA; www.n-of-one.com). For the purpose of our analysis, “cell cycle pathway” aberrations included CDKN2A/B, CDKN2C or RB1 alterations. Similarly, “phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway” aberrations included alterations of AKT2, FBXW7, NF1, PIK3CA, PIK3R1, PTEN or RICTOR. “DNA repair gene” abnormalities included alterations in ATM, BAP1, BRCA1/2, CHEK2, FANCA or MLH1. We have evaluated whether certain genomic alterations were actionable or not based on the availability of a drug that is approved or in clinical trials that targets that aberration with low 50% inhibitory concentration (IC50) or an antibody that primarily targets that abnormality.
ACKNOWLEDGMENTS
Philip R. Cohen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
FUNDING
This project was supported in part by the Joan and Irwin Jacobs Fund.
CONFLICTS OF INTEREST
Razelle Kurzrock has research funds from Genentech, Merck Serono, Pfizer, Guardant, Foundation Medicine, and Sequenom, consultant fees from Sequenom, and an ownership interest in Novena, Inc. Sheryl K. Elkin, Brett N. Tomson, Erica Marchlik, and Jennifer Levin Carter are employees of N-of-One, Inc., Lexington, MA. Philip R. Cohen has no conflicts.
REFERENCES
1. Hughes MP, Hardee ME, Cornelius LA, Hutchins LF, Becker JC, Gao L. Merkel cell carcinoma: epidemiology, target, and therapy. Curr Dermatol Rep. 2014; 3:46–53.
2. Marek L, Grzanka A, Chmielowska E, Jankowski M, Schwartz RA, Czajkowski R. Merkel cell carcinoma: an illustrative case and review. Postepy Dermatol Alergol. 2014; 31:325–328.
3. Schwartz JL, Wong SL, McLean SA, Hayman JA, Lao CD, Kozlow JH, Malloy KM, Bradford CR, Frohm ML, Fullen DR, Lowe L, Bichakjian CK. NCCN guidelines implementation in the multidisciplinary Merkel cell carcinoma program at the University of Michigan. J Natl Compr Cancer Netw. 2014; 12:434–441.
4. Raju S, Vazirnia A, Totri C, Hata TR. Treatment of Merkel cell carcinoma of the head and neck: a systematic review. Dermatol Surg. 2014; 40:1273–1283.
5. Prewett SL, Ajithkumar T. Merkel cell carcinoma: current management and controversies. Clinica Oncology. 2015; 27:436–444.
6. Munoz IP, Masferrer JP, Vegas JO, Montalvo MSM, Diaz RJ, Casas AMP. Merkel cell carcinoma from 2008 to 2012: reaching a new level of understanding. Cancer Treatment Reviews. 2013; 39:421–429.
7. Nghiem P, Bhatia S, Daud A, Friedlander P, Kluger H, Kohrt H, Kudchadkar R, Lipson E, Lundgren L, Margolin K, Reddy S, Shantha E, Sharfman W, et al. Activity of PD-1 blockade with pembrolizumab as first systemic therapy in patients with advanced Merkel cell carcinoma [Abstract 22LBA: 18th ECCO-40th ESMA European Cancer Congress, Vienna, Austria, 25-29 September 2015]. European J Cancer. 2015; 51:S720–S721.
8. Moshiri AS, Nghiem P. Milestones in the staging, classification, and biology of Merkel cell carcinoma. J Natl Compr Canc Netw. 2014; 12:1255–1262.
9. Miller NJ, Bhatia S, Parvathaneni U, Iyer JG, Nghiem P. Emerging and mechanism-based therapies for recurrent or metastatic Merkel cell carcinoma. Curr Treat Options Oncol. 2013; 14:249–263.
10. Aldabagh B, Joo J, Yu SS. Merkel cell carcinoma: current status of targeted and future potential for immunotherapies. Semin Cutan Med Surg. 2014; 33:76–82.
11. Tothill R, Estall V, Rischin D. Merkel cell carcinoma: emerging biology, current approaches, and future directions. Am Soc Clin Oncol Educ Book. 2015; 35:e519–e526.
