
INTRODUCTION
Cancer is a complex group of diseases with many possible causes. It can be partly explain as a result of a progressive accumulation of mutations in cellular DNA, which provides a selective growth advantage to cancer cells and facilitates metastasis. Hotspot mutations are frequently present within oncogenes while some other aberrations are found in tumor suppressor genes in common solid tumors. The deregulation of certain signaling pathways, together with chromosomal abnormalities, has been identified in different solid tumors. Different oncogenic events have been described in cancer including mainly mutations in the RAS/RAF/MAPK and the PIK3/PTEN/AKT pathways. Therefore, mutations affecting the coding sequences of these specific genes are the hallmark of the disease and are currently targeted in clinical trials [1].
Our knowledge of cancer genomics has been enabled by the genome sequencing and other high-throughput omics technologies, leading to the discovery of new targets [2]. The development of targeted drugs has allowed for a more precise and personalized therapy, something which could be of major benefit to the patients. This drug sensitivity approach is reinforced by the efficacy shown in clinical trials using epidermal growth factor receptor (EGFR) and BRAF tyrosine kinase inhibitors (TKIs) [3–6]. The discovery of activating mutations located in the tyrosine kinase domains of EGFR has expanded the therapeutic options of lung cancer patients since they can be treated by EGFR-TKIs [7]. In metastatic colorectal cancer (mCRC) patients whose tumors are wild type for all KRAS/NRAS alleles, the administration of monoclonal antibodies against EGFR, such as cetuximab and panitumumab, in combination with conventional chemotherapy, substantially improves survival [8–10]. The presence of KRAS and NRAS mutations acts as a negative predictor to sensitivity to anti-EGFR monoclonal antibody therapy and, therefore, has caused an important change in the treatment of mCRC. The presence of the BRAF V600E activating mutation, found in approximately half of the diagnosed melanomas, is a turning point in the treatment of the metastatic disease through BRAF-TIKs [3, 11]. The use of targeted drugs against the oncogenic alterations of the KRAS gene and/or its downstream components (e.g. BRAF, MEK) seems to be a promising approach to cancer therapeutics either alone or in combination with other targeted agents [12–14].
Somatic mutation analysis has become a useful tool in selecting personalized therapies for many solid tumors. Mutation profiling can assist in the prognosis, prediction and treatment of solid tumors. Thus, molecular stratification for genotype-directed therapy could be required [15]. The mass spectrometry technique, matrix-assisted laser desorption/ionization-time of flight, has been used to assess point mutations across different solid tumors [16]. The Sequenom MassARRAY technology, in combination with a commercial kit called OncoCarta v1.0, was used to screen 238 somatic mutations across 19 oncogenes. This mutation panel interrogates somatic changes in oncogenes with known responses or resistance-targeted therapy. Custom assays can be also incorporated into the whole design, permitting the detection of specific target genes.
The goal of this study was to characterize the presence of common somatic mutations affecting known oncogenes in resected solid tumors that could provide potential therapeutic targets.
RESULTS
Patient characteristics
The median age of the patients was 58 years. The study included individuals with advanced-stage tumors who had received at least one line of treatment (67.4%).
The different tumor types representing the 197 enrolled patients were colorectal cancer (n= 75), breast cancer (n=73), ovarian cancer (n=10), lung cancer (n=9, 8 adenocarcinoma and 1 squamous), endometrial cancer (n=8) and other tumor types (n=20), including cervical, gastric, pancreatic, melanoma, anal, appendiceal, esophageal, renal, oral cavity and thyroid tumors. Formalin-fixed paraffin-embedded (FFPE) primary tumor samples were obtained for 123 (62.4%) subjects with nodal and/or metastatic tumor samples being available for a further 73 (37.1%) patients. The clinical characteristics of the patients have been included in Table 1. Colorectal and breast carcinoma were the two most represented tumor types with 75 and 73 cases enrolled, respectively (Supplementary Table S1).
Table 1: Classification of the samples studied by age and clinical characteristics (N=197)
Clinical characteristic |
N (%) |
|---|---|
Age (years) |
|
Median (Range) |
58 (27-88) |
Gender |
|
Female |
129 (65.5) |
Male |
68 (34.5) |
Tumor type |
|
Colorectal cancer |
75 (38.1) |
Breast cancer |
73 (37.1) |
Ovarian cancer |
10 (5.1) |
Lung cancer |
9 (4.6) |
Endometrial cancer |
8 (4.0) |
Others* |
20 (10.1) |
Unknown |
2 (1.0) |
Prior therapy |
|
No treatment |
52 (26.4) |
One line of treatment |
70 (35.5) |
Two lines of treatment |
32 (16.2) |
Three or more lines of treatment |
31 (15.7) |
Unknown |
12 (6.0) |
Origen of the samples |
|
Primary tumor |
123 (62.4) |
Metastasis |
73 (37.1) |
Unknown |
1 (0.5) |
*Others include cervical (4), gastric (4), pancreas (4), melanoma (2), anal (1), appendiceal (1), esophageal (1), renal (1), oral cavity (1) and thyroid (1) cancer
Mutational detection
A total of 197 samples were subjected to a hotspot mutation screening of 25 known cancer genes using the OncoCarta Panel v1.0 (Sequenom, San Diego, CA) and two customized panels. Mutations with frequencies higher than 10% were detected with high accuracy. One hundred and thirty-four oncogenic mutations were detected in 97 (49.2%) patients, and these mutations were found in the KRAS, PIK3CA, KIT, MET, RET, NRAS, EGFR, BRAF, CDK4, GNAS, ABL1, AKT1, AKT3, PDGFRA, IDH1, ERBB2 and ERBB3 genes (Figure 1 and Supplementary Figure S1). A total of 49 different oncogenic mutations were identified, 33 (80.5%) of them base transitions. The RAS/RAF/MAPK and the PIK3/AKT pathways were the most frequently mutated with 50 (51.5%) and 35 (36.1%) tumors mutated, respectively. Mutations in the KRAS gene were detected in 40/97 (41.2%) patients whereas mutations in the PIK3CA gene were detected in 30/97 (30.9%) patients. See Supplementary Table S2. Furthermore, 31 patients had mutations in at least two genes (32.0%), 2 of them carriers of synchronous mutations within the PIK3CA oncogene. Moreover, 3 of the samples carried more than two different mutations.

