
INTRODUCTION
Metastatic disease is responsible for over 90% of cancer-related deaths [1]. Due to a growing insight in the molecular mechanisms driving cancer evolution and identification of specific molecular aberrations involved, an increasing number of patients is now considered candidate for treatment with so called targeted agents [2, 3]. However, when it comes to therapy decision making, intra-patient heterogeneity should be taken into account. Here we discuss the molecular heterogeneity within the circulating tumor cell (CTC) compartment in various tumor types. Furthermore, we review the causes and consequences of this heterogeneity and the clinical perspective.
Intra-tumor heterogeneity
Advances in DNA sequencing techniques and comparison of tumor samples obtained from different sites and at different time points, have revealed an extensive view on clonal evolution and intra tumor heterogeneity (ITH). During tumor development, cancer cells acquire various aberrations, including both passenger (neutral) and driver (advantageous) mutations. Due to selection and clonal expansion, multiple genetically distinct subclones can emerge that often evolve following a pattern of branched evolution, which has been described for various solid tumor types [4–15]. This branched evolution comprises multiple subclones that have a phenotypic advantage within a particular environment and evolve simultaneously resulting in ITH, whereas a linear evolutionary pattern describes a random genetic drift where fitter clones outgrow ancestral clones, resulting in a relatively homogeneous tumor at any given moment [16]. Exome sequencing of multiple tumor foci from clear-cell renal carcinomas revealed that only one-third of the identified driver aberrations were present in every region analyzed from an individual tumor, suggesting these to be early founder aberrations. In contrast, 71% of driver mutations were heterogeneous between tumor regions, although appearing clonally dominant within individual regions, showing branched evolution with spatially separated dominant subclones [6].
During the development of metastatic disease, tumor cells shed from the primary tumor are able to travel to distant organ sites to seed metastatic tumors [17]. Moreover, in breast, prostate, and pancreatic cancers, it has been shown that these cells disseminate long before metastatic colonization becomes clinical evident [18, 19]. Both early and late dissemination, as well as polyclonal and bidirectional seeding between different tumor sites, and parallel evolution have been described [20, 21]. Hence, different tumor sites will consist of unique evolutionary landscapes, leading to inter-metastasis heterogeneity [12, 21–23].
Although clonal diversity can be resolved by spatial sampling [7] in combination with deep-sequencing of tumor tissue to determine (sub)clonality of certain mutations [9–11, 24, 25], the field is shifting towards single cell sequencing (SCS) studies to shed light on this heterogeneity. SCS allows to study rare tumor cell populations and clonal expansion, and is already widely used in hematopoietic cancers, including Acute Myeloid Leukemia [26, 27]. However, in solid tumors, patients often exhibit multiple lesions composed of genetically diverse subclones that evolve in parallel over time [28, 29], hampering the evaluation of targetable aberrations in a patient’s metastatic disease [22, 30, 31]. Hence, single tumor biopsies fail to represent the clonal landscape of the overall tumor burden. Moreover, changing biology and resistance patterns, influenced by prior therapies, stresses the need for repeated sampling of a patients tumor burden, to expose the molecular landscape at various moments in time [23, 32].
Circulating tumor cells
CTCs are shed into the peripheral blood from various tumor deposits and represent the actual tumor mass as was demonstrated by comparative analysis of CTCs, primary tumors, and metastases in various tumor types [33–37]. CTC capturing systems have revealed that aggressive tumors release thousands of cancer cells into the circulation each day [38–41], although most CTCs only persist for a short time in the circulation, with an estimated half-life between 1 and 24 hours [38, 42, 43]. It is assumed, however, that CTCs with an intermediate phenotype between epithelial and mesenchymal have the highest plasticity and can survive in the circulation [44–46]. Although CTCs are a frequent phenomenon in cancer, only a small fraction (< 0.01%) eventually succeed in forming metastasis [47, 48]. This was further demonstrated with the identification of specific subsets of CTCs with tumor-initiating capacity [39, 40, 49, 50].
In general, CTCs are relatively rare, representing only one in more than a million blood cells [40]. Still, CTC count of patients with metastatic cancer is a strong prognostic factor for overall survival in several tumor types [51–60]. Moreover, changes in CTC counts during treatment are used as a marker for therapy response [42, 55, 61–64]. Genotyping of circulating tumor (ct)DNA, derived from tumor deposits and lysed CTCs, also has the potential to serve as a marker for tumor burden, therapy response, and even therapy resistance patterns, when followed longitudinally [32, 65–68]. Moreover, mutation levels in plasma can reflect the multifocal clonal hierarchy of tissue biopsies from a patient with metastatic breast cancer during therapy [23]. Compared to CTCs, ctDNA is easier and less laborious to obtain. Nonetheless, CTCs represent pure and intact tumor cells. Molecular analysis on DNA, RNA, and protein level [33, 69], as well as functional cellular characteristics can only be interrogated in CTCs [39]. In addition, molecular analysis of CTCs enables researchers to detect the presence of multiple mutations within the same cell, in order to decipher tumor heterogeneity and map clonal evolution. When combining genomic and transcriptomic evaluation of CTCs, a potential linkage between mutational status and pathway activation can be observed [70].
CTCs can be analyzed both as pure cells as well as enriched fractions. Mutation detection of DNA extracted from CTC-enriched samples demonstrated activating mutations in the EGFR, KRAS, and AR genes in patients suffering from lung cancer, colorectal cancer (CRC), and castration-resistant prostate cancer (CRPC) respectively [65, 71, 72]. Additionally, RNA analysis of enriched CTC fractions have been performed using reverse transcription PCR (RT-PCR) amplification of tumor-specific transcripts, such as AR splice variant 7 in CRPC, and translocations like EML4–ALK in lung cancer and TMPRSS2–ERG in prostate cancer [42, 73–75]. However, sequencing of enriched fractions iscomplicated by low levels of tumor-specific templates and contamination by abundant leukocyte-derived sequences, limiting the sensitivity and specificity [76, 77]. Advances in next generation sequencing (NGS) strategies and computational analyses help resolve this challenge. Nevertheless, single CTC sequencing strategies can provide a direct insight into CTC heterogeneity by identifying co-existing mutations within a cell. Heitzer and colleagues, profiled individual CTCs isolated from patients with metastatic CRC, using array-Comparative Genomic Hybridization (CGH) and targeted panel sequencing of 68 genes. Various genomic aberrations in CTCs were found, indicative for their subclonal origin from specific areas of the original tumor [33].
Overall, cancer presents a problem of continuous spatial and temporal complexity, particularly due to selection pressures such as anti-cancer drugs, that may promote dominance of previously minor or dormant lineages [78]. It is important to note that subclonal diversity is viewed as a snapshot, and only serial analysis of CTCs can clarify the much needed dynamic view of tumor genomes, as pointed out in Figure 1. Both in metastasis research, as well as in clinical practice, it is important to know whether a minor subclone is emerging or has been outcompeted by the dominant subclone [16]. Longitudinal CTC studies have been performed to investigate the clonal changes in both phenotypical and molecular profiles associated with disease evolution and therapy resistance [79–81]. Hence, CTCs might reflect the characteristics of the current status of the biologically and clinically relevant subclones irrespective of a detailed anatomical distribution, and should ideally be suited to provide dynamic assessments of tumor characteristics in patients with metastatic disease. Even more since repeated sampling of multiple metastatic lesions is an invasive procedure and often not feasible.
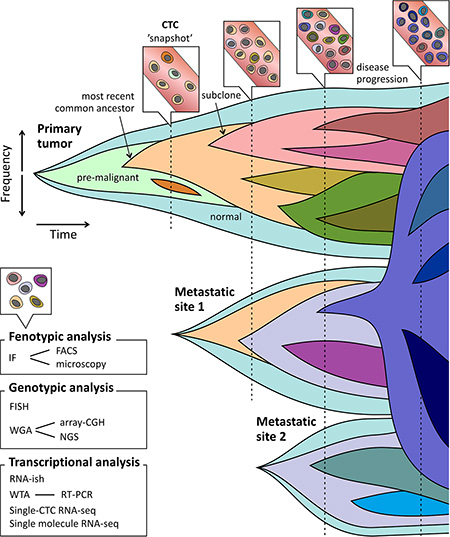
Figure 1: CTCs as snapshot of the evolving tumor landscape. Clonal evolution depicted as emergence of clones after acquisition of driver mutations. New (sub) clones derive from ancestral clones following linear and branched evolution. Outgrowth and repression (therapeutic or outcompeting) of these subclones can lead to emergence and disappearance of driver mutations respectively. Seeding and re-seeding of tumor cells causes development of changing tumor landscapes at multiple sites. Selective therapy pressure can lead to outgrowth of resistant clones at time of disease progression. CTCs sampling can function as a snapshot of the overall tumor bulk (primary tumor and metastases). When profiling CTCs at multiple time points emerging and decreasing subclones can be unveiled. Techniques to profile CTCs include phenotypical and molecular analyses.
Although increasingly sophisticated technologies have become available to detect and isolate CTCs, as is already extensively reviewed [82–88], further progress in CTC research is needed to envision heterogeneity and clonal evolution within the CTC compartment. Major questions in CTC research implicate the clonal relationship between CTCs and the number of CTCs that have to be analyzed in order to capture the overall profile of the dominant disease driving (sub)clones in a patient suffering from widespread metastatic disease. In this review, we will focus on the genomic and transcriptional heterogeneity found in the CTC compartment, and its significance for clinical decision making.