12. Erstad DJ, Cusack JC Jr. Mutational analysis of Merkel cell carcinoma. Cancers. 2014; 6:2116–2136.
13. Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu YM, Dhanasekaran SM, Palanisamy N, Siddiqui J, Cao X, Su F, Wang R, Xiao H, Kunju LP, et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Research. 2015; 75:3720–7.
14. Wong SQ, Waldeck K, Vergara IA, Schröder J, Madore J, Wilmott JS, Colebatch AJ, De Paoli-Iseppi R, Li J, Lupat R, Semple T, Arnau GM, Fellowes A. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Research. 2015; 75:5228–34.
15. Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Molecular Cancer Therapeutics. 2015; 14:847–856.
16. Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, Stafstrom K, Moshiri A, Yelistratova L, Levinsohn J, Chan TA, Nghiem P, Lifton RP, et al. Mutational landscape of MCPyV-positive and MCPyV-negative merkel cell carcinomas with implications for immunotherapy. Oncotarget. 2016; 7:3403–15. doi: 10.18632/oncotarget.6494.
17. Brunner M, Thurnher D Pammer J, Heiduschka G, Petzelbauer P, Schmid C, Schneider S, Erovic BM. Expression of hedgehog signaling molecules in Merkel cell carcinoma. Head Neck. 2010; 32:333–340.
18. Xie H, Liu T, Wang N, Bjornhagen V, Hoog A, Larsson C, Lui WO, Xu D. TERT promoter mutations and gene amplification: promoting mutations and gene amplification: promoting TERT expression in Merkel cell carcinoma. Oncotarget. 2014; 5:10048–10057. doi: 10.18632/oncotarget.2491.
19. Lassacher A, Heitzer E, Kerl H, Wolf P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J Invest Dermatol. 2008; 128:1788–1796.
20. Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res. 2011; 17:4806–4813.
21. Cimino PJ, Robirds DH, Tripp SR, Pfeifer JD, Abel HJ, Duncavage EJ. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Modern Pathol. 2014; 27:1073–1087.
22. Sahi H, Savola S, Sihto H, Koljonen V, Bohling T, Knuutila S. RB1 gene in Merkel cell carcinoma: hypermethylation in aall tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. Acta Pathologica Microbiologica Et Immunologica Scandinavica. 2014; 122:1157–1166.
23. Cohen PR, Kurzrock R. Merkel cell carcinoma with a suppressor of fused (SUFU) mutation: case report and potential therapeutic implications. Dermatol Ther (Heidelb). 2015; 5:129–143.
24. Hafner C, Houben R, Baeurle A, Ritter C, Schrama D, Landthaler M, Becker JC. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS One. 2012; 7:e31255.
25. Nardi V, Song Y, Santamaria-Barria JA, Cosper AK, Lam Z, Faber AC, Boland GM, Yeap BY, Bergethon K, Scialabba VL, Tsao H, Settleman J, Ryan DP, et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res. 2012; 18:1227–11236.
26. Swick BL, Srikantha R, Messingham KN. Specific analysis of KIT and PDGFR-alpha expression and mutational status in Merkel cell carcinoma. J Cutan Pathol. 2013; 40:623–630.
27. Kartha RV, Sundram UN. Silent mutations in kit and pdgfra and coexpression of receptors with SCF and PDGFA in Merkel cell carcinoma: implications for tyrosine kinase-based tumorigenesis. Mod Pathol. 2008; 21:96–104.
28. Van Gele M, Leonard JH, van Roy N, Cook AL, de Paepe A, Speleman F. Frequent allelic loss at 10q23 but low incidence of pten mutation in Merkel cell carcinoma. Int J Cancer. 2001; 92:409–413.
29. Larramendy ML, Koljonen V, Bohling T, Tukiainen E, Knuutila S. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary Merkel cell carcinomas. Mod Pathol. 2004; 17:561–567.
30. Xie H, Lee L, Caramuta S, Hoog A, Browaldh N, Bjornhagen V, Larsson C, Lui WO. MicroRNA expression patterns related to Merkel cell polyomavirus infection in human Merkel cell carcinoma. J Invest Dermatol. 2014; 134:507–517.
31. Loader DE, Feldmann R, Baumgartner M, Breier F, Schrama D, Becker JC, Steiner A. Clinical remission of Merkel cell carcinoma after treatment with imatinib. J Am Acad Dermatol. 2013; 69:e181–e183.