Figure 1: Genomic mutations found across the different solid tumor types enrolled in the study. Selected genes are mutated in at least one tumor sample. Samples with mutations are shown in black. A. Colorectal samples; B. Breast cancer samples; C. Ovarian cancer samples; D. Lung cancer samples; E. Endometrial cancer samples, F. Other tumor samples (oral cavity, cervical, melanoma, gastric, anal, renal, pancreatic, appendiceal, esophageal and thyroid cancers). The histogram represents the percentage of gene mutation across the different tumor types. Colorectal cancer and breast cancer are represented with dots and with lines, respectively, while all other tumors are represented together in black.
Twenty of the 31 cases with co-occurrence mutations (64.5%) were initially diagnosed with colorectal cancer. First, the co-occurrence of mutations within KRAS and PIK3CA was found in 8 (25.8%) patients. KRAS mutations were mainly located within exon 2, affecting G12 and G13 amino-acids, whereas PIK3CA mutations were mainly located in the helical domain, in positions 420, 452 and 546. Second, the mutations found in KIT and PIK3CA were found in 6 (19.4%) patients. These mutations affected amino-acids D52 and E839 in KIT and E542, E545 and H1047 in PIK3CA. Interestingly, mutation E839K in KIT appeared exclusively with the PIK3CA E452K mutation. Last, the co-mutations in KIT and RET were present in 4 (12.9%) patients. These mutations were D52N in the KIT gene and C634W in the RET gene (Table 2 and Figure 2).
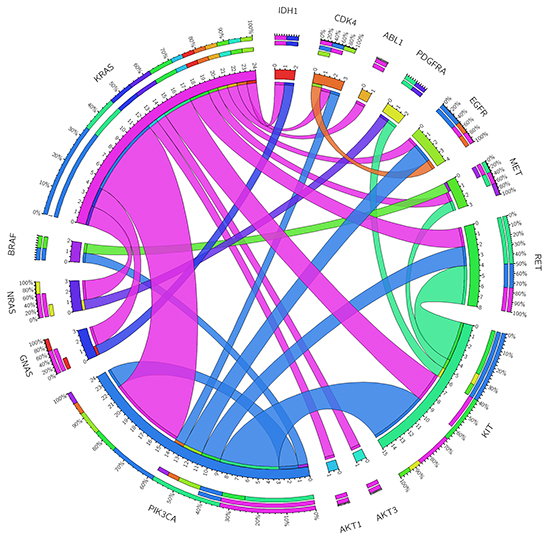
Figure 2: Genomic co-occurrence mutations found across those tumor samples with two or more mutations. The length of the arc corresponds to the frequency of mutations in the first gene, and the width of the ribbon corresponds to the percentage of patients who also had a mutation in the second gene. This diagram was obtained using the Circos software (http://mkweb.bcgsc.ca/tableviewer/visualize/).
Table 2: Samples with co-occurrence of mutations
Sample |
Type of Cancer |
Most frequently mutated genes |
Least frequently mutated genes |
||||||
|---|---|---|---|---|---|---|---|---|---|
KRAS |
PIK3CA |
KIT |
RET |
Gene |
Mutation |
Gene |
Mutation |
||
INV063 |
Breast, liver mets |
G12D |
ABL1 |
Y253H |
|||||
INV110 |
Rectal |
G13D |
AKT1 |
E17K |
|||||
INV086 |
Colon |
G13D |
AKT3 |
G171R |
|||||
INV034 |
Cervix |
G12D |
GNAS |
R201H |
|||||
INV198 |
Colon |
G12D |
GNAS |
R201H |
|||||
INV161 |
Colon |
G12C |
KIT |
D52N |
|||||
INV005 |
Rectal, lung mets |
G12D |
MET |
R970C |
|||||
INV017 |
Rectal |
A146V |
NRAS |
G12S |
|||||
INV186 |
Colon |
G13D |
NRAS |
G13D |
|||||
INV016 |
Colon |
G12D |
E542K |
D52N |
C634W |
||||
INV163 |
Colon |
G13D |
E542K & H1047R |
D52N |
|||||
INV042 |
Breast |
Q61R |
C420R |
CDK4 |
R24C |
EGFR |
P772_H773InsV |
||
INV028 |
Colon |
G12D |
E542K |
||||||
INV059 |
Colon |
G12S |
E542K |
||||||
INV084 |
Colon |
G13D |
Q546R |
||||||
INV181 |
Colon |
G12V |
Y1021C |
||||||
INV185 |
Colon |
G12C |
G1049R |
||||||
INV001 |
Colon |
G12D |
C634Y |
||||||
INV045 |
Colon |
C420R |
BRAF |
V600E |
|||||
INV054 |
Colon |
G1049R |
EGFR |
D770_N771>AGG |
|||||
INV177 |
Breast |
E545K |
D52N |
||||||
INV036 |
Cervix |
E542K |
E839K |
||||||
INV141 |
Colon, liver mets |
H1047R |
D52N |
C634W |
|||||
INV126 |
Breast |
E542K |
E839K |
||||||
INV088 |
Breast, pleural mets |
E545K & G1049R |
|||||||
INV134 |
Breast, lung mets |
L576P |
MET |
N375S |
|||||
INV055 |
Rectal |
D52N |
PDGFRA |
D842V |
|||||
INV081 |
Colon |
D52N |
C634W |
||||||
INV071 |
Breast, pleural mets |
D52N |
C634W |
||||||
INV011 |
Kidney |
MET |
R970C |
BRAF |
L597S |
||||
INV023 |
Ovary |
NRAS |
G13D |
PDGFRA |
D1071N |
||||
Most and least frequently mutated genes in samples with co-occurrence mutations
mets: metastasis
Bold represent the most frequent associations: KRAS + PIK3CA; PIK3CA + KIT and KIT + RET
The concordance between the OncoCarta Panel v1.0 and the customized panels was 90.0%. Moreover, the concordance between the OncoCarta Panel v1.0 and Junior NGS technology was 88.0% (data not shown). In the present study, taking into account the mutations with frequencies higher than 10%, the sensibility and specificity were 79.0% and 93.5%, respectively. Those samples with non-concordant results had low allelic frequency mutations.
