
INTRODUCTION
Juvenile myelomonocytic leukemia (JMML) is a rare aggressive leukemia of early childhood, characterized by excessive proliferation of monocytic and granulocytic cells, which can infiltrate organs, including spleen, liver, gastrointestinal tract, and lung. [1]. Around 90% of newly diagnosed JMML patients are characterized by driver mutations in Ras signaling pathway genes including PTPN11, KRAS, NRAS, CBL and NF1 [2, 3]. Using whole exome sequencing, secondary mutations in SETBP1 and JAK3 were recently found in 17% of children with JMML, conferring poor prognosis to mutated patients [4]. Moreover, for SETBP1 an increased frequency of mutations was discovered (30.3%), considering also subclonal mutations identified using droplet digital polymerase chain reaction (ddPCR) [5].
SETBP1 is a proto-oncogene over-expressed in myeloid progenitor cells leading to enhance self-renewal capacity through direct transcriptional activation of HOXA9 and HOXA10 [6]. SETBP1 is known to bind the SET nuclear oncoprotein [7] with an inhibitory effect on protein phosphatase type 2a (PP2A) [8].
Although the role of SETBP1 in leukemogenesis remains elusive, deleterious mutations of SETBP1 have been reported to occur with high frequency in adults with atypical chronic myeloid leukemia (aCML) [9], chronic myelomonocytic leukemia (CMML) and secondary acute myeloid leukemia (AML) [10–13]; by contrast, mutations have been found only rarely in pediatric myeloid malignancies. JAK3 encodes for a member of the Janus family of tyrosine kinases essential for signal transduction through the JAK/STAT pathway shown to be crucial for the development of lymphoid cells, in particular mature T and Natural Killer (NK) lymphocytes [14]. Functional JAK3 mutations in the PTK (Pseudokinase) domains have been identified in acute megakaryoblastic leukemia (AMKL), T-cell prolymphocytic leukemia and NK T-cell lymphoma [15].
Here, we report the incidence of mutation events in the functional domains of SETBP1 and JAK3 in a cohort of JMML AIEOP (Italian Pediatric Hematology and Oncology Association) patients, together with data pointing to a propagating capacity in mutated clones.
RESULTS AND DISCUSSION
Known driver mutations in the Ras signaling pathway were found in 84% of 70 patients analyzed in this study; 36% were mutated in RAS (NRAS or KRAS), 43% in PTPN11, 4.2% in CBL and one patient had a diagnosis of NF1 (Neurofibromatosis syndrome type1), whereas 11% of patients did not harbor any mutations of the aforementioned genes (Figure 1A).
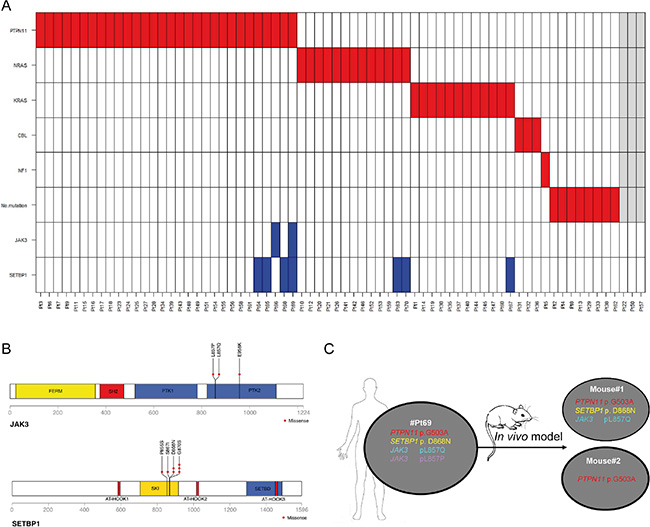
Figure 1: (A) Mutation profile of 70 JMML patients. Sixty-seven patients were screened for PTPN11, NRAS, KRAS, CBL mutations, clinical signs of NF1 and secondary SETBP1 and JAK3 mutations. Asterisk indicates the presence of a heterozygous CBL mutation. In grey are 3 patients for which the driver mutation status is not known but were included in the SETBP1 and JAK3 mutation screening. (B) Distribution of missense alterations at the functional domains of JAK3 and SETBP1 proteins. Altered amino-acids identified in our cohort are highlighted: plus sign in JAK3 p959 amino-acid indicates that the amino-acid was altered only at the genomic level; red arrows point to the novel p.P855S and p.S867I mutations in the SKI domain of SETBP1. (C) Scheme of the in vivo assay indicating the patient sample xenografted. SH2: Src homology 2 domain; PTK1 and PTK2: pseudo-kinase domain; SKI: v-ski sarcoma viral oncogene homolog domain; SETBD: SET-binding domain.