32. National Cancer Institute. A phase II trial of sti-571/imatinib (Gleevec) (Nsc-716051) in neuroendocrine carcinoma of the skin (Merkel cell carcinoma). Available online: http://clinicaltrials.gov/show/nct00068783 (accessed on 24 October 2015).
33. Davids MS, Charlton A, Ng SS, Chang ML, Laubscher K, Dar M, Hodge J, Soong R, Goh BC. Response to a novel multitargeted tyrosine kinase inhibitor pazopanib in metastatic Merkel cell carcinoma. J Clin Oncol. 2009; 27:e97–e100.
34. Lodish MB, Stratakis CA. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Practice & Research Clinical Endocrinology & Metabolism. 2010; 24:439–449.
35. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, et al. RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group: Everolimus for advanced pancrease nonendocrine tumors. N Engl J Med. 2011; 364:514–523.
36. Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, Merghoub T, Ribas A, Chapman PB, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Research. 2014; 74:2340–2350.
37. Said R, Hong DS, Warneke CL, Lee JJ, Wheler JJ, Janku F, Naing A, Falchook GS, Fu S, Piha-paul S, Tsimberidou AM, Kurzrock R. P53 mutations in advanced cancers: clinical characteristics, outcomes, and correlations between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013; 4:705–714. doi: 10.18632/oncotarget.974.
38. Schwaederle M, Lazar V, Validire P, Hansson J, Lacroix L, Soria JC, Pawitan Y, Kurzrock R. VEGF-A expression correlates with TP53 mutations in non-small cell lung cancer: implications for antiangiogenesis therapy. Cancer Res. 2015; 75:1187–1190.
39. Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Letters. 2013; 340:1–8.
40. Pike KG, Malaqu K, Hummersone MG, Menear KA, Duggan HM, Gomez S, Martin NM, Ruston L, Pass SL, Pass M: Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Letters. 2013; 23:1212–1216.
41. Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Mol Cancer Ther. 2015; 14:395–406.
42. Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A Tsimberiduo AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, LU KH, Kurzrock R. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012; 30:777–782.
43. Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberdou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Reports. 2014; 6:377–387.
44. O’Sullivan, Coyne G, Chen A, Kummar S. Delivery on the promise: poly ADP ribose polymerase inhibition as targeted anticancer therapy. Curr Opin Oncol. 2015; 27:475–481.
45. Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol. 2015; 33:1397–1406.
46. Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: a new era of targeted therapy. Maturitas. 2015; 81:5–9.
47. Steffen JD, Brody JR, Armen RS, Pascal JM. Structural implications for selective targeting of PARPs. Front Oncol. 2013; 3:301.
48. Tutt A, Robinson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, et al. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010; 376:235–244.
49. Bao Z, Cao C, Geng X, Tian B, Wu Y, Zhang C, Chen Z, Li W, Shen H, Ying S. Effectiveness and safety of poly (ADP-ribose) polymerase inhibitors in cancer therapy: a systematic review and meta-analysis. Oncotarget. 2015; doi: 10.18632/oncotarget.5367. [Epub ahead of print].
50. Angevin E, Lopez-Martin JA, Linn CC, Gschwend JE, Harzstark A, Castellano D, Soria JC, Sen P, Chang J, Shi M, Kay A, Escudier B. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res. 2013; 19:1257–1268.
51. Corrado A, Ferrari SM, Politti U, Mazzi V, Miccoli M, Materazzi G, Antonelli A, Ulisse S, Fallahi P, Miccoli P. Aggressive thyroid cancer: targeted therapy with sorafenib. Minerva Endocrinol. 2015; Jun 26. [Epub ahead of print].
52. Taipale M, Krykbaeva I, Whitesell L, Santagata S, Zhang J, Liu Q, Gray NS, Lindquist S. Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Nat Biotechnol. 2013; 31:630–637.
53. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371:1005–1015.
54. Ewald F, Grabinski N, Grottke A, Windhorst S, Norz D, Carstensen L, Staufer K, Hofmann BT, Diehl F, David K, Schumacher U, Nashan B, Jucker M. Combined targeting of AKT and mTOR using MK-2206 and RAD001 is synergistic in the treatment of cholangiocarcinoma. Int J Cancer. 2013; 133:2065–2076.