Association with clinical characteristics
Association with clinical characteristics was performed for the two most represented tumor types.
Colorectal cancer
Overall, mutations were detected in 48 of 75 (64.2%) available FFPE tumors, predominantly in primary tumor samples (37/48, 77.1%) (Table 3 and Supplementary Table S1). Specifically, KRAS, PIK3CA and KIT mutations were detected in 31/48 (64.6%), 11/48 (22.9%), 8/48 (16.7%) tumor specimens, respectively. There was (66/75) 88.0% concordance for FFPE tumoral mutation status between the OncoCarta Sequenom panel and the next generation sequencing (NGS) Junior (Roche). Mutations in five of the samples found by NGS were not detected by Sequenom (KRAS p.G12C 10.8%, p.Q61K 47.1% and p.A146T 11.6%, and p.Q61L 13.7% and BRAF p.V600E 12.3%; percentages represent the frequency of mutant alleles). Four of them were close to the 10% threshold established. Among the other 4 samples, mutations were detected only by the Sequenom technology (KRAS p.G12D 15.0% and p.G13D 15.2%, NRAS p.G13D 21.4% and PIK3CA p.H1047R 23.0%).
Table 3: Mutation distribution across colorectal cancer samples
Sample |
Location |
Type |
Gene |
Mutation |
%M |
Gene |
Mutation |
%M |
Gene |
Mutation |
%M |
Gene |
Mutation |
%M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
INV001 |
Left colon |
Primary |
KRAS |
G12D |
42.7 |
RET |
C634Y |
10.5 |
||||||
INV004 |
Left colon |
Primary |
KRAS |
A146V |
23.3 |
|||||||||
INV014 |
Left colon |
Primary |
KRAS |
G12D |
35.3 |
|||||||||
INV086 |
Left colon |
Primary |
KRAS |
G13D |
39.8 |
AKT3 |
G171R |
15.1 |
||||||
INV104 |
Left colon |
Primary |
KRAS |
G12V |
31.6 |
|||||||||
INV164 |
Left colon |
Primary |
KRAS |
G12D |
33.7 |
|||||||||
INV060 |
Left colon |
Metastasis |
KRAS |
Q61R |
36.9 |
|||||||||
INV008 |
Left colon |
Primary |
PIK3CA |
G1049R |
14.5 |
|||||||||
INV054 |
Left colon |
Primary |
PIK3CA |
G1049R |
10.6 |
EGFR |
D770_N771>AGG |
11.1 |
||||||
INV138 |
Left colon |
Metastasis |
PIK3CA |
G1049R |
10.7 |
|||||||||
INV081 |
Left colon |
Primary |
KIT |
D52N |
27.6 |
RET |
C634W |
31.8 |
||||||
INV161 |
Left colon |
Primary |
KIT |
D52N |
14.6 |
KRAS |
G12C |
24.1 |
||||||
INV141 |
Left colon |
Metastasis |
KIT |
D52N |
47.2 |
PIK3CA |
H1047R |
23.0 |
RET |
C634W |
42.4 |
|||
INV154 |
Left colon |
Metastasis |
MET |
N375S |
34.2 |
|||||||||
INV180 |
Left colon |
Primary |
AKT1 |
E17K |
36.6 |
|||||||||
INV186 |
Left colon |
Primary |
KRAS |
G13D |
15.2 |
NRAS |
G13D |
21.4 |
||||||
INV196 |
Left colon |
Primary |
KRAS |
G12V |
14.3 |
|||||||||
INV201 |
Left colon |
Primary |
NRAS |
G12D |
24.1 |
|||||||||
INV016 |
Right colon |
Primary |
KRAS |
G12D |
10.0 |
KIT |
D52N |
10.4 |
PIK3CA |
E542K |
13.4 |
RET |
C634W |
23.5 |
INV026 |
Right colon |
Primary |
KRAS |
G12D |
35.0 |
|||||||||