Mutation hotspot regions covering the regions encoding for the SKI homologous domain of SETBP1 and for the PTK domain of JAK3 were included in the mutation analysis (Figure 1B). Seven out of 70 patients (10%) carried SETBP1 mutations and 2 out of 70 (2.8%) mutations in JAK3. One patient harbored a single JAK3 mutation and the other one carried mutations in both SETBP1 and JAK3 (Figure 1A) in two different JAK3 mutated clones (p.L857Q and p.L857P).
The two JAK3 mutations, both predicted to be deleterious by SIFT and PolyPhen bioinformatics tools, were found in distinct sub-clones. Among SETBP1 mutations, two new mutations in the SKI domain (p.P855S, p.S867I), not previously described, were identified. These new mutations cluster in the evolutionary, highly conserved region that bears also the known mutations of SETBP1 and were predicted to be deleterious and damaging for the protein function by SIFT and PolyPhen-2 (Figure 1B, Supplementary Table 1). The new mutations cluster in the same SKI-homologous domain of SETBP1 protein as previously reported mutations, supposedly altering the protein function by decreasing its stability [9].
All mutations identified here were detected at genomic and transcriptome level, except for the mutation at JAK3 p.E958K. Indeed, #Pt66 showed a genomic mutation and only wild-type expression of JAK3 at diagnosis (Supplementary Figure S1). Moreover, the BM sample at relapse of this patient did not show any sign of the genomic JAK3 alteration, indicating lack of clonal propagation of the JAK3 mutated clone (Supplementary Figure S1). The wild-type JAK3 expression at diagnosis and the absence of the mutation at relapse suggest that a functional role at the protein level cannot be attributed to this mutation. These observations indicate that, at least in our cohort of patients, the role of mutated JAK3 in JMML is very limited.
In our cohort of JMML patients, secondary mutations of SETBP1 and JAK3 were exclusively found in association with mutated PTPN11 or RAS (p = 0.0027 and p = 0.002, respectively) (Table 1) and correlated with older age (median 56.8 months vs 23.67 months, p = 0.037, Table 1). This observation is in line with a general age-related accumulation of mutations and with the low average number of mutations per sample in JMML patients, compared to those reported in other human cancers, being JMML a disease of young patients [4].
Table 1: Clinical and genetic characteristics of the patients included in the study
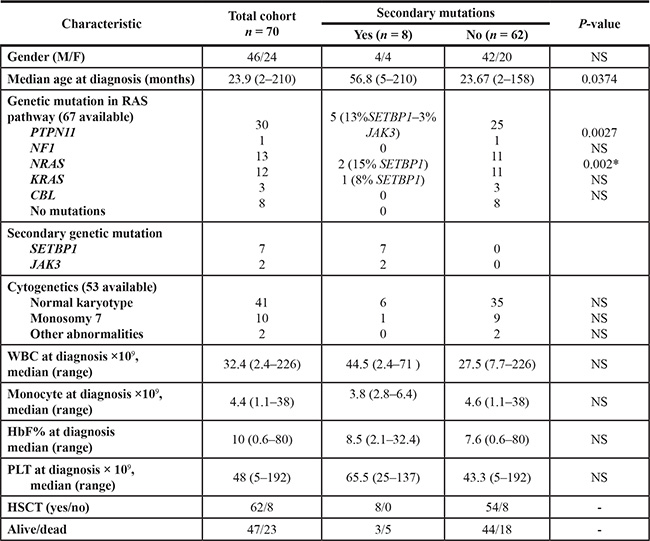
To compare the frequency of mutations with that of other clinical and biological parameters, categorical variables were analyzed using Fisher’s exact test and continuous variables with Mann-Whitney U test. HbF: hemoglobin F; HSCT hematopoietic stem cells transplantation; PLT platelets; WBC white blood cells. *p-value was calculated considering NRAS and KRAS as a single variable.
White blood count (WBC), monocyte count, platelet count and fetal hemoglobin levels (HbF) did not correlate with the presence of secondary mutations in univariate analysis (Table 1). Survival analysis showed a 10-year OS probability of 0% for patients with secondary mutations and of 60% (SE = 7.5) for patients without mutations in SETBP1 and JAK3 (p = 0.3) (Supplementary Figure S5).