GENOTYPIC CTC HETEROGENEITY
A growing number of research articles have been published demonstrating genotypic heterogeneity in the circulating compartment, emphasizing the need for studies analyzing multiple purified CTC samples. This can be performed focusing on several types of aberrations such as gene rearrangements, mutations, and CNA profiles. Here we compare the results regarding genomic variation in CTCs of various tumor types (summarized in Table 1). We found that in many patients rearrangements as well as specific and global mutation profiles were highly heterogeneous. Concerning CNA profiles, homogeneity in overall profiles was reported frequently, although in both breast and prostate cancer intra-patient variation was observed. Furthermore, changes in CNA profiles over time were documented and in depth analysis of copy number profiles of specific genes in various tumor types demonstrated extensive heterogeneity.
Table 1: Genomic heterogeneity in CTCs
#CTC |
#Pts |
Isolation |
Analysis |
Targets |
Heterogeneity |
Ref. |
|---|---|---|---|---|---|---|
Lung cancer |
||||||
n.s. |
32 |
MF (ISET) |
FA-FISH |
ALK rearrangements |
18 ALK+ patients exhibited between 7 and 24 CTCs/ml, mean percentage of ALK-rearranged CTCs was 63% (range 28-100%). All ALK– patients had < 4 rearranged CTCs. |
89 |
n.s. |
5 |
MF (ISET) |
FA-FISH |
ALK rearrangements |
5 patients showed ALK-gene rearrangements in all CTCs (100%), while in the primary tumor only half of the tumor cells show these rearrangements. |
91 |
177 |
1 |
microfluidics + cytospin |
FISH |
ALK rearrangements |
25% of the total 177 CTC of 1 patient harbored ALK-gene rearrangements, and 54% of the 200 primary tumor cells did. |
90 |
n.s. |
8 |
MF (ISET) |
FA-FISH |
ROS1 rearrangements |
ROS1 rearrangements were detected in the CTCs of all 4 ROS+ patients. ROS1 copy number was heterogeneous within these CTCs and increased at time of disease progression. |
95 |
8 |
1 |
CS + MM |
WES |
CNA; mutations; indels |
CNA show inter-CTC homogeneity, and represent the metastatic tumor. SCLC and NSCLC can be differentiated based on CNA-profile. Mutations and indels were highly heterogeneous in all CTCs. |
35 |
8 + pools |
2 |
CS + DEPArray |
WGS; TAS |
CNA; TP53, RB1 mutations |
CNA strongly correlated, but 1 of 6 CTC harbored substantial CNA differences. TP53 and RB1 mutations were homogeneous. |
112 |
1 pool |
4 |
microfluidics |
Allele-specific PCR |
EGFR mutations and CNA |
Temporal heterogeneity in EGFR mutations. Genotypes of enriched CTC fractions evolved during therapy, with consistent presence of the primary EGFR activating mutation and the emergence of a drug-resistant mutation. |
65 |
Colorectal cancer |
||||||
37 |
6 |
CS + MM |
aCGH; Panel |
CNA; 68 CRC-related gene panel |
Multiple CRC related CNA and mutations were found in CTC and tissue samples. Various CTC-specific mutations were detected, but retraced at subclonal level by ultra-deep sequencing of the tissue samples. Inter-CTC heterogeneity, with some private mutations. |
33 |
741 |
33 |
CS + MM |
qPCR; TAS |
EGFR CNA; PIK3CA, KRAS, and BRAF mutations |
CN-gain of EGFR was found in 27% of CTCs of 3 patients, 1 patient had KRAS mutations in 33% of CTCs, 39% of CTCs of 4 patients harbored PIK3CA mutations. |
105 |
126 |
31 |
CS + MM |
TAS |
TP53, KRAS and BRAF mutations |
CTCs were analyzed of 18 patients. 6 patients harbored heterogeneous CTC populations. |
107 |
pools |
21 |
DGC + DEPArray |
TAS; PyroSeq |
KRAS mutations |
In 1 patient, 3 pools of CTCs had different mutational statuses, two mutations were found in the first pool and another mutation in a second pool of isolated CTCs. |
108 |
pools |
2 |
CS enriched |
qPCR |
KRAS mutations |
Temporal heterogeneity: enriched CTC fractions exhibited different mutational status of KRAS during treatment. |
109 |
Prostate cancer |
||||||
n.s. |
49 |
CS |
On-chip FISH |
ERG rearrangements;PTEN and AR CNA |
FISH on CTCs revealed homogenous ERG rearrangements but heterogeneous AR amplifications and PTEN deletions. |
96 |
n.s. |
77 |
CS + cytospin |
FISH |
AR and MYC CNA |
There was considerable variability in the morphology of CTCs in individual patients. 1 patient showed heterogeneity of FISH patterns, with AR amplification in a subset of CTCs, but all with high copy number gain for MYC. |
117 |
n.s. |
7 |
DGC + cytospin |
FISH |
BRCA1 CNA |
In 4 of 7 patients, BRCA1 losses appeared in a fraction of CTCs. |
118 |
pools |
9 |
IE/FACS |
aCGH |
CNA |
CTCs from all patients revealed a wide range of CNA. Replicate CTC isolates where comparable showing gains in the CCND1 and AR locus. |
114 |
41 |
1 |
HD-CTC + MM |
WGS |
CNA |
Three different clonal lineages were found. Clone B was present subclonally at first blood draw, but demonstrated outgrowth in the third blood draw. A third clone emerged at fourth blood draw. |
80 |
19 + 10 |
2 |
MagSweeper + MM |
WES |
Somatic SNV |
Although non-uniform coverage, a heterogeneous mutation profile was detected in single CTCs. When pooling the CTC data, found SNVs were comparable to the primary tumor. |
34 |
Breast cancer |
||||||
261 + pools |
42 |
CS + DEPArray |
aCGH; qPCR; TAS |
CNA; ERBB2 CNA; PIK3CA mutations |
2 patients had heterogeneous PIK3CA mutational status in their single and pooled CTCs. 10 of 16 patients harboring PIK3CA mutations showed molecular heterogeneity based on CNA. ERBB2 amplification was uniformly detected in all CTCs of 7 patients. |
101 |
26 |
12 |
CS + flow sorting (MoFlo XDP) |
aCGH; qPCR; TAS |
CNA; CCND1 CNA; PIK3CA mutations |
CNA were found breast cancer related in all CTCs, but differences in CNA between related CTCs were present in all cases. 1 patient harbored a mutation in exon 20 of the PIK3CA gene in both CTCs and 1 patient harbored another PIK3CA mutation in 1 of 1 CTCs. |
100 |
147 + pools |
26 |
CS + DEPArray |
TAS |
PIK3CA hotspots |
11 of 26 patients were found to harbor a heterogeneous PIK3CA mutational status in their CTC compartment. |
102 |
115 + pools |
18 |
CS + DEPArray |
TAS |
PIK3CA hotspots |
3 patients were homogeneously mutated in all CTCs. 1 patient was found to have three different PIK3CA mutations. |
104 |
185 |
17 |
MagSweeper + MM |
TAS |
PIK3CA hotspots |
1 patient harbored a heterogeneous CTC compartment based on PIK3CA status. |
103 |
11 + pools |
2 |
CS + DEPArray |
TAS |
TP53 mutations |
In one patient, 2 of 6 single CTC harbored two different TP53 mutations. In the second patient, 3 of 5 single and 5 of 6 clusters of CTCs showed a TP53R110delC mutation. |
111 |
402 |
3 |
DGC + cytospin |
IF/FISH (BioView) |
EGFR CNA |
10 of 91 ALDH1+/HPSE+ cells showed EGFR amplification. This was 19 of 311 in the ALDH1–/HPSE+ population. |
50 |
31 + pools |
1 |
CS ór DGC + MM |
WGS; aCGH |
CNA |
CNA show homogeneity within all isolated CTCs. |
36 |
n.s. |
3 |
IE/FACS |
aCGH |
CNA |
Temporal heterogeneity: Serial testing of enriched CTC populations revealed numerous additional CNA beyond the baseline profile. |
116 |
Melanoma |
||||||
24 + 18 |
2 |
Microfluidic + LCM |
TAS |
BRAF mutations |
Consistency in the BRAFV600E mutation, and in accordance with the primary tumor. |
110 |
15 |
7 |
IM + MM |
CGH |
CNA |
In 5 of 6 patients with ≥ 1 isolated CTC, hierarchical clustering showed a clonal origin. |
115 |
Multiple cancers |
||||||
n.s. |
20 |
IM + cytospin |
FISH |
CNA |
6 patients had a homogeneous pattern of aneusomy in all CTCs. In 10 patients a heterogeneous pattern was observed, including 6 cases with two distinct clones. |
49 |
Abbreviations: aCGH, array comparative genomic hybridization; CNA, copy number alterations; CS, CellSearch enrichment; CTC, circulating tumor cell; DGC, density gradient centrifugation; FA-FISH, filter adapted fluorescent in situ hybridization; HD-CTC, high-definition CTC assay; IE/FACS, immunomagnetic enrichment and fluorescence-activated cell sorting; IF, immunofluorescence; IM, immunomagnetic enrichment; LCM, laser capture microscopy; MF, microfiltration; MM, micromanipulation; TAS, targeted amplicon sequencing; qPCR, quantitative polyclonal chain reaction; WES, whole exome sequencing; WGS, whole genome sequencing; n.s., not specified.