INV028 |
Right colon |
Primary |
KRAS |
G12D |
14.0 |
PIK3CA |
E542K |
14.4 |
||||||
INV031 |
Right colon |
Primary |
KRAS |
G12D |
38.6 |
|||||||||
INV059 |
Right colon |
Primary |
KRAS |
G12S |
23.4 |
PIK3CA |
E542K |
19.4 |
||||||
INV066 |
Right colon |
Primary |
KRAS |
G13D |
40.9 |
|||||||||
INV084 |
Right colon |
Primary |
KRAS |
G13D |
24.6 |
PIK3CA |
Q546R |
12.7 |
||||||
INV163 |
Right colon |
Primary |
KRAS |
G13D |
27.0 |
KIT |
D52N |
20.0 |
PIK3CA |
E542K |
14.6 |
PIK3CA |
H1047R |
23.1 |
INV082 |
Right colon |
Metastasis |
KRAS |
A59T |
19.5 |
|||||||||
INV045 |
Right colon |
Primary |
BRAF |
V600E |
15.7 |
PIK3CA |
C420R |
23.3 |
||||||
INV181 |
Right colon |
Primary |
KRAS |
G12V |
31.1 |
PIK3CA |
Y1021C |
72.0 |
||||||
INV185 |
Right colon |
Primary |
KRAS |
G12C |
15.0 |
PIK3CA |
G1049R |
15.0 |
||||||
INV193 |
Right colon |
Primary |
KRAS |
G12S |
26.9 |
|||||||||
INV197 |
Right colon |
Primary |
BRAF |
V600E |
11.5 |
|||||||||
INV198 |
Right colon |
Primary |
KRAS |
G12D |
67.5 |
GNAS |
R201H |
31.1 |
IDH1 |
R132C |
44 |
|||
INV017 |
Rectum |
Primary |
KRAS |
A146V |
10.8 |
NRAS |
G12S |
46.0 |
||||||
INV110 |
Rectum |
Primary |
KRAS |
G13D |
36.5 |
AKT1 |
E17K |
33.9 |
||||||
INV005 |
Rectum |
Metastasis |
KRAS |
G12D |
49.4 |
MET |
R970C |
48.9 |
||||||
INV018 |
Rectum |
Primary |
NRAS |
Q61R |
23.8 |
|||||||||
INV020 |
Rectum |
Primary |
KIT |
D52N |
19.8 |
|||||||||
INV055 |
Rectum |
Primary |
KIT |
D52N |
27.8 |
PDGFRA |
D842V |
20.5 |
||||||
INV030 |
Rectum |
Metastasis |
KIT |
D52N |
16.4 |
|||||||||
INV147 |
Rectum |
Primary |
EGFR |
G719S |
22.7 |
|||||||||
INV079 |
Unknown |
Primary |
KRAS |
G12D |
58.3 |
|||||||||
INV145 |
Unknown |
Primary |
KRAS |
A146T |
16.7 |
|||||||||
INV184 |
Unknown |
Primary |
MET |
N375S |
27.5 |
|||||||||
INV190 |
Unknown |
Metastasis |
KRAS |
G12C |
33.4 |
|||||||||
INV191 |
Unknown |
- |
KRAS |
G12V |
15.4 |
%M represent the percentage of mutant alleles in each reported gene
Breast cancer
Overall, mutations were detected in 34 of 73 (46.6%) available FFPE tumors, predominantly in the metastatic tissue (23/34, 67.6%) (See Table 4 and Supplementary Table S1). Specifically, PIK3CA and KIT mutations were detected in 16 (47.0%), and 8 (23.5%), respectively, of the tumor specimens. There was (70/73) 96.0% concordance for FFPE tumoral mutation status between the OncoCarta Sequenom panel and the NGS Junior. Two samples showed PIK3CA mutations in NGS but not in Sequenom (p.E542K, 29% and p.H1047L 11%; percentages represent the frequency of mutant alleles). The last reported mutation is close to the threshold of detection by Sequenom technology. Finally, the last, fourth, sample showed AKT1 mutation in NGS, but not in Sequenom (p.E17K 44.8%; percentage represents the frequency of mutant alleles).