55. Dumble M, Crouthamel MC, Zhang SY, Schaber M, Levy D, Robell K, Liu Q, Figueroa DJ, Minthorn EA, Seefeld MA, Rouse MB, Rabindran SK, Heerding DA, et al. Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with MEK inhibitor. PLoS One. 2014; 9:e100880.
56. Rocca A, Farolfi A, Bravaccini S, Schirone A, Amadori D. Palbociclib [PD 0332991]: targeting the cell cycle machinery in breast cancer. Expert Opin Pharmacol. 2014; 15:407–420.
57. Wheler JJ, Tsimberdou AM, Falchook GS, Zinner RG, Hong DS, Fok JY, Fu S, Piha-Paul SA, Naing A, Kurzrock R. Combining erlotinib and cetuximab is associated with activity in patients with non-small cell lung cancer (including squamous cell carcinomas) and wild-type EGFR or resistant mutations. Molecular Cancer Therapeutics. 2013; 12:2167–2175.
58. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66:8109–8115.
59. Howard SMj, Yanez DA, Stark JM. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genetics. 2015; 11:e1004943.
60. Tentori L, Leonetti C, Muzi A, Dorio AS, Porru M, Dolci S, Campolo F, Vernole P, Lacal PM, Praz F, Graziani G. Influence of MLH1 on colon cancer sensitivity to poly(ADP-ribose) polymerase inhibitor combined with irinotecan. Int J Oncol. 2013; 43:210–218.
61. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015; 372:2509–2520.
62. Koo J, Yue P, Deng X, Khuri FR, Sun SY. mTOR complex 2 stabilizes Mcl-a protein by suppressing its glycogen synthase kinase 3-dependent and SCF-FBXW7-mediated degradation. Mol Cell Biol. 2015; 35:2344–2355.
63. Li S, Oh YT, Yue P, Khuri FR, Sun SY. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene. 2015; Apr 20. [Epub ahead of print].
64. Messersmith WA, Shapiro GI, Clearh JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, English PA, McLachlan KR, Kern KA, et al. A phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-0308401. Clin Cancer Res. 2015; 21:60–67.
65. Dantas-Barbosa C, Bergthold G, Daudigeos-Dubus E, Blackus H, Boylan JF, Ferreira C, Puget S, Abely M, Vassal G, Grill J, Geoerger B. Inhibition of the NOTCH pathway using gamma-secretase inhibitor RO4929097 has limited antitumor activity in established glial tumors. Anticancer Drugs. 2015; 26:272–283.
66. Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, Moss PA, Taylor AM, Stankovic T. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010; 116:4578–4587.
67. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013; 368:2385–2394.
68. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, Pfister DG, Cohen EE, Janisch L, Nauling F, Hong DS, Ng CS, Ye L, et al. Activity of XL184 (Cobazantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011; 29:2660–2666.
69. Euhus D. Genetic testing today. Ann Surg Oncol. 2014; 21:3209–3215.
70. VonHoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey HM, Lum BL, Darbonne WC, et al. Inhibition of the hedgehog pathway in advanced basal cell carcinoma. N Engl J Med. 2009; 361:1164–1172.
71. Femia AP, Soares PV, Luceri C, Lodovici M, Giannini A, Caderni G. Sulindac, 3, 3′-diindolylmethane and curcumin reduce carcinogenesis in the Pirc rat, an Apc-driven model of colon carcinogenesis. BMC Cancer. 2015; 15:611.
72. Bi X, Pohl N, Dong H, Yang W. Selenium and sulindac are synergistic to inhibit intestinal tumorigenesis in Apc/p21 mice. J Hematol Oncol. 2013; 6:8.
73. Andea AA, Patel R, Ponnazhagan S, Isayeva T, Kumar S, Siegal GP. Detection of Merkel cell polyomavirus in formalin-fixed, paraffin-embedded tissue of Merkel cell carcinoma and correlation with prognosis. Rom J Morphol Embryol. 2014; 55:1057–1062.
74. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008; 73:1849–1850.
75. Van Gele M, Kaghad M, Leonard JH, Van Roy N, Naeyaert JM, Geerts ML, Van Belle S, Cocquyt V, Bridge J, Solot R, De Wolf-Peeters C, De Paepe A, Caput D, et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer. 2000; 82:823–826.