Rearrangements
Several research groups studied rearrangements of the ALK gene in CTCs using Fluorescence in situ hybridization (FISH) [89–91]. In non-small cell lung cancer (NSCLC), EML4-ALK fusion is present in approximately 3–7% of cancers, and these patients are eligible for targeted treatment with crizotinib and ceritinib [92–94]. Pailler and colleagues demonstrated that percentages of ALK-rearranged CTCs ranged between 28% and 100% in patients with ALK-positive tumors, and varied within these patients during crizotinib therapy. This suggests that the ALK-rearranged CTC population might be a consequence of clonal selection from a specific subpopulation of primary tumor cells, and that outgrowth of this subpopulation can be an indication for therapy resistance [89]. Percentages of ALK-rearranged CTCs were confirmed by two other studies. In a first report, one-fourth of the total 177 CTCs of one patient harbored ALK rearrangements [90] and in the other, 100% of the CTCs of 5 patients were ALK-rearranged [91], whereas in the primary tumor tissue this was around 50% in both studies. Furthermore, ROS1 rearrangements were found in CTCs of four patients with lung cancer [95]. FISH has also been used to analyze ERG rearrangement in prostate cancer CTCs [96, 97]. TMPRSS2ERG gene fusion was either homogeneously present in all CTCs of one patient or absent [96]. Although presence of this ERG rearrangement demonstrates a significant association with PSA response to abiraterone in this study, TMPRSS2-ERG status could not predict a decline in PSA or other clinical outcomes in response to abiraterone therapy in a clinical trial evaluating enriched CTC populations [97].
Hotspot mutations
In breast cancer, PIK3CA is mutated in up to 25% of patients, with mutation frequencies rising to 40% in the hormone receptor-positive subgroups [98, 99]. Analyzing the PIK3CA genotype has clinical relevance with respect to drug resistance, e.g. against HER2-targeted therapy. Hence, various studies are performed investigating the PIK3CA mutational status in CTCs. In a first study, two single CTCs per patient were analyzed [100]. In two patients PIK3CA mutations were found in all CTCs of these patients (resp. 1 and 2 CTCs). In a similar, but much larger study, PIK3CA mutations were detected in 16 patients, two of whom harboring a heterogeneous mutational status in their single and pooled CTCs [101]. De Laere and colleagues profiled CTCs of 26 hormone receptor positive patients, ranging between 4 and 311 CTCs per patient. In 19 cases (73%) PIK3CA mutations were detected. Of these, six cases were found almost homogeneously mutant for one specific mutation, whereas another six patients were extensively heterogeneous with subclones harboring one or multiple PIK3CA mutations [102]. In contrast, another study detected PIK3CA mutations in only one out of 17 patients, which might be due to different patient selection [103]. Single CTCs of 24 samples (containing 2–50 CTCs) of 12 patients were examined for presence of PIK3CA mutations. In one patient an exon 9 mutation was detected in two out of nine serial samples, both at a heterogeneous level [103]. Pestrin et al. identified PIK3CA mutations in CTCs in 6 out of 18 patients [104]. In three cases with multiple CTCs analyzed, all CTCs were homogeneously mutant. One patient had a heterogeneous mutational status, with 3 out of 16 single CTCs harboring three different PIK3CA mutations [104]. When combining aforementioned studies, from a total of 47 PIK3CA mutated patients, 15 had a heterogeneous circulating compartment with mutated CTCs present at a subclonal level. Also in a study on CRC, PIK3CA mutations were present at a subclonal level in four patients; one of whom harbored two different PIK3CA mutations in separate CTCs [105].
Since PIK3CA, BRAF, KRAS, and PTEN are relevant genes in predicting resistance to anti-EGFR therapy [106], mutations in these genes are frequently studied using CTCs. A recent study isolated 37 single CTCs from six patients with metastatic CRC for sequencing of a 68 CRC-associated gene panel to determine mutational landscapes in CTCs and the corresponding primary tumors and metastases [33]. Point mutations in APC, KRAS, PIK3CA, and TP53 in the primary tumors were also present in the single CTCs. However, 20 ‘branch’ mutations were found exclusively in CTCs, although targeted ultra-deep sequencing revealed the presence of 17 of these mutations at subclonal level in either the primary tumor or metastases [33]. Two more studies performed targeted sequencing of BRAF, KRAS, and TP53 of respectively 741 and 126 single CTCs [105, 107]. The first study detected the presence of KRAS mutations in one-third of CTCs of one patient [105], while in the other, 6 out of 18 patients demonstrated a heterogeneous CTC compartment regarding these genes [107]. Moreover, two studies examined heterogeneity of KRAS mutations in pools of CTCs [108, 109]. Fabbri et al. reported one patient harboring three pools of CTCs with different mutational statuses. Two specific KRAS mutations were detected in the first pool, and another KRAS mutations was found in a second pool of pure CTCs [108]. Also, temporal heterogeneity was shown as enriched CTC fractions exhibiting different mutational status of KRAS during treatment [109]. However, one can argue on the sensitivity of mutation detection in enriched samples containing low CTC-counts, as often seen during therapy. Furthermore, mutational analysis was performed on multiple single CTCs collected from two patients with stage-IV melanoma. All CTCs were consistently BRAFV600E mutated analogous to the primary tumor [110].
In a study towards TP53 mutations, single and pooled CTCs of two patients with metastatic triple-negative inflammatory breast cancer, known for harboring a TP53 mutation in their primary tumor, were recovered for molecular analysis [111]. In the first patient, 2 of 6 single CTC harbored two different TP53 mutations, one of these was also found in the pool of 14 CTCs. In the second patient, 3 of 5 single and 5 of 6 clusters of CTCs had a TP53R110delC mutation. In contrast, TP53 and RB1 were homogeneous in all CTCs of lung cancer patients [112].
Temporal heterogeneity was demonstrated in pools of pure CTCs from patients with NSCLC receiving tyrosine kinase inhibitors. Serial analysis showed emergence of activating mutations in the gene encoding the EGFR conferring a mechanism of acquired resistance to therapy [65]. EGFR mutation detection was also performed on enriched CTC samples. In 4 out 31 cases, multiple EGFR mutations were documented, suggesting possible CTC heterogeneity [113]. However, the actual mutational landscape and subclonality can only be detected in single CTC samples or multiple pools of pure CTCs.
Global mutational profile
A recent study applied whole exome sequencing (WES) of 19 single CTCs from a patient with metastatic prostate cancer [34]. Although non-uniform coverage, a heterogeneous mutation profile was detected in single CTCs. To compensate for the low coverage and random polymerase errors that did occur in individual CTCs, single-CTC data was pooled. Half of the somatic SNV in CTCs could be detected in the primary and metastatic sites, whereas the rest were CTC-specific mutations [34]. Moreover, Ni et al. determined single nucleotide variation landscapes in CTCs of four patients with lung cancer by single-cell exome sequencing [35]. The exome data showed extensive variation from cell to cell and presence of ‘private’ CTC mutations, not detected in tissue samples. The authors raise the question of false discovery due to interfering technical errors compatible with the MALBAC method used [35].
Copy number alterations
Methods used to study genome-wide CNA include array-CGH and whole genome or exome sequencing. In prostate cancer, a wide range of CNA in pools of pure CTCs were detected in nine patients, using array-CGH. But more specifically, CTCs showed uniform copy number gains in both the AR and CCND1 locus [114]. In one study where two single breast cancer CTCs per patient were analyzed for CNA, all CTCs displayed a typical breast cancer related copy number profile [100], with six patients harboring CCND1 amplification in both CTCs. Yet, differences in CNA between CTC couples were to a greater or lesser extent visible in all cases. Furthermore, multiple CTCs of 16 patients with breast cancer were analyzed using array-CGH. Ten of these patients showed molecular heterogeneity based on CNA. Although, in seven cases were ERBB2 amplification was detected, it was homogeneous in all CTCs [101].
However, in multiple studies in various tumor types, homogeneity in the copy number profile was demonstrated. WES was applied to lung cancer CTCs in two studies [35, 112]. Five out of six patients had highly homogeneous copy number profiles, although one patient harbored substantial CNA heterogeneity [112]. In another study, the copy number profiles of the single CTCs were highly similar and shared most of the same CNAs as the primary and metastatic tumor cells. Furthermore, CNA patterns were indicative for specific lung cancer subtypes [35]. A recent study isolated 37 single CTCs from six patients with metastatic CRC for copy number profiling with array-CGH [33]. In general, many of the CTCs shared a number of gains and losses with the primary and metastatic lesions. However, they also observed private copy number changes in CTCs as well as heterogeneity between CTCs [33]. To define CNA in melanoma, the genomes of 15 individually isolated CTCs from seven patients were analyzed by single-cell CGH [115]. All of the analyzed CTCs displayed multiple chromosomal changes and carried aberrations typical for melanoma. In five of six cases with multiple CTCs isolated, hierarchical clustering of the CTCs showed a clonal relationship [115].