Table 4: Mutation distribution across breast cancer samples
Sample |
Molecular subtype |
Histology |
Type |
Gene |
Mut |
%M |
Gene |
Mut |
%M |
Gene |
Mut |
%M |
Gene |
Mut |
%M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
INV174 |
Luminal A |
Ductal |
Primary |
KIT |
D52N |
15.3 |
|||||||||
INV096 |
Luminal A |
Ductal |
Metastasis |
KIT |
D52N |
23.8 |
|||||||||
INV095 |
Luminal A |
Ductal |
Metastasis |
KIT |
K550_K558del |
17.6 |
|||||||||
INV107 |
Luminal A |
Lobular |
Metastasis |
PIK3CA |
N345K |
12.5 |
|||||||||
INV033 |
Luminal A |
Ductal |
Metastasis |
PIK3CA |
E542K |
32.0 |
|||||||||
INV117 |
Luminal A |
Ductal |
Metastasis |
PIK3CA |
M1043I |
15.5 |
|||||||||
INV072 |
Luminal A |
Lobular |
Metastasis |
PIK3CA |
H1047R |
15.8 |
|||||||||
INV205 |
Luminal A |
Ductal |
Metastasis |
PIK3CA |
H1047R |
37.7 |
|||||||||
INV169 |
Luminal B |
Ductal |
Primary |
AKT1 |
E17K |
20.4 |
|||||||||
INV170 |
Luminal B |
Ductal |
Metastasis |
AKT1 |
E17K |
59.9 |
|||||||||
INV128 |
Luminal B |
Ductal |
Metastasis |
EGFR |
H773_V774insH |
30.9 |
|||||||||
INV071 |
Luminal B |
Lobular |
Metastasis |
KIT |
D52N |
14.3 |
RET |
C634W |
48.3 |
||||||
INV073 |
Luminal B |
Ductal |
Metastasis |
KIT |
Y553_Q556del |
11.3 |
|||||||||
INV155 |
Luminal B |
Not specified |
Primary |
MET |
N375S |
35.1 |
|||||||||
INV173 |
Luminal B |
Ductal |
Metastasis |
PIK3CA |
E542K |
37.8 |
|||||||||
INV126 |
Luminal B |
Lobular |
Primary |
PIK3CA |
E542K |
12.0 |
KIT |
E839K |
14.4 |
||||||
INV105 |
Luminal B |
Ductal |
Metastasis |
PIK3CA |
E545K |
57.4 |
|||||||||
INV177 |
Luminal B |
Ductal |
Primary |
PIK3CA |
E545K |
59.3 |
KIT |
D52N |
10.0 |
||||||
INV077 |
Luminal B |
Ductal |
Metastasis |
PIK3CA |
E545K |
30.4 |
|||||||||
INV088 |
Luminal B |
Ductal |
Metastasis |
PIK3CA |
E545K |
66.3 |
PIK3CA |
G1049R |
19.2 |
||||||
INV101 |
Luminal B |
Ductal |
Primary |
PIK3CA |
H1047R |
20.5 |
|||||||||
INV092 |
Luminal B |
Ductal |
Primary |
RET |
C634W |
31.3 |
|||||||||
INV042 |
Basal like |
Ductal |
Primary |
PIK3CA |
C420R |
12.1 |
CDK4 |
R24C |
13.2 |
EGFR |
P772_H773insV |
13.2 |
KRAS |
Q61R |
12.4 |
INV057 |
Basal like |
Ductal |
Primary |
PIK3CA |
H1047R |
18.9 |
|||||||||
INV044 |
Basal like |
Lobular |
Metastasis |
PIK3CA |
H1047R |
19.5 |
|||||||||
INV069 |
Her2 |
Ductal |
Metastasis |
PIK3CA |
M1043I |
49.6 |
|||||||||
INV074 |
Her2 |
Tubule-lobular |
Metastasis |
PDGFRA |
D842V |
32.2 |
|||||||||
INV094 |
Her2 |
Ductal |
Primary |
KRAS |
G12D |
14.8 |
|||||||||
INV134 |
Her2 |
Ductal |
Metastasis |
KIT |
L576P |
13.3 |
MET |
N375S |
17.6 |
||||||
INV070 |
Her2 |
Ductal |
Metastasis |
CDK4 |
R24H |
10.4 |
|||||||||
INV063 |
Her2 |
Ductal |
Metastasis |
ABL1 |
Y253H |
12.2 |
KRAS |
G12D |
22.6 |
Mut, mutation; %M represent the percentage of mutant alleles in each reported gene
Personalized therapy
A total of 101 patients could benefit from targeted therapies. Seventy-five of the patients presented potential actionable mutations, whereas an additional 26 patients with colorectal cancer had KRAS wild type status. Among these 101 patients, 28 received genotype-directed therapy (28.0%), including 20 colorectal cancer patients that received clinically available agents. Five of these 20 colorectal cancer patients (25.0%) received anti-EGFR therapy, whereas the rest (15 patients) received other available therapies.
The remaining 8 patients were enrolled in clinical trials. These patients had breast or gynecological malignancies. Seven of them carried a mutation in the PI3KCA gene, and one had a mutation in the ERBB2 gene. Among the PI3KCA mutation carriers, 5 received PI3K/AKT inhibitors. The other 3 received other target drugs, including an anti-IGF1 therapy in one case and an anti-ERBB3 therapy in two patients (See Supplementary Figure S2).
A total of 73 patients who could possibly have benefitted from targeted therapies were not treated. The most common reasons for not offering targeted therapies according to the mutations found were diverse. Consequently, twenty-five patients (34.3%) followed standard therapies. Another 19 (26%) patients did not progress during the study period and did not require a new treatment. The rest 29 (39.7%) were not eligible due to co-morbidities, poor performance status, concurrent secondary neoplasm or loss of follow up.
DISCUSSION
Many different solid tumors contain hotspot mutations within oncogenes that confer a relevant susceptibility or resistance to targeted anticancer therapies. A comprehensive characterization of several cancer genomes has been made possible as a result of the development of NGS technologies. At present, however, these techniques are still not fully cost-effective for the medium-sized clinical laboratory. The analysis of key cancer-driving mutations using mass-spectrometry is a cost-effective, sensitive high throughput approach for identifying mutations of clinical relevance to molecular-based therapy [17].