To evaluate the functional importance of co-occurring mutations in JMML, in particular the PTPN11 and SETBP1 mutations, we tested them in a colony assay as previously reported [16]. In this assay, patient cells (n = 2) were differentiated in vitro into distinct lineages and subsequently tested for the permissiveness of these two mutations. All colonies of immature (LTC-IC) and more mature (BFU-E, CFU-GM and B- and T-lineage) hematopoietic cell types were positive for the presence of both mutations, as revealed by Sanger sequencing except for the colonies of PHA-activated T-cells. Like PTPN11, also SETBP1 mutations are apparently a hit in hematopoietic progenitor cells even if the SETBP1 mutation is secondary to the PTPN11 mutation (Supplementary Table 3).
To assess the in vivo propagating capacity of cells harboring JAK3 and SETBP1 mutations, mononuclear cells isolated from the spleen of #Pt69 harboring PTPN11p.G503A, SETBP1p.D868N and JAK3p.L857Q, p.L857P mutations were intravenously transplanted (107 per transplant) in two NSG (NOD SCID gamma) mice (Figure 1C). Nine months after transplantation, mice were sacrificed and both bone marrow (BM) and spleen cells were analyzed by flow-cytometry for huCD45/huCD33 markers (being positive at diagnosis). By this method, engraftment of huCD45/huCD33 cells was observed in the BM of mouse #1, whereas flow-cytometry did not detect huCD45/huCD33 cells neither in the spleen nor in BM of mouse #2 (Supplementary Figure S2). Ultra-deep 454 sequencing analysis (Roche Applied Science, Penzberg, Germany), using human-PTPN11 specific primers, revealed the presence of the mutated and wild type PTPN11 alleles in both BM and spleen of mouse#1, as well as in DNA from BM of mouse#2. DNA of the spleen of mouse#2 was not available. Mutant allele frequencies (MAF) of PTPN11 were 50%, corresponding to heterozygous mutations in all cells (Supplementary Figure S3). Mutation analysis of JAK3 and SETBP1, however, showed a different mutational pattern. Only 2 out of 3 mutations (the SETBP1 mutation and only one of the 2 JAK3 mutations) were identified in the BM and spleen of mouse #1, whereas the clone harboring the JAK3p.L857P mutation was undetectable, suggesting a distinctive propagation capacity of mutated JAK3 clones (Supplementary Figure S3). In the DNA of mouse#2 with positivity for the PTPN11 mutation, JAK3 and SETBP1 mutations were not detected. The latter is suggestive of a reduced propagating capacity of cells lacking the secondary mutations, although the absence of BM engraftment in mouse#2 may also be ascribed to biological variability of the model (Supplementary Figure S3). In line with increased propagating capacity of clones with additional mutations, two recent papers by Stieglitz et al., [17] and Caye et al., [18] reported an association with worse outcome of JMML patients carrying > 1 mutation (secondary alterations). Our intriguing results of xenotransplanted JMML specimens are, however limited to a small number of mice, a problem frequently faced in studies of JMML patients where access to large biological samples is often a limiting factor.
Increased propagating capacity of clones harboring SETBP1 mutations was suggested to involve transcriptional activation of the HOXA9 [6]. In line with this notion, we found that JMML patients carrying a SETBP1 mutation had increased expression of HOXA9 as compared with patients harboring the wild-type protein (Supplementary Figure S4). Moreover, we found in mutated patients up-regulation of the RNA-binding protein encoded by RPBMS (Supplementary Figure S4), which physically interacts with SMAD proteins, key mediators of TGF-β signaling [19]. The over-expression of RPBMS enhances Smad-dependent transcriptional activity in a TGF-β-dependent manner, causing an alteration in the TGF-β pathway in patients with SETBP1 mutations, as reported for aCML patients [9]; further studies are needed to understand the physiological activity of SETBP1 in JMML patients.
In conclusion, this study led to the identification of secondary mutations in SETBP1 (10%) and JAK3 (2, 8%) in a cohort of patients with JMML already present at diagnosis. The frequency of mutated SETBP1 was slightly higher in the AIEOP cohort (10%) compared to the cohorts from Japan (6%) [12] and a COG study (7%) [5]. In the latter study, using ddPCR the frequency of SETBP1 mutations increases 4-fold (30%) comprising a high number of patients with mutated sub-clones. In the AIEOP cohort, we identified two new mutations in the SETBP1 SKI domain.