Sampling at multiple time-points to evaluate genetic evolution based on CNA profiles was performed in three studies [36, 80, 116]. Dago and colleagues thoroughly analyzed CNA of multiple single CTCs of one patient with prostate cancer by WGS at various time points. Three different clonal lineages were found. One specific clone was present at subclonal level at the first blood draw, but demonstrated outgrowth at time of the third blood draw. A third clone only emerged at the fourth time point [80]. Both array-CGH and WGS were applied for copy number analyses in one patient with breast cancer harboring extensive numbers of CTCs [36]. CNA demonstrated high similarities between the 31 single and 21 pools of CTCs ranging between 5 and 100 CTCs. Furthermore, a high degree of analogy was also found with CNA in primary and metastatic tissue samples [36]. In a large breast cancer cohort, array-CGH of CTCs revealed a wide range of CNA, including those known for breast cancer [116]. In one patient, where multiple sampling was performed, CTCs of the second blood draw revealed numerous additional CNA beyond the baseline profile, while the third sample, divided in two pools, was comparable with itself and the second. Interestingly, the patient initially responded to her cancer treatment, but subsequently developed disease progression. In two other cases temporal homogeneity was documented between first and second blood draw. Furthermore, CTCs and the primary tumor were moderately and highly correlated, respectively [116].
Then, various studies have thoroughly analyzed CNA of specific target genes using FISH. In 4 patients with lung cancer, ROS1 copy numbers were heterogeneous between CTCs [95]. In prostate cancer, FISH was applied to study CNA of AR, BRCA1, MYC, and PTEN [96, 117, 118]. Leversha and colleagues report a considerable variability in CTCs of individual patients. In one patient, a subset of CTCs showed AR amplification, whereas all CTCs had high copy number gain for MYC [117]. A similar heterogeneity in AR amplifications and loss of the tumor suppressor gene PTEN was detected by Attard et al. when profiling 49 patients suffering CRPC [96]. FISH analysis further revealed BRCA1 losses appearing in minute fractions of CTCs in four of seven patients [118]. In breast cancer, fluorescent cell sorting was combined with FISH to analyze EGFR amplification in CTCs [50]. 11% and 6% of CTCs from ALDH1 positive and negative populations respectively, harbored EGFR amplification [50]. Furthermore, EFGR copy number gain was found in 37% of CTCs of three patients with CRC, based on array-CGH data [105].
TRANSCRIPTIONAL CTC HETEROGENEITY
While in diploid cells chromosomal DNA molecules are present with only two copies, a single cell harbors thousands of copies of each mRNA transcript, which facilitates single-cell RNA approaches [119]. Yet, single cell RNA studies are affected by transcriptional bursting or pulsing [120, 121]. This phenomenon can account for the high variability in gene expression between cells in isogenic populations, and therefore transcriptional heterogeneity should be evaluated with caution. On the other hand, variability in gene expression may also contribute to resistance of sub-populations of cancer cells to chemotherapy [122]. Gene-expression studies in single CTCs may be essential for determining the nature and extent of tumor heterogeneity, linking phenotypic differences with genetic and epigenetic aberrations. However, preserving RNA is more difficult than DNA and concerns have been raised about the impact of sample processing on CTC expression profiles [123]. Hence, several devices have been developed for direct and fast isolation of CTCs using a microfluidic approach [37, 75, 81, 124–126].
Single cell expression profiling is performed using RNA-in situ hybridization (ish), RT-PCR, and RNA-sequencing (seq). While RNA-ish has the advantage of direct analysis of the RNA without whole transcriptome amplification, expression of far more genes can be evaluated using RT-PCR or RNA-seq. Differentiating the changes in gene expression that are biologically relevant from those caused by technical and biological noise remains a significant hurdle for single-cell transcriptome studies. Hence, single cell mRNA-seq protocols are being developed with improved transcriptome coverage, high reproducibility, and low technical variation [127, 128].
Hereafter, we review various publications on transcriptional heterogeneity in CTCs. Often, patient-specific global expression profiles were observed. However, when looking in detail, significant heterogeneity between CTCs is found regarding specific transcripts, which is often linked to therapy selection or response. Table 2 gives an overview of the experimental details of these studies.
Table 2: Transcriptional heterogeneity in CTCs
#CTC |
#Pts |
Isolation |
Analysis |
Targets |
Heterogeneity |
Ref. |
|---|---|---|---|---|---|---|
Breast cancer |
||||||
n.s. |
17 |
HBCTC-Chip |
RNA-ish; RNA-seq-DGE |
EMT markers |
Heterogeneous fractions of Epithelial (E), Mesenchymal (M), and EM-CTCs; In TNBC more homogeneous pools of M-CTCs. Temporal heterogeneity: at progressive disease, 10 patients harbored emerging numbers of M-CTCs. |
81 |
105 |
35 |
MagSweeper + MM |
qRT-PCR |
87 cancer-associated genes |
Two major subgroups of CTCs, i.e. high expression of EMTgenes and high metastasis-associated genes. Heterogeneity based on CTCs not clustering by patient-ID and 8 patients having CTCs in both clusters. |
132 |
15 pools + 14 clusters |
10 |
negCTC-iChip + MM |
RNA-Seq |
Whole transcriptome |
Based on global gene expression level, all isolated CTCs clustered closely by patient of origin. Based on JUP and 31 cluster-associated genes, CTC-clusters could be differentiated from pooled single CTCs. |
124 |
~400 |
20 |
IM (Maintrac) + AP |
PCR + gelelectro-phoresis |
HER2, EpCAM, Vimentin, and NANOG |
Expression patterns changed after surgery, with emerging of a sub-population of EpCAM positive CTC expressing NANOG and/or vimentin. |
130 |
Prostate cancer |
||||||
77 |
13 |
negCTC-iChip + MM |
RNA-seq |
Whole transcriptome |
Single CTCs from nine individual patient with at least 3 CTCs analyzed, showed considerably higher intra-patient heterogeneity in their transcriptional profiles compared to single cells from prostate cancer cell lines. |
126 |
20 |
4 |
MagSweeper + MM |
RNA-seq |
Whole transcriptome |
All CTCs, except two, cluster in a patient specific manner. 181 cancer-specific genes were overexpressed in the CTCs, compared to normal tissue. Specific transcripts, e.g. related to CRPC or ERG-fusion, were detected homogeneously within the same patients. |
131 |
48 |
2 |
MagSweeper + Nanowell |
RNA-seq |
KLK3 (PSA) mRNA |
KLK3 expression was variable between the 26 individual CTCs, for which a sufficient number of genes including KLK3 were covered. |
34 |
38 |
8 |
MF + MM |
qRT-PCR |
84 EMT-related genes |
Heterogeneous upregulation of EMT-associated gene expression, especially in CRPC. |
129 |
pools |
21 |
IM (AdnaTest) |
qRT-PCR |
AR full length + AR-V7 |
Temporal heterogeneity: 1 out 9 patients converted to AR-V7 positive, at progression on Taxane. While 7 out 12 patient who were at baseline AR-V7 positive became negative at progression. |
73 |
Pancreatic cancer |
||||||
265 |
15 |
HBCTC-Chip |
RNA-ish; RNA-Seq-DGE |
WNT2 |
RNA-ish showed heterogeneity of WNT2 expression in CTCs and the primary tumor. This was confirmed by RNA-seq with DGE, showing rare WNT2 RNA reads in the enriched CTC sample and the primary tumor. |
37 |
Melanoma |
||||||
6 |
1 |
MagSweeper + MM |
RNA-seq |
Whole transcriptome |
CTCs show a uniform upregulation of melanoma markers, including MAGE as well as uniform up- or downregulation of certain plasma membrane proteins. |
128 |
Multiple cancers |
||||||
7, 29, 77 |
n.s. |
negCTC-iChip + MM |
RNA-seq |
Whole transcriptome |
High expression of stromal-derived ECM proteins in > 15% of CTC samples. One glycoprotein was expressed in 100% of pancreatic CTCs compared to 31% of breast and 9% of prostate CTCs. |
125 |
Abbreviations: CTC, circulating tumor cell; DGE, digital gene extraction; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; IM, immunomagnetic enrichment; MF, microfiltration; MM, micromanipulation; qRT-PCR, quantitative reverse transcription polyclonal chain reaction; RNA-ish, RNA in situ hybridization; RNA-seq, RNA sequencing; n.s., not specified.