Sequenom technology has been recently approved for clinical diagnosis, allowing mutation frequencies of as low as 1% to be detected. Although in the present study, mutations with frequencies higher than 10% were considered to be positive, samples were deeply evaluated for their tumor content, and only sections containing more than 30% tumor cells were considered in order to detect targetable aberrations. This threshold percentage was established by others as an accurate and detectable level of rare alleles [17–19]. The present approach focused only on oncogenes hotspots and did not contemplate other mutations or tumor suppressors. Furthermore, the infrequent variations might not have any association with therapy. Therefore, this methodology makes it possible for a medium-sized laboratory to analyse multiple key hotspot mutations rapidly (within 3 days) and without complex bioinformatics analysis tools at a moderate price. At present, NGS technology is becoming more accessible, and the analysis is being simplified. Sequenom technology, however, remains a good validation technology and is optimal when only hotspots are pursued.
In the present study, we have characterized the mutation status of 25 known cancer genes in a large series of 197 solid tumors from various anatomical sites using the Sequenom Platform. The mutation sites included in the Sequenom OncoCarta Panel v1.0 assay are frequently seen in many different types of solid tumors and are clinically actionable. Mutations in 17 different genes at 49 different nucleotide positions were detected in 97 of our cancer patients, of which 28 received targeted therapies. Thus, the overall rate of success in matching patients to personalized treatments was 28 out of 97 (28.0%), similar to other recently published studies [20–22]. This rate includes 20 CRC samples treated both by anti-EGFR, as well as other available therapies. The remaining 8 patients treated were enrolled in clinical trials, most of them against PI3K/AKT inhibitors, in accordance with other publications [23]. In the present series, the KRAS and the PIK3CA genes were the most frequently mutated genes in 41.2% and 30.9% of the mutated patients, respectively. Mutations in these genes disrupt many different and overlapping signaling pathways, including the PI3K/AKT and ERK/MAPK, influencing important cellular processes. Cross-validation of detected mutations was feasible by two customized mass-spectrometry panels and NGS Junior 454 Roche technology with a concordance rate of 90.0% and 88.0%, respectively. Concordance was considered when the same alleles at similar mutation frequencies were detected by the two different panels or techniques. MassARRAY technology’s high sensibility and specificity made the results obtained with this platform highly reproducible.
Colorectal and breast cancer were the two most represented tumor types with 75 and 73 cases enrolled, respectively. Among colorectal cancer samples, mutations were detected in 64.0% of the analyzed tumors, a similar ratio to those previously published [17, 24–26]. In the colorectal cancer set, KRAS (42.5%), PIK3CA (17.8%) and KIT (10.9%) were the most frequently mutated genes. Frequencies for both KRAS and PIK3CA were similar to the COSMIC database and to those of other publications (http://cancer.sanger.ac.uk/cosmic and http://www.cbioportal.org/) (See Supplementary Table S3) [17, 24, 27]. Furthermore, sporadic mutations appeared across RET, BRAF, EGFR, AKT1, AKT3, MET, NRAS, PDGFRA, IDH1 and ERBB3 [24].
Among breast cancer samples, mutations were detected in 46.6% of the analyzed tumors, specifically in PIK3CA and KIT. Mutations among other genes were present in less than 5%, a rate similar to those of the COSMIC database and other studies such as The Cancer Genome Atlas Network (See Supplementary Table S3) [21, 28–30]. PIK3CA mutations were found in 7 (46.7%) luminal B, 4 (26.7%) luminal A, 3 (20.0%) basal-like and 1 (6.7%) HER2 subtypes (Breast cancer subtypes according to Perou and colleagues, 2000) [31]. Nevertheless, half of all the HER2 subtype tumors carried at least one mutation, and PIK3CA mutations were more frequently found in estrogen receptor-positive cancers compared to triple negative breast cancer [28].
At present, KIT mutations are without clinical implications in the current therapeutical approach to colorectal and breast cancer.
The present work focused on individuals with advanced solid tumors and potential candidates to phases I/II clinical trials due to initial treatment failure. Variations in frequencies between our data and other reports may be attributed to advanced tumor selection and the number of samples analyzed.
Interestingly, one third of the patients with mutated tumors had two genes altered, of which two thirds were initially diagnosed as colorectal cancer. Two patients carried synchronous mutations within the PIK3CA oncogene. Among breast cancer samples, co-occurrence appeared mainly in PIK3CA and KIT. In the colorectal cancer cases, however, co-mutation was observed most frequently in the KRAS and PIK3CA genes. The KRAS, NRAS and BRAF mutations in colorectal cancer are normally mutually exclusive. Conversely, the coexistence of mutations in KRAS and PIK3CA has been described in a significant percentage of colorectal tumors, confirming the parallel activation of ERK/MAPK and PI3K/AKT signaling convergent pathways [15, 32].
Remarkably, the co-occurrence of mutations within KRAS and PIK3CA was the most common, in 8 (25.8%) patients. KRAS mutations were mainly located within exon 2, affecting the functionally G12 and G13 amino-acids. Co-existent PIK3CA mutations were mainly located in the helical domain, in positions 420, 452 and 546. The coexistence of PIK3CA and KRAS mutations has been shown in several different tumors types including lung, colorectal, pancreatic and ovarian cancer [33–35].
Mutations found in KIT and PIK3CA were found in 6 (19.4%) patients, having an effect on amino-acids D52 and E839 in KIT and E542, E545 and H1047 in PIK3CA. Interestingly, mutation E839K in KIT appeared exclusively with the PIK3CA E452K mutation. Finally, co-mutations in KIT and RET were present in 4 (12.9%) patients. These mutations were D52N in the KIT gene and C634W in the RET gene. The co-occurrence of mutations in KIT and PIK3CA or RET has been described very little. Results obtained from The Cancer Genome Atlas Network for both colorectal and breast cancer showed the co-existence of mutations in these genes, although in low proportions (4.93% for PIK3CA and KIT and 1.23% for KIT and RET).