The frequency of mutated JAK3 was higher among JMML patients from Japan (10%) [12] compared with that (2.6%) detected in the AIEOP cohort. Similarly to the SETBP1 mutation, also the frequency of JAK3 mutations at diagnosis of JMML might increase using ddPCR, this technique being capable to detect also minor mutated sub-clones. The only transcribed JAK3 mutation found in our study was tertiary to PTPN11 and SETBP1. A prerequisite for the former mutations of JAK3 to occur is, however, excluded, since in the study from Japan JAK3 mutations occur also in combination with mutated RAS [12]. Our findings provide evidence for the secondary nature of SETBP1 and JAK3 mutations, although SETBP1 mutations occurred in early cancer-initiating precursor cells, and increased propagating capacity of mutated clones maybe due to their role as transcription regulator. The effective role of these mutations in particular of JAK3 in the pathogenesis of JMML remains to be elucidated.
MATERIALS AND METHODS
Patients
Seventy patients enrolled in the EWOG-MDS 2006 study, with a diagnosis of JMML were included in this study (Supplementary Table 1). Written informed consent was obtained from patients’ parents or legal guardians before sample collection in accordance with the Declaration of Helsinki.
Diagnosis of JMML was based on the standard criteria proposed by the JMML Working Group during the second International JMML Symposium [20]. Presence of mutations of PTPN11, RAS and CBL was determined as part of the diagnostic work-up of all patients included in the study.
Sequencing
Genomic DNA was extracted from total BM cells of patients at diagnosis of JMML using Gentra Puregene DNA Purification Kit (Qiagen, Monza, Italy) following the manufacturer’s protocol.
All samples were amplified and sequenced with primers listed in Supplementary Table 2, covering the entire regions encoding for the SKI homologous domain of SETBP1 and for the PTK domain of the JAK3 gene, where the majority of mutations for both genes have been found. PCR amplification was performed using 50 ng of genomic DNA as template followed by bidirectional Sanger sequencing.
Total RNA was extracted using Trizol reagents (Invitrogen-Life Technologies Karlsruhe, Germany) and was reverse transcribed with SuperScript II system (Invitrogen-Life Technologies) using random hexamer primers, according to the manufacturer’s instructions. Target sequences for SETBP1 and JAK3 were PCR amplified and sequenced by Sanger sequencing.
Xenograft mouse model
Mononuclear cells (1 × 107 per transplant) from the spleen of a JMML patient carrying a PTPN11 driver mutation who underwent splenectomy were intravenously injected into NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/Sz) mice. Animal experiments were approved by the appropriate authorities. After transplantation, mice were regularly examined for human cell load by flow cytometry gating for huCD45 and huCD33. After 9 months, mice were sacrificed and cells were harvested from BM and spleen. DNA was analyzed for PTPN11 mutations by ultra-deep sequencing with 454 GS Junior System using human PTPN11 specific fusion primers with adaptor and MID as requested by the manufacturer (Roche Applied Science, Penzberg, Germany).
Colony assay
Epstein–Barr virus-induced B-lymphoblastoid cell line (B-LCL), phytohemagglutinin (PHA)-activated T-lymphoblastoid cells (PHA-activated T cells), erythroid burst-forming units (BFU-E), CFU-GM, and LTC-IC colonies were obtained as previously reported [16].
Statistical analysis
To compare the frequency of mutations with that of other clinical and biological parameters, categorical variables were analyzed using Fisher’s exact test, while continuous variables were analyzed with Mann-Whitney U test. Probabilities of overall survival (OS) were calculated by the Kaplan-Meier method and compared with the Log-Rank test using Bioconductor packages (www.r-project.org).
Gene expression analysis
Total RNA was extracted using Trizol, and quality was checked by the use of Agilent 2100 Bioanalyzer. Microarray data were obtained using 3′ IVT Express Kit and HG-U133 Plus 2.0 array (Affymetrix, Santa Clara, CA, USA). Gene expression data of this study were part of a previous study deposited in GSE14858 [21]. For this study we selected samples that had been screened for SETBP1 mutations; p-values were determined using Welch’s t-test.
Authors’ contributions
Contributions: SB and PdF: designed the study and performed the experiments; LHM: performed xenograft assays; FV and RM: provided patient clinical data and performed survival analysis; SB, FL and GtK: wrote the manuscript; MZ: provided patient samples; GB, FL, CD and GtK: supervised the study; all authors reviewed the manuscript and contributed to specific sections.
GRANT SUPPORT
This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) (SB and FL), Cariparo (GteK and SB) and Fondazione ‘Sofia Luce Rebuffat’ (MZ).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. British journal of haematology. 2008; 140:610–624.
2. Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, Mehta P, Schmid I, Kropshofer G, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nature genetics. 2010; 42:794–800.
3. Loh ML. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. British journal of haematology. 2011; 152:677–687.
4. Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, Kon A, Sanada M, Chiba K, Tanaka H, Makishima H, Wang X, Xu Y, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nature genetics. 2013; 45:937–941.
5. Stieglitz E, Troup CB, Gelston LC, Haliburton J, Chow ED, Yu KB, Akutagawa J, Taylor-Weiner AN, Liu YL, Wang YD, Beckman K, Emanuel PD, Braun BS, et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood. 2015; 125:516–24.
6. Oakley K, Han Y, Vishwakarma BA, Chu S, Bhatia R, Gudmundsson KO, Keller J, Chen X, Vasko V, Jenkins NA, Copeland NG, Du Y. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012; 119:6099–6108.
7. Minakuchi M, Kakazu N, Gorrin-Rivas MJ, Abe T, Copeland TD, Ueda K, Adachi Y. Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia-associated protein SET. European journal of biochemistry. 2001; 268:1340–1351.
8. Trimarchi T, Ntziachristos P, Aifantis I. A new player SETs in myeloid malignancy. Nature genetics. 2013; 45:846–847.
9. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, Antolini L, Mologni L, Donadoni C, Papaemmanuil E, Schnittger S, Kim DW, Boultwood J, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nature genetics. 2013; 45:18–24.
10. Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, Gelsi-Boyer V, de Botton S, Vey N, Preudhomme C, Clavert A, Delabesse E, Park S, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013; 27:1401–1403.
11. Thol F, Suchanek KJ, Koenecke C, Stadler M, Platzbecker U, Thiede C, Schroeder T, Kobbe G, Kade S, Loffeld P, Banihosseini S, Bug G, Ottmann O, et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia. 2013; 27:2072–2075.
12. Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, Ng KP, Gudmundsson KO, Vishwakarma BA, Jerez A, Gomez-Segui I, Takahashi M, Shiraishi Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nature genetics. 2013; 45:942–946.
13. Inoue D, Kitaura J, Matsui H, Hou HA, Chou WC, Nagamachi A, Kawabata KC, Togami K, Nagase R, Horikawa S, Saika M, Micol JB, Hayashi Y, et al. SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia. 2015; 29:847–57.
14. Bergmann AK, Schneppenheim S, Seifert M, Betts MJ, Haake A, Lopez C, Maria Murga Penas E, Vater I, Jayne S, Dyer MJ, Schrappe M, Duhrsen U, Ammerpohl O, et al. Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes, chromosomes and cancer. 2014; 53:309–316.
15. Degryse S, de Bock CE, Cox L, Demeyer S, Gielen O, Mentens N, Jacobs K, Geerdens E, Gianfelici V, Hulselmans G, Fiers M, Aerts S, Meijerink JP, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014; 124:3092–3100.
16. De Filippi P, Zecca M, Novara F, Lisini D, Maserati E, Pasquali F, Rosti V, Carlo-Stella C, Zavras N, Cagioni C, Zuffardi O, Pagliara D, Danesino C, et al. The strange case of the lost NRAS mutation in a child with juvenile myelomonocytic leukemia. Pediatr Blood Cancer. 2012; 59:580–2.
17. Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, Esquivel E, Yu A, Seepo S, Olsen SR, Rosenberg M, Archambeault SL, Abusin G, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nature genetics. 2015; 47:1326–1333.
18. Caye A, Strullu M, Guidez F, Cassinat B, Gazal S, Fenneteau O, Lainey E, Nouri K, Nakhaei-Rad S, Dvorsky R, Lachenaud J, Pereira S, Vivent J, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nature genetics. 2015; 47:1334–1340.
19. Sun Y, Ding L, Zhang H, Han J, Yang X, Yan J, Zhu Y, Li J, Song H, Ye Q. Potentiation of Smad-mediated transcriptional activation by the RNA-binding protein RBPMS. Nucleic acids research. 2006; 34:6314–6326.
20. Chan RJ, Cooper T, Kratz CP, Weiss B, Loh ML. Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leukemia research. 2009; 33:355–362.
21. Bresolin S, Zecca M, Flotho C, Trentin L, Zangrando A, Sainati L, Stary J, de Moerloose B, Hasle H, Niemeyer CM, Te Kronnie G, Locatelli F, Basso G. Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. Journal of clinical oncology. 2010; 28:1919–1927.