Metastasis-associated gene expression
In prostate cancer RT-PCR of 84 EMT-related genes was applied to analyze multiple single CTCs of 8 patients [129]. Heterogeneous upregulation of EMT-associated gene expression was found, especially in CRPC. RT-PCR was also used to target vimentin, EpCAM, and stem cell gene NANOG mRNA for EMT evaluation in approximately 400 breast CTCs [130]. Temporal heterogeneity was shown as expression patterns changed after surgery, with emerging of a sub-population of EpCAM positive CTC expressing NANOG and/or vimentin. Yu et al. applied RNA-ish for scoring the relative abundance of epithelial versus mesenchymal transcripts within individual breast cancer CTCs of 15 patients, both during therapy or at time of progression [81]. Clear heterogeneity was shown, with various proportions of CTCs that were mesenchymal. Moreover, relative changes during treatment in the expression of epithelial and mesenchymal markers in CTCs correlated with response and prognosis. For one patient, single CTCs were analyzed with RNA-ish over 7 time points and two different treatment regimens. An increased number of mesenchymal CTCs was repeatedly detected in the samples taken at time of disease progression [81]. Additionally, single molecule RNA-seq was applied on CTCs to identify signaling pathways that contribute to EMT, and 45 enriched genes were identified [81]. In metastatic pancreatic cancer, RNA-ish was used for detection of CTC-specific transcripts of Wnt2, which is known for its role in tumor sphere formation and metastasis initiation [37]. Wnt2 transcripts were identified in 23 out of 66 (35%) cytokeratin-positive CTCs from 2 out of 8 patients. Heterogeneity was also shown in the primary tumors. The small number of Wnt2-positive cells was consistent with RNA-seq analysis, which showed rare Wnt2 RNA reads in both enriched CTCs and primary tumors [37]. This demonstrates Wnt2-positive CTCs are present at subclonal level and represent a rare subset of the primary tumor population. Ting and colleagues isolated 7, 29, and 77 single CTCs from patients with pancreas, breast, and prostate cancer respectively [125]. In more than 15% of all CTC samples, CTCs exhibit a very high expression of stromal-derived extracellular matrix (ECM) genes, which have an important role in metastatic spread. One specific ECM glycoprotein gene was expressed at high levels in 100% of pancreatic CTCs compared to 31% of breast and 9% of prostate CTCs [125].
Global gene expression profiling
Recently, genome-wide expression profiling of single cells using NGS has been achieved [127, 128]. In a study regarding patients with metastatic breast cancer, a homogeneous global expression pattern was shown, with all CTCs clustering together patient wise, except for two patients [124]. Furthermore, in advanced melanoma, some highly expressed transcripts in single CTCs were detected [128]. Although slight differences in gene expression, CTCs show a uniform and high upregulation of cell-cycle and melanoma specific markers, as well as uniform up- or downregulation of certain plasma membrane proteins [128]. The same single cell mRNA-seq protocol was used for CTCs isolated from patients with metastatic prostate cancer [131]. High rates of RNA degradation consistent with apoptosis amongst CTCs was noted, although prostate-specific and cancer-specific transcripts could still be elucidated. 181 genes were overexpressed in the CTCs compared to normal prostate tissue [131]. Unsupervised clustering revealed that all CTCs, except two, cluster in a patient specific manner. Specific transcripts, e.g. related to CRPC or ERG-fusion, were detected homogeneously within the same patients [131]. In another RNA-seq study on prostate cancer, hierarchical clustering analysis also demonstrated patient-specific CTCs clustering, separated from cancer cell lines. However, single CTCs from nine individual patients with at least 3 CTCs analyzed, showed considerably higher heterogeneity in their transcriptional profiles compared to single cells from prostate cancer cell lines [126]. Moreover, RT-PCR of a panel of 87 cancer genes demonstrated heterogeneity among individual breast cancer CTCs, separating them into two major subgroups based on 31 highly expressed genes [132]. This was in contrast to several breast cancer cell lines tested.
Prostate cancer specific gene expression
Isolated single CTCs were tested for expression level of the PSA gene KLK3 [34]. The expression profile of KLK3 was heterogeneous between the 26 out 48 selected individual CTCs, for which sufficient part of the transcriptome was covered [34]. Besides, expression patterns of AR splice variants have been studied at a single cell level using either RT-PCR or RNA-seq [73, 126]. One study demonstrated that more than half of all patients had multiple AR splice variants present within different CTCs and that a subpopulation of single CTCs had simultaneous expression of several AR splice variants [126]. These results are in line with other data showing that acquisition of AR-independent alterations conferring resistance to antiandrogen therapies is very heterogeneous in patients with CRPC [79]. Temporal heterogeneity between multiple enriched CTC samples from 21 patients with prostate cancer was shown by emerging of AR-V7 in one out nine patients treated with taxane chemotherapy. In contrast, seven out of twelve patients who were AR-V7 positive at baseline, only harbored full length AR at time of progression [73]. Relations between therapy response and presence of variants are increasingly studied [74, 97], although usually not at multiple time points or with multiple CTC samples, which is needed to study tumor evolution.
DISCUSSION
Technical considerations
Studies across multiple tumor types have demonstrated the feasibility of analyzing molecular profiles of single CTCs. Although technical improvements are needed, it becomes clear that CTC profiling contributes to our understanding of tumor heterogeneity, disease evolution (through serial sampling), and clinical management. To maximize the potential of CTC profiling, key issues in CTC research must be addressed regarding both technical and biological challenges.
Evolution in multiple-marker and marker-independent CTC enrichment has already increased yield and diversity of CTCs [50, 81, 133], although it is not as extensively validated as EpCAM enrichment strategies. Furthermore, efforts have been made to improve both amplification methods [134–136] and sequencing techniques [34, 127, 128] as well as subsequent data interpretation and bioinformatics [10, 137, 138], reviewed in more detail by Van Loo and Voet [24]. This all contributes to more reliable detection of aberrations and evaluation of heterogeneity in CTC research.
A major question in CTC research remains how many CTCs should be profiled to account for heterogeneity. Often, the molecular characteristics of only a few CTCs out of the entire pool of CTCs from a patient have been adequately analyzed [34, 100]. As a consequence their diversity remains largely unknown. In primary breast cancer for example, single-molecule sequencing indicated that many of the diverse mutations occur at low frequencies (< 10%) in the tumor mass [139]. Navin demonstrated, using a power analysis, that detection of a 10% subclone would require sequencing at least 20 single cells to achieve a 0.87 detection power [140]. Besides, subclonality can be evaluated using multiple small pools of pure CTCs (Figure 2) and determining the variant allele frequencies. Herewith technical errors typical for single cell research [24] can be reduced, although more CTCs need to be available and isolated. Furthermore, in depth comparative research towards CTCs and multiple metastases [31] should clarify whether the whole tumor burden contributes equally to the CTC pool or if some subclones might be underrepresented or absent.
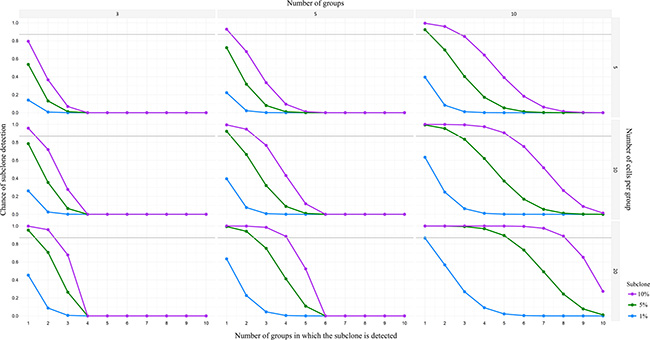
Figure 2: Power analysis for detection of minor subclones in pools of CTC. Chances of detection of minor subclones (i.e. 1%, 5%, or 10%), calculated with a power of 0.87, for three different number of groups (i.e. 3, 5, or 10 groups) and three different number of cells per group (i.e. 5, 10, or 20 cells). As depicted in the lower right graph (10 groups of 20 cells), there is a 90% change of detecting a 1% subclone in 1 out of 10 groups, or detecting a 5% subclone in 5 out of 10 groups, or detecting a 10% subclone in 8 out of 10 groups.
Clinical implications and future perspectives
Currently, biomarkers predicting therapy response are frequently assessed using primary tumor biopsies, reflecting only parts of a patient’s disease at a specific moment in time [141]. It is well-known that targetable molecules can change during the course of the disease. CTCs have shown to be useful in understanding and predicting acquired resistance to therapies, and might in the future be used to circumvent this. In lung cancer, serial analysis identified emergence of activating mutations in the EFGR gene in some patients receiving EGFR-targeting therapy, conferring a mechanism of acquired resistance to therapy [65]. Moreover, clonal selection of ALK-rearranged CTCs during crizotinib therapy was detected in patients with lung cancer [89]. Serial RNA analysis of prostate CTCs demonstrated emergence of AR-V7 during taxane chemotherapy [73], and TMPRSS2-ERG status in CTCs is a predictive biomarker of abiraterone acetate sensitivity in CRPC [97]. Hence, repeated CTC sampling may have the potential to guide optimal therapy regimens depending on the evolving molecular profile of the tumor burden within an individual patient. However, CTC characterization is currently only performed in clinical trials [142]. Therefore, efforts to increase clinical utility, have to be made. A comprehensive analysis of multiple patient samples, including CTCs, cfDNA, and tissue samples, on both RNA and DNA level can provide a holistic view of a patient’s (sub)clonal landscape. The development of multi-compartment molecular databases of large patient cohorts will enable the creation of algorithms able to predict outcome at a more individual patient level [3, 143, 144].