These facts suggest that cancer development may progress due to accumulation of different somatic driver mutations, affecting different pathways. At the same time, the presence of several mutations across different genes may point out tumor heterogeneity and suggest the presence of subclones. It is the detection of different clones, some of which may show resistance to therapies, a major concern, that is changing standard therapeutic approaches.
The present study aimed at identifying key alterations that may represent important targets for novel therapies. We used mass-spectrometry, an effective and high throughput approach, which successfully detected frequent cancer mutations in degraded DNA isolated from FFPE samples and provided some advantages in terms of minimizing cost and time. This technology, in combination with the OncoCarta Panel v1.0, covers up to 95% of known druggable markers for an efficient mutation screening in clinical research trials and has an elevated grade of concordance with NGS technologies.
MATERIALS AND METHODS
Patient selection and data collection
The design of the study was exploratory and prospective. A total of 213 consecutive and non-related cancer cases were recruited from September 2013 to December 2014 at the Hematology and Medical Oncology Unit of the Clinic University Hospital in Valencia, Spain. Patient eligibility criteria included clinical and histological diagnoses of advanced solid cancer or potential candidates to phases I/II clinical trials due to initial treatment failure and at least one biopsiable lesion.
Clinical information, including age, sex, tumor type, location and treatments were collected (See Table 1). All study subjects gave written, informed consent, and the study was approved by the Biomedical Research Institute INCLIVA Ethics Committee.
Formalin-fixed paraffin-embedded (FFPE) tissues were evaluated for their tumor content, and sections containing more than 30% tumor cells were defined and cut by an expert pathologist. Genomic DNA was isolated from 4 unstained sections of 20 μm and diluted to a final solution of 10ng/μl. This was done using two extraction kits: Recover All Total Nucleic Acid Isolation kit (Ambiom, Life Technologies) and the QIAamp DNA FFPE tissue kit (QIAGEN). DNA concentration was quantified in samples by NanoDrop (NanoDrop Technologies, Wilmington, DE, USA).
Sixteen cases did not yield DNA of sufficient quantity, and were excluded from further analyses, leaving 197 samples in the study.
Sequenom MassARRAY somatic mutation genotyping
The Sequenom MassARRAY and OncoCarta Panel v1.0 were used following the manufacturer’s protocol (Sequenom, San Diego, CA, USA; (http://agenabio.com/oncocarta-panel)). The panel consisted of 24 multiplex assays capable of detecting 238 mutations in 19 oncogenes. This procedure was a rapid, cost-effective method of identifying key cancer driving mutations across a large number of samples because it avoided complex bioinformatic analyses and assays were performed within two days. The amount of DNA added to the polymerase chain reaction was 20 ng per reaction. DNA was amplified using the OncoCarta PCR primer pools. Unincorporated nucleotides were inactivated by shrimp alkaline phosphatase (SAP), and a single base extension reaction was performed using extension primers that hybridize immediately adjacent to the mutations and a custom mixture of nucleotides. Salts were removed by the addition of a cation exchange resin. Multiplexed reactions were spotted onto SpectroCHIP II arrays, and DNA fragments were resolved by MALDI-TOF on the Compact Mass Spectrometer (Sequenom, San Diego, CA). Two additional customized mutation panels were used. These panels were designed in collaboration with the Cancer Genomics Group at the Vall d’Hebron Institute of Oncology and included, in 12 multiplexes, a total of 107 somatic mutations in 15 genes. These two panels included 49 additional positions in 6 additional genes. Therefore, a total of 287 different positions in 25 oncogenes were checked (See Supplementary Table S4).
Next generation sequencing (NGS)
The Junior 454 Roche sequencing technology was used by the Genotyping and Genetic Diagnosis Unit (UCIM) following the manufacturer’s protocol. This sequencing technology was used to analyze hotspot mutations in the AKT1, BRAF, EGFR, KRAS, NRAS and PIK3CA genes. A complete list of all the informed mutations is provided in Supplementary Table S5.
Statistical analyses
Data were analyzed using the Sequenom MassARRAY Typer Analyser 4.0 Software to visualize the mass spectra for mutations and to determine the frequency of mutant and wild-type alleles. The lower threshold for mutation detection has been between 5-10% [17–19]. In order to reduce putative false positives we set the threshold at 10%. More specifically, only mutations with frequencies higher than 10% were taken as positive results. Mutations were manually reviewed by use of visual and raw spectrum patterns. Two different personnel in the laboratory scored mutations, and no discrepancies were observed. Analyses were performed using IBM SPSS Statistics for Windows, Version 19.0. Armonk, NY: IBM Corp (IBM Corp. Released 2010).
ACKNOWLEDGMENTS
We would like to thank the expert personnel at the Genotyping and Epigenetics Laboratory and the Genotyping and Genetic Diagnosis Unit at the Central Biomedical Research Unit (UCIM) of the University of Valencia.
FUNDING
This work was supported in part by grants from the Spanish government Ministerio de Salud Carlos III (FIS PI 12/02767); the Generalitat Valenciana Prometeo (Prometeo/2013/005); and Fondos FEDER. MI-V is funded by the Ministerio de Salud Carlos III under a Sara Borrell contract (CD15/00153). TF is funded by the Ministerio de Salud Carlos III under a Rio Hortega Program (CM13/00193). GR is funded by the Ministerio de Salud Carlos III under a Miguel Servet contract (CPII14-00013). MJLL-C is a pre-doctoral fellow from the Generalitat Valenciana (Prometeo/2013/005).