76. Heath M, Jaimes N, Lemos B, Mostaghimi A, Wang LC, Penas PF, Nghiem P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008; 58:375–381.
77. Lill C, Schneider S, Item CB, Loewe R, Houben R, Halbauer D, Heiduschka G, Brunner M, Thurnher D. P53 mutation is a rare event in Merkel cell carcinoma of the head and neck. Eur Arch Otorhinolaryngol. 2011; 268:1639–1646.
78. Houben R, Dreher C, Angermeyer S, Borst A, Utikal J, Haferkamp S, Peitsch WK, Schrama D, Hesbacher S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J Invest Dermatol. 2013; 133:2453–2460.
79. Papadaki H, Finkelstein SD, Kounelis S, Bakker A, Swalsky PA, Kapadia SB. The role of p53 mutation and protein expression in primary and recurrent adenoid cystic carcinoma. Hum Pathol. 1996; 27:567–572.
80. Wie W, Wang Y, Yu X, Ye L, Jinag Y, Cheng Y. Expression of TP53, BCL-2, and VEGFA genes in esophagus carcinoma and its biological significance. Med Sci Monit. 2015; 21:3016–3022.
81. Ghiasi N, Habibagahi M, Rosli R, Ghaderi A, Yusoff K, Hosseine A, Abdullah S, Jaberipour M. Tumor suppressive effects of WEE1 gene silencing in breast cancer cells. Asian Pac J Cancer Prev. 2014; 14:6605–6611.
82. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674.
83. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Reviews Cancer. 2009; 9:153–166.
84. Kato S, Schwaederle M, Daniels GA, Piccioni D, Kesari S, Bazhenova L, Shima K, Parker BA, Fanta P, Kurzrock R. Cyclin-dependent kinase pathway aberrations in diverse malignancies: clinical and molecular characteristics. Cell Cycle. 2015; 14:1252–1259.
85. Schwaederle M, Daniels GA, Piccioni DE, Fanta PT, Schwab RB, Shimabukuro KA, Parker BA, Kurzrock R. Cyclin alterations in diverse cancers: outcome and co-amplification network. Oncotarget. 2015; 6:3033–3042. doi: 10.18632/oncotarget.2848.
86. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006; 24:1770–1783.
87. Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clinical Cancer Research. 2013; 19:5320–5328.
88. Veija T, Sarhadi VK, Koljonen V, Bohling T, Knuutila S. Hotspot mutations in polyomavirus positive and negative Merkel cell carcinomas. Cancer Genetics. 2015; S2210-776200234–3.
89. Pipas JM, Levine AJ. Role of T antigen interactions with p53 in tumorigenesis. Semin Cancer Biol. 2001; 11:23–30.
90. Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, Fu S, Luthra R, Garrido-Laguna I, Kurzrock R. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced tumors. PloS One. 2011; 6:e22769.
91. Vidwans SJ, Turski ML, Janku F, Garrido-Laguna I, Munoz J, Schwab R, Subbiah V, Rodon J, Kurzrock R. A framework for genomic biomarker actionabiity and its use in clinical decision making. Oncoscience. 2014; 1:614–623. doi: 10.18632/oncoscience.90.
92. Kurzrock R, Giles FJ. Precision oncology for patients with advanced cancer: the challenges of malignant snowflakes. Cell Cycle. 2015; 14:2219–2221.
93. Wheler J, Lee JJ, Kurzrock R. Unique molecular landscapes in cancer: implications for individualized, curated drug combinations. Cancer Res. 2014; 74:7181–7184.
94. Wheler JJ, Parker BA, Lee JJ, Atkins JT, Janku F, Tsimberidou AM, Zinner R, Subbiah V, Fu S, Schwab R, Moulder S, Valero V, Schwaederle M, et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: implications for current treatment paradigms. Oncotarget. 2014; 5:2349–2354. doi: 10.18632/oncotarget.1946.
95. Harms PW, Collie AMBC, Hovelson DH, Cani AK, Verhaegen ME, Patel RM, Fullen DR, Omata K, Dlugosz AA, Tomlins SA, Billings SD. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod Pathol. 2016; doi: 10.1038/modpathol.2015.154. [Epub ahead of print].