A key issue remains to what extent heterogeneity in the circulating compartment affects therapy outcome and whether one should take a minor subclone into account if it comes to treatment selection. The analysis of subclonal heterogeneity may help clinicians understand why patients do not respond homogeneously to targeted drugs. Furthermore, longitudinal molecular analysis of individual CTCs can uncover clonal evolution caused by therapy pressure [32, 78, 145]. In a patient with CRPC, sequentially progressive on chemo and targeted therapy, comparable CTC clones were observed before the start and during standard chemotherapy. However, subsequent clinical response to targeted therapy was associated with the drastic depletion of the fist clone and emergence of a second clone, while a third tumor lineage was detected at time of disease progression [80]. As acquired drug resistance and disease relapse is common, drugs may only ablate specific subpopulations of tumor cells, allowing resistant cells to grow, evolve and seed new tumor foci that may not respond to cytotoxic or targeted therapies [32, 78, 145]. Hence, a tremendous potential of CTCs lies in profiling them over the entire clinical course to study the evolutionary history of tumors and to optimize clinical trial design. In the TRACERx trial (NCT01888601), primary tumors of 842 NSCLC patients will be sequenced, as well as cfDNA and CTCs, obtained at multiple time points during therapy. To evaluate the effect of clonal heterogeneity and selection pressure on clinical outcomes, and to identify targetable driver events, repeated tumor sampling will be performed at time of disease recurrence. These patients will be eligible for the DARWIN trial (NCT02183883). This trial aims at evaluating whether targeting driver events, detected by the TRACERx trial, has a different clinical outcome in patients harboring the driver dominantly compared to subclonally.
In conclusion, molecular characterization of CTCs provides the opportunity to repeatedly assess the biological features of cancer during the evolution of the disease. Therefore, CTCs may facilitate the development of new therapeutic strategies and enable clinicians to tailor therapy to an individual patient in a longitudinal fashion. The relevance of CTC heterogeneity as a cause or consequence of resistance to targeted therapy is yet to be unveiled. Hence, a tremendous potential of CTCs lies in single-cell profiling techniques that will contribute to understanding the predictive value of driver molecular aberrations in subclones of CTCs and emergence of resistant populations on targeted therapy.
ACKNOWLEDGMENTS AND FUNDING
This work is supported by grants of the Foundation Against Cancer (LD, SVL), the Flemish League against Cancer (LD, SVL, and MP, DP, AB), and the University of Antwerp (BDL, SVL). AB is supported by an Emmanuel van der Schueren fellowship of the Flemish League against Cancer and a FWO-umbrella grant of the University of Antwerp.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest regarding the publication of this paper.
Abbreviations
General: CGH, comparative genomic hybridization; CNA, copy number alteration; CRC, colorectal cancer; CRPC, castration resistant prostate cancer; CTC, circulating tumor cell; DGE, digital gene extraction; EMT, epithelial-to-mesenchymal transition; FISH, fluorescent in situ hybridization; ITH, intra tumor heterogeneity; NCSLC, non-small cell lung cancer; NGS, next generation sequencing; SCS, single cell sequencing; SNV, single nucleotide variation; WGA, whole genome amplification; WGS, whole genome sequencing; WTA, whole transcriptome amplification; Genes: ALK, anaplastic lymphoma kinase; APC, adenomatous polyposis coli; BRAF, v-Raf murine sarcoma viral oncogene homolog B; BRCA1, breast cancer 1; CCND1, cyclin-D1; EGFR, epidermal growth factor receptor; EML4, echinoderm microtubule-associated protein-like 4; ERBB2, avian erythroblastosis oncogene B 2; ERG, ETS-related gene; KLK3, kallikrein 3; KRAS, Kirsten rat sarcoma viral oncogene homolog; MYC, V-myc avian myelocytomatosis viral oncogene homolog; PIK3CA, phosphatidylinositol-4, 5-bisphosphate 3-kinase; PTEN, phosphatase and tensin homolog; RB1, retinoblastoma 1; ROS1, ROS Proto-Oncogene 1; TMPRSS2, transmembrane protease serine 2; TP53, tumor protein 53; Markers: ALDH, aldehyde dehydrogenase; AR, androgen receptor; EpCAM, epithelial cell adhesion molecule; HER2, receptor tyrosine-protein kinase erbB-2; PSA, prostate-specific antigen.
REFERENCES
1. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006; 6:449–458.
2. Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, Lopez-Bigas N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015; 27:382–396.
3. Dienstmann R, Jang IS, Bot B, Friend S, Guinney J. Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov. 2015; 5:118–123.
4. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010; 467:1109–1113.
5. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467:1114–1117.
6. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014; 46:225–233.
7. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–892.
8. Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, et al. Genome remodelling in a basal-like breast cancer metastasis, xenograft. Nature. 2010; 464:999–1005.
9. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993.
10. Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, et al. The life history of 21 breast cancers. Cell. 2012; 149:994–1007.
11. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486:395–399.
12. Bashashati A, Ha G, Tone A, Ding J, Prentice LM, Roth A, Rosner J, Shumansky K, Kalloger S, Senz J, Yang W, McConechy M, Melnyk N, et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J Pathol. 2013; 231:21–34.
13. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013; 153:666–677.
14. Nordentoft I, Lamy P, Birkenkamp-Demtroder K, Shumansky K, Vang S, Hornshoj H, Juul M, Villesen P, Hedegaard J, Roth A, Thorsen K, Hoyer S, Borre M, et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014; 7:1649–1663.
15. Diaz-Cano SJ, Blanes A, Rubio J, Matilla A, Wolfe HJ. Molecular evolution and intratumor heterogeneity by topographic compartments in muscle-invasive transitional cell carcinoma of the urinary bladder. Lab Invest. 2000; 80:279–289.
16. Hiley C, de Bruin EC, McGranahan N, Swanton C. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 2014; 15:453.
17. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011; 147:275–292.
18. Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, Klein CA. Systemic spread is an early step in breast cancer. Cancer Cell. 2008; 13:58–68.
19. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012; 148:349–361.
20. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009; 9:302–312.
21. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM, Hognas G, Annala M, Kivinummi K, Goody V, Latimer C, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015; 520:353–357.
22. Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, Adejola N, Gurel M, Hicks J, Meeker AK, Halushka MK, Simons JW, Isaacs WB, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest. 2013; 123:4918–4922.
23. Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D, Ali HR, Shah P, Contente-Cuomo T, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015; 6:8760.
24. Van Loo P, Voet T. Single cell analysis of cancer genomes. Curr Opin Genet Dev. 2014; 24:82–91.
25. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012; 30:413–421.
26. Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, Fulton R, Magrini V, Schmidt H, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012; 366:1090–1098.
27. Hughes AE, Magrini V, Demeter R, Miller CA, Fulton R, Fulton LL, Eades WC, Elliott K, Heath S, Westervelt P, Ding L, Conrad DF, White BS, et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014; 10:e1004462.
28. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012; 12:323–334.
29. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015; 27:15–26.
30. Amir E, Miller N, Geddie W, Freedman O, Kassam F, Simmons C, Oldfield M, Dranitsaris G, Tomlinson G, Laupacis A, Tannock IF, Clemons M. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J Clin Oncol. 2012; 30:587–592.
31. Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, Ligon KL, Tabernero J, Seoane J, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015; 5:1164–1177.
32. Diaz LA, Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537–540.
33. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Hofler G, Eisner F, Sill H, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013; 73:2965–2975.
34. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R, Trombetta JJ, Lu D, Tallapragada N, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014; 32:479–484.
35. Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, Zong C, Bai H, Chapman AR, Zhao J, Xu L, An T, Ma Q, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A. 2013; 110:21083–21088.
36. Heidary M, Auer M, Ulz P, Heitzer E, Petru E, Gasch C, Riethdorf S, Mauermann O, Lafer I, Pristauz G, Lax S, Pantel K, Geigl JB, et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014; 16:421.
37. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, Ulman MJ, Xega K, Contino G, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012; 487:510–513.
38. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004; 10:8152–8162.
39. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, Klein C, Saini M, Bauerle T, Wallwiener M, Holland-Letz T, Hofner T, Sprick M, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013; 31:539–544.
40. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004; 10:6897–6904.
41. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007; 450:1235–1239.
42. Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, Inserra EJ, Ulman M, Springer S, Nakamura Z, Moore AL, Tsukrov DI, Kempner ME, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010; 2:25ra23.
43. Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010; 16:116–122.
44. Giordano A, Gao H, Anfossi S, Cohen E, Mego M, Lee BN, Tin S, De Laurentiis M, Parker CA, Alvarez RH, Valero V, Ueno NT, De Placido S, et al. Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol Cancer Ther. 2012; 11:2526–2534.
45. Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, Mavroudis D, Georgoulias V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010; 288:99–106.
46. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013; 19:1438–1449.
47. Fidler IJ. Metastasis: guantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2'-deoxyuridine. J Natl Cancer Inst. 1970; 45:773–782.
48. Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res. 1975; 35:218–224.
49. Fehm T, Sagalowsky A, Clifford E, Beitsch P, Saboorian H, Euhus D, Meng S, Morrison L, Tucker T, Lane N, Ghadimi BM, Heselmeyer-Haddad K, Ried T, et al. Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin Cancer Res. 2002; 8:2073–2084.
50. Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, Goodman JC, Groves MD, Marchetti D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013; 5:180ra148.
51. Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E, Lilja H, Schwartz L, Larson S, Fleisher M, Scher HI. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007; 13:7053–7058.
52. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ, Raghavan D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008; 14:6302–6309.
53. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004; 351:781–791.
54. Bidard FC, Peeters DJ, Fehm T, Nole F, Gisbert-Criado R, Mavroudis D, Grisanti S, Generali D, Garcia-Saenz JA, Stebbing J, Caldas C, Gazzaniga P, Manso L, et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014; 15:406–414.
55. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen LW, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26:3213–3221.
56. Rao CG, Chianese D, Doyle GV, Miller MC, Russell T, Sanders RA, Jr., Terstappen LW. Expression of epithelial cell adhesion molecule in carcinoma cells present in blood and primary and metastatic tumors. Int J Oncol. 2005; 27:49–57.
57. Naito T, Tanaka F, Ono A, Yoneda K, Takahashi T, Murakami H, Nakamura Y, Tsuya A, Kenmotsu H, Shukuya T, Kaira K, Koh Y, Endo M, et al. Prognostic impact of circulating tumor cells in patients with small cell lung cancer. J Thorac Oncol. 2012; 7:512–519.
58. Krebs MG, Sloane R, Priest L, Lancashire L, Hou JM, Greystoke A, Ward TH, Ferraldeschi R, Hughes A, Clack G, Ranson M, Dive C, Blackhall FH. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J Clin Oncol. 2011; 29:1556–1563.
59. Nichols AC, Lowes LE, Szeto CC, Basmaji J, Dhaliwal S, Chapeskie C, Todorovic B, Read N, Venkatesan V, Hammond A, Palma DA, Winquist E, Ernst S, et al. Detection of circulating tumor cells in advanced head and neck cancer using the CellSearch system. Head Neck. 2012; 34:1440–1444.
60. Han L, Chen W, Zhao Q. Prognostic value of circulating tumor cells in patients with pancreatic cancer: a meta-analysis. Tumour Biol. 2014; 35:2473–2480.
61. Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009; 27:3742–3748.
62. Olmos D, Arkenau HT, Ang JE, Ledaki I, Attard G, Carden CP, Reid AH, A'Hern R, Fong PC, Oomen NB, Molife R, Dearnaley D, Parker C, et al. Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience. Ann Oncol. 2009; 20:27–33.
63. Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, Doyle GV, Matera J, Allard WJ, Miller MC, Fritsche HA, Hortobagyi GN, Terstappen LW. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol. 2005; 23:1420–1430.
64. Budd GT, Cristofanilli M, Ellis MJ, Stopeck A, Borden E, Miller MC, Matera J, Repollet M, Doyle GV, Terstappen LW, Hayes DF. Circulating tumor cells versus imaging—predicting overall survival in metastatic breast cancer. Clin Cancer Res. 2006; 12:6403–6409.
65. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008; 359:366–377.
66. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–1209.
67. Seiden MV, Kantoff PW, Krithivas K, Propert K, Bryant M, Haltom E, Gaynes L, Kaplan I, Bubley G, DeWolf W, Sklar J. Detection of circulating tumor cells in men with localized prostate cancer. J Clin Oncol. 1994; 12:2634–2639.
68. McBride DJ, Orpana AK, Sotiriou C, Joensuu H, Stephens PJ, Mudie LJ, Hamalainen E, Stebbings LA, Andersson LC, Flanagan AM, Durbecq V, Ignatiadis M, Kallioniemi O, et al. Use of cancer-specific genomic rearrangements to quantify disease burden in plasma from patients with solid tumors. Genes Chromosomes Cancer. 2010; 49:1062–1069.
69. Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. 2008; 8:329–340.
70. Klein CA, Seidl S, Petat-Dutter K, Offner S, Geigl JB, Schmidt-Kittler O, Wendler N, Passlick B, Huber RM, Schlimok G, Baeuerle PA, Riethmuller G. Combined transcriptome and genome analysis of single micrometastatic cells. Nat Biotechnol. 2002; 20:387–392.
71. Mostert B, Jiang Y, Sieuwerts AM, Wang H, Bolt-de Vries J, Biermann K, Kraan J, Lalmahomed Z, van Galen A, de Weerd V, van der Spoel P, Ramirez-Moreno R, Verhoef C, et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J Cancer. 2013; 133:130–141.
72. Jiang Y, Palma JF, Agus DB, Wang Y, Gross ME. Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer. Clin Chem. 2010; 56:1492–1495.
73. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015; 1:582–591.
74. Onstenk W, Sieuwerts AM, Kraan J, Van M, Nieuweboer AJ, Mathijssen RH, Hamberg P, Meulenbeld HJ, De Laere B, Dirix LY, van Soest RJ, Lolkema MP, Martens JW, et al. Efficacy of Cabazitaxel in Castration-resistant Prostate Cancer Is Independent of the Presence of AR-V7 in Circulating Tumor Cells. Eur Urol. 2015.
75. Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen PI, Morgan B, Trautwein J, Kimura A, Sengupta S, Stott SL, et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med. 2013; 5:179ra147.
76. Markou A, Strati A, Malamos N, Georgoulias V, Lianidou ES. Molecular characterization of circulating tumor cells in breast cancer by a liquid bead array hybridization assay. Clin Chem. 2011; 57:421–430.
77. Sieuwerts AM, Mostert B, Bolt-de Vries J, Peeters D, de Jongh FE, Stouthard JM, Dirix LY, van Dam PA, Van Galen A, de Weerd V, Kraan J, van der Spoel P, Ramirez-Moreno R, et al. mRNA and microRNA expression profiles in circulating tumor cells and primary tumors of metastatic breast cancer patients. Clin Cancer Res. 2011; 17:3600–3618.
78. Kreso A, O'Brien CA, van Galen P, Gan OI, Notta F, Brown AM, Ng K, Ma J, Wienholds E, Dunant C, Pollett A, Gallinger S, McPherson J, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013; 339:543–548.
79. Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, Ulman M, Smas ME, Lord JB, Brannigan BW, Trautwein J, Bander NH, Wu CL, Sequist LV, et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov. 2012; 2:995–1003.
80. Dago AE, Stepansky A, Carlsson A, Luttgen M, Kendall J, Baslan T, Kolatkar A, Wigler M, Bethel K, Gross ME, Hicks J, Kuhn P. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS One. 2014; 9:e101777.
81. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013; 339:580–584.
82. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011; 192:373–382.
83. Alix-Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013; 59:110–118.
84. Maheswaran S, Haber DA. Circulating tumor cells: a window into cancer biology and metastasis. Curr Opin Genet Dev. 2010; 20:96–99.
85. Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Molecular analysis of circulating tumour cells-biology and biomarkers. Nat Rev Clin Oncol. 2014; 11:129–144.
86. Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014; 14:623–631.
87. Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014; 4:650–661.
88. Heitzer E, Auer M, Ulz P, Geigl JB, Speicher MR. Circulating tumor cells and DNA as liquid biopsies. Genome Med. 2013; 5:73.
89. Pailler E, Adam J, Barthelemy A, Oulhen M, Auger N, Valent A, Borget I, Planchard D, Taylor M, Andrere F, Soria JC, Vielh P, Besse B, Farace F. Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK-positive non-small-cell lung cancer. J Clin Oncol. 2013; 31:2273–2281.
90. Khoo BL, Warkiani ME, Tan DS, Bhagat AA, Irwin D, Lau DP, Lim AS, Lim KH, Krisna SS, Lim WT, Yap YS, Lee SC, Soo RA, et al. Clinical validation of an ultra high-throughput spiral microfluidics for the detection and enrichment of viable circulating tumor cells. PLoS One. 2014; 9:e99409.
91. Ilie M, Long E, Butori C, Hofman V, Coelle C, Mauro V, Zahaf K, Marquette CH, Mouroux J, Paterlini-Brechot P, Hofman P. ALK-gene rearrangement: a comparative analysis on circulating tumour cells and tumour tissue from patients with lung adenocarcinoma. Ann Oncol. 2012; 23:2907–2913.
92. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008; 8:11–23.
93. Scagliotti G, Stahel RA, Rosell R, Thatcher N, Soria JC. ALK translocation and crizotinib in non-small cell lung cancer: an evolving paradigm in oncology drug development. Eur J Cancer. 2012; 48:961–973.
94. Khozin S, Blumenthal GM, Zhang L, Tang S, Brower M, Fox E, Helms W, Leong R, Song P, Pan Y, Liu Q, Zhao P, Zhao H, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res. 2015; 21:2436–2439.
95. Pailler E, Auger N, Lindsay CR, Vielh P, Islas-Morris-Hernandez A, Borget I, Ngo-Camus M, Planchard D, Soria JC, Besse B, Farace F. High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Ann Oncol. 2015; 26:1408–1415.
96. Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, A’Hern R, Levink R, Coumans F, Moreira J, Riisnaes R, Oommen NB, Hawche G, Jameson C, et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009; 69:2912–2918.
97. Danila DC, Anand A, Sung CC, Heller G, Leversha MA, Cao L, Lilja H, Molina A, Sawyers CL, Fleisher M, Scher HI. TMPRSS2-ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration-resistant prostate cancer patients treated with abiraterone acetate. Eur Urol. 2011; 60:897–904.
98. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, Wilson RK. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
99. Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005; 24:1477–1480.
100. Neves RP, Raba K, Schmidt O, Honisch E, Meier-Stiegen F, Behrens B, Mohlendick B, Fehm T, Neubauer H, Klein CA, Polzer B, Sproll C, Fischer JC, et al. Genomic high-resolution profiling of single CKpos/CD45neg flow-sorting purified circulating tumor cells from patients with metastatic breast cancer. Clin Chem. 2014; 60:1290–1297.