CONFLICTS OF INTEREST
The authors state that there are no conflicts of interest.
REFERENCES
1. Dancey JE, Bedard PL, Onetto N, Hudson TJ (2012) The genetic basis for cancer treatment decisions. Cell 148: 409-420.
2. Vucic EA, Thu KL, Robison K, Rybaczyk LA, Chari R, Alvarez CE, Lam WL (2012) Translating cancer ‘omics’ to improved outcomes. Genome Res 22: 188-195.
3. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507-2516.
4. Yamaguchi M, Harada K, Ando N, Kawamura T, Shibagaki N, Shimada S (2011) Marked response to imatinib mesylate in metastatic acral lentiginous melanoma on the thumb. Clin Exp Dermatol 36: 174-177.
5. Costa C, Molina MA, Drozdowskyj A, Gimenez-Capitan A, Bertran-Alamillo J, Karachaliou N, Gervais R, Massuti B, Wei J, Moran T, Majem M, Felip E, Carcereny E, et al. (2014) The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res 20: 2001-2010.
6. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, et al. (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361: 958-967.
7. Marks JL, Broderick S, Zhou Q, Chitale D, Li AR, Zakowski MF, Kris MG, Rusch VW, Azzoli CG, Seshan VE, Ladanyi M, Pao W (2008) Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thorac Oncol 3: 111-116.
8. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351: 337-345.
9. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25: 1658-1664.
10. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, et al. (2013) Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369: 1023-1034.
11. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Jr., Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380: 358-365.
12. Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66: 3992-3995.
13. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Sileni VC, Lebbe C, Mandala M, et al. (2014) Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N Engl J Med.
14. Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. (2014) Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N Engl J Med.
15. Dienstmann R, Rodon J, Barretina J, Tabernero J (2013) Genomic medicine frontier in human solid tumors: prospects and challenges. J Clin Oncol 31: 1874-1884.
16. Voss JS, Holtegaard LM, Kerr SE, Fritcher EG, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR (2013) Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol 44: 1216-1222.
17. Fumagalli D, Gavin PG, Taniyama Y, Kim SI, Choi HJ, Paik S, Pogue-Geile KL (2010) A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer 10: 101.
18. Fang DD, Zhang CC, Gu Y, Jani JP, Cao J, Tsaparikos K, Yuan J, Thiel M, Jackson-Fisher A, Zong Q, Lappin PB, Hayashi T, Schwab RB, et al. (2013) Antitumor Efficacy of the Dual PI3K/mTOR Inhibitor PF-04691502 in a Human Xenograft Tumor Model Derived from Colorectal Cancer Stem Cells Harboring a Mutation. PLoS One 8: e67258.
19. Dutton-Regester K, Irwin D, Hunt P, Aoude LG, Tembe V, Pupo GM, Lanagan C, Carter CD, O’Connor L, O’Rourke M, Scolyer RA, Mann GJ, Schmidt CW, et al. (2012) A high-throughput panel for identifying clinically relevant mutation profiles in melanoma. Mol Cancer Ther 11: 888-897.
20. Ong M, Carreira S, Goodall J, Mateo J, Figueiredo I, Rodrigues DN, Perkins G, Seed G, Yap TA, Attard G, de Bono JS (2014) Validation and utilisation of high-coverage next-generation sequencing to deliver the pharmacological audit trail. Br J Cancer 111: 828-836.
21. Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, Lacroix-Triki M, Lacroix L, Cohen P, Gentien D, Adelaide J, Dalenc F, Goncalves A, et al. (2014) Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 15: 267-274.
22. Johnson DB, Dahlman KH, Knol J, Gilbert J, Puzanov I, Means-Powell J, Balko JM, Lovly CM, Murphy BA, Goff LW, Abramson VG, Crispens MA, Mayer IA, et al. (2014) Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist 19: 616-622.
23. Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, Routbort M, Piha-Paul SA, Janku F, Ueno N, Hong D, De Groot J, Ravi V, et al. (2015) Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol 33: 2753-2762.
24. Gavin PG, Colangelo LH, Fumagalli D, Tanaka N, Remillard MY, Yothers G, Kim C, Taniyama Y, Kim SI, Choi HJ, Blackmon NL, Lipchik C, Petrelli NJ, et al. (2012) Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res 18: 6531-6541.
25. Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH, Kania K, Viale A, Oschwald DM, Vacic V, Emde AK, Cercek A, Yaeger R, et al. (2014) Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol 15: 454.
26. (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330-337.
27. Dienstmann R, Markman B, Tabernero J (2012) Application of monoclonal antibodies as cancer therapy in solid tumors. Curr Clin Pharmacol 7: 137-145.
28. Santarpia L, Qi Y, Stemke-Hale K, Wang B, Young EJ, Booser DJ, Holmes FA, O’Shaughnessy J, Hellerstedt B, Pippen J, Vidaurre T, Gomez H, Valero V, et al. (2012) Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res Treat 134: 333-343.
29. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, et al. (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486: 400-404.
30. (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61-70.
31. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. (2000) Molecular portraits of human breast tumours. Nature 406: 747-752.
32. Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A (2014) Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov 4: 1269-1280.
33. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S, et al. (2011) PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 10: 558-565.
34. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11: 753-762.
35. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, Zakowski MF, Rusch V, Sima CS, Ladanyi M, Kris MG (2012) Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther 11: 485-491.