101. Polzer B, Medoro G, Pasch S, Fontana F, Zorzino L, Pestka A, Andergassen U, Meier-Stiegen F, Czyz ZT, Alberter B, Treitschke S, Schamberger T, Sergio M, et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol Med. 2014; 6:1371–1386.
102. De Laere B, Peeters D, Salgado RF, Vermeulen P, van Dam P, Dirix LY, Van Laere S. Exploring the intra-patient PIK3CA mutational heterogeneity of circulating tumour cells by massive parallel sequencing in patients with metastatic hormone receptor-positive breast cancer. Annals of Oncology. 2015; 26:iii15.
103. Deng G, Krishnakumar S, Powell AA, Zhang H, Mindrinos MN, Telli ML, Davis RW, Jeffrey SS. Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer. 2014; 14:456.
104. Pestrin M, Salvianti F, Galardi F, De Luca F, Turner N, Malorni L, Pazzagli M, Di Leo A, Pinzani P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol Oncol. 2015; 9:749–757.
105. Gasch C, Bauernhofer T, Pichler M, Langer-Freitag S, Reeh M, Seifert AM, Mauermann O, Izbicki JR, Pantel K, Riethdorf S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem. 2013; 59:252–260.
106. De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011; 12:594–603.
107. Steinert G, Scholch S, Niemietz T, Iwata N, Garcia SA, Behrens B, Voigt A, Kloor M, Benner A, Bork U, Rahbari NN, Buchler MW, Stoecklein NH, et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014; 74:1694–1704.
108. Fabbri F, Carloni S, Zoli W, Ulivi P, Gallerani G, Fici P, Chiadini E, Passardi A, Frassineti GL, Ragazzini A, Amadori D. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013; 335:225–231.
109. Kalikaki A, Politaki H, Souglakos J, Apostolaki S, Papadimitraki E, Georgoulia N, Tzardi M, Mavroudis D, Georgoulias V, Voutsina A. KRAS genotypic changes of circulating tumor cells during treatment of patients with metastatic colorectal cancer. PLoS One. 2014; 9:e104902.
110. Hou S, Zhao L, Shen Q, Yu J, Ng C, Kong X, Wu D, Song M, Shi X, Xu X, OuYang WH, He R, Zhao XZ, et al. Polymer nanofiber-embedded microchips for detection, isolation, and molecular analysis of single circulating melanoma cells. Angew Chem Int Ed Engl. 2013; 52:3379–3383.
111. Fernandez SV, Bingham C, Fittipaldi P, Austin L, Palazzo J, Palmer G, Alpaugh K, Cristofanilli M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res. 2014; 16:445.
112. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, Polanski R, Burt DJ, Simpson KL, Morris K, Pepper SD, Nonaka D, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014; 20:897–903.
113. Marchetti A, Del Grammastro M, Felicioni L, Malatesta S, Filice G, Centi I, De Pas T, Santoro A, Chella A, Brandes AA, Venturino P, Cuccurullo F, Crino L, et al. Assessment of EGFR mutations in circulating tumor cell preparations from NSCLC patients by next generation sequencing: toward a real-time liquid biopsy for treatment. PLoS One. 2014; 9:e103883.
114. Magbanua MJ, Sosa EV, Scott JH, Simko J, Collins C, Pinkel D, Ryan CJ, Park JW. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer. 2012; 12:78.
115. Ulmer A, Schmidt-Kittler O, Fischer J, Ellwanger U, Rassner G, Riethmuller G, Fierlbeck G, Klein CA. Immunomagnetic enrichment, genomic characterization, and prognostic impact of circulating melanoma cells. Clin Cancer Res. 2004; 10:531–537.
116. Magbanua MJ, Melisko M, Roy R, Sosa EV, Hauranieh L, Kablanian A, Eisenbud LE, Ryazantsev A, Au A, Scott JH, Park JW. Molecular profiling of tumor cells in cerebrospinal fluid and matched primary tumors from metastatic breast cancer patients with leptomeningeal carcinomatosis. Cancer Res. 2013; 73:7134–7143.
117. Leversha MA, Han J, Asgari Z, Danila DC, Lin O, Gonzalez-Espinoza R, Anand A, Lilja H, Heller G, Fleisher M, Scher HI. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin Cancer Res. 2009; 15:2091–2097.
118. Bednarz N, Eltze E, Semjonow A, Rink M, Andreas A, Mulder L, Hannemann J, Fisch M, Pantel K, Weier HU, Bielawski KP, Brandt B. BRCA1 loss preexisting in small subpopulations of prostate cancer is associated with advanced disease and metastatic spread to lymph nodes and peripheral blood. Clin Cancer Res. 2010; 16:3340–3348.
119. Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat Methods. 2014; 11:22–24.
120. Bahar Halpern K, Tanami S, Landen S, Chapal M, Szlak L, Hutzler A, Nizhberg A, Itzkovitz S. Bursty gene expression in the intact mammalian liver. Mol Cell. 2015; 58:147–156.
121. Chubb JR, Liverpool TB. Bursts and pulses: insights from single cell studies into transcriptional mechanisms. Curr Opin Genet Dev. 2010; 20:478–484.
122. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010; 141:69–80.
123. Peeters DJ, De Laere B, Van den Eynden GG, Van Laere SJ, Rothe F, Ignatiadis M, Sieuwerts AM, Lambrechts D, Rutten A, van Dam PA, Pauwels P, Peeters M, Vermeulen PB, et al. Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from CellSearch enriched blood samples using dielectrophoretic cell sorting. Br J Cancer. 2013; 108:1358–1367.
124. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014; 158:1110–1122.
125. Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, Aceto N, Bersani F, Brannigan BW, Xega K, Ciciliano JC, Zhu H, MacKenzie OC, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014; 8:1905–1918.
126. Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J, Arora KS, Desai N, Dahl DM, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015; 349:1351–1356.
127. Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 2010; 5:516–535.
128. Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, Schroth GP, Sandberg R. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012; 30:777–782.
129. Chen CL, Mahalingam D, Osmulski P, Jadhav RR, Wang CM, Leach RJ, Chang TC, Weitman SD, Kumar AP, Sun L, Gaczynska ME, Thompson IM, Huang TH. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate. 2013; 73:813–826.
130. Pizon M, Zimon D, Carl S, Pachmann U, Pachmann K, Camara O. Heterogeneity of circulating epithelial tumour cells from individual patients with respect to expression profiles and clonal growth (sphere formation) in breast cancer. Ecancermedicalscience. 2013; 7:343.
131. Cann GM, Gulzar ZG, Cooper S, Li R, Luo S, Tat M, Stuart S, Schroth G, Srinivas S, Ronaghi M, Brooks JD, Talasaz AH. mRNA-Seq of single prostate cancer circulating tumor cells reveals recapitulation of gene expression and pathways found in prostate cancer. PLoS One. 2012; 7:e49144.
132. Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One. 2012; 7:e33788.
133. Gorges TM, Tinhofer I, Drosch M, Rose L, Zollner TM, Krahn T, von Ahsen O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012; 12:178.
134. Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012; 338:1622–1626.
135. Swennenhuis JF, Reumers J, Thys K, Aerssens J, Terstappen LW. Efficiency of whole genome amplification of single circulating tumor cells enriched by CellSearch and sorted by FACS. Genome Med. 2013; 5:106.
136. Binder V, Bartenhagen C, Okpanyi V, Gombert M, Moehlendick B, Behrens B, Klein HU, Rieder H, Ida Krell PF, Dugas M, Stoecklein NH, Borkhardt A. A new workflow for whole-genome sequencing of single human cells. Hum Mutat. 2014; 35:1260–1270.
137. Voet T, Kumar P, Van Loo P, Cooke SL, Marshall J, Lin ML, Zamani Esteki M, Van der Aa N, Mateiu L, McBride DJ, Bignell GR, McLaren S, Teague J, et al. Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res. 2013; 41:6119–6138.
138. Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, Wu H, Ye X, Ye C, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012; 148:873–885.
139. Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014; 512:155–160.
140. Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015; 25:1499–1507.
141. Dancey JE, Bedard PL, Onetto N, Hudson TJ. The genetic basis for cancer treatment decisions. Cell. 2012; 148:409–420.
142. Bidard FC, Fehm T, Ignatiadis M, Smerage JB, Alix-Panabieres C, Janni W, Messina C, Paoletti C, Muller V, Hayes DF, Piccart M, Pierga JY. Clinical application of circulating tumor cells in breast cancer: overview of the current interventional trials. Cancer Metastasis Rev. 2013; 32:179–188.
143. Eggermont AM, Caldas C, Ringborg U, Medema R, Tabernero J, Wiestler O. Cancer Core Europe: a consortium to address the cancer care-cancer research continuum challenge. Eur J Cancer. 2014; 50:2745–2746.
144. Lawler M, Siu LL, Rehm HL, Chanock SJ, Alterovitz G, Burn J, Calvo F, Lacombe D, Teh BT, North KN, Sawyers CL. All the World's a Stage: Facilitating Discovery Science and Improved Cancer Care through the Global Alliance for Genomics and Health. Cancer Discov. 2015; 5:1133–1136.
145. Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez-Farre X, Munoz M, Russnes HG, Helland A, Rye IH, Borresen-Dale AL, Maruyama R, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014; 6:514–527.