
INTRODUCTION
Micro-RNAs (miR) are small non-coding RNAs that act as key regulators of cellular and supracellular (for example immune response) processes under normal and diseased conditions. The importance of miRs in cancer has been underlined by controlling the expression of target mRNAs to facilitate carcinogenesis [1]. Cancer promoter or suppressor function of miRs depends on their direct target genes. As an example, it has been showed that miR155 [2], miR21 [3], miR300 [4] promote breast cancer, while miR7 [5], miR16 [6], miR30a [7] are involved in its suppression. Recently, miR301 has attracted much attention due its significant contribution to different biological process including differentiation, proliferation, survival, apoptosis in pancreatic- [8], hepatocellular- [9], lung-, colorectal- [10], breast cancer [11] and in sickle cell disease [12].
Several miRs target signaling molecules like TGF-β, Wnt, EGF and Akt, and they serve as nodes for signaling pathways that regulate the central cellular processes such as cell survival, proliferation, transcription, translation, cell cycle, differentiation and metabolism [13]. PI3K-Akt signaling pathway is hyper-activated in majority of cancers because of the frequent mutation of tumor suppressor gene, PTEN [14]. PTEN is a central negative regulator of PI3K-Akt signal transduction by dephosphorylating PI(3,4,5)P3 and inhibiting downstream signals [14]. Although PTEN has well-delineated function in the cytoplasm, it has also been found in the nucleus, under certain conditions. Nuclear function of PTEN may be in opposition to its cytoplasmic function [15].
PI3K is a heterodimeric complex containing a separate regulatory- (P85) and catalytic subunit (P110) which phophorylates the inositol ring of PI(4,5)P2 at third positions to create PI(3,4,5)P3 and that serves as an anchorage site for signaling molecules [16]. This function of PI3-Kinase is essential for the recruitment of particular proteins containing pleckstrin-homology (PH) domain or FYVE domain to the cellular membrane [17]. Akt/PKB, a primary downstream signal-integrator and transducer carries the PH domain, and is recruited to the plasma membrane [18, 19]. Membrane recruitment and binding to PI(3,4,5)P3 causes conformational changes in Akt, resulting in exposure of its phoshorylation sites T308 and S473 which are then phosphorylated by phosphoinositide dependent kinase (PDK) and mTOR/Rictor [18–20]. The PI3K/Akt signaling cascade affects (indirectly) the regulation of over 800 downstream signaling molecules that play a role in cell survival, growth, migration, proliferation and cell death [18, 19].
Recently, we have reported that Akt and its nuclear localization play an important role in maintenance and proliferation of cancer stem-like cells through the counter-regulation of p21Waf1/Cip1 and p27kip1 [21]. Nuclear and cytoplasmic Akt pools play partially opposing roles. Nuclear Akt directly inhibits the activity of CDK2-inhibitor p27kip1, whereas it indirectly inhibits the activity of anti-apoptotic molecules such as Bcl2 (by increasing the activity of CDK2). Akt also may inhibit forkhead transcription factor FKHR and promote the activation of anti-apoptotic molecules [22]. The function of miR301 in regulation of PI3K-Akt signaling pathway is poorly understood; therefore in the present study we have investigated the effect of miR301 on Akt mediated biological functions in breast cancer cells, when miR301 expression was decreased upon Akt-overexpression.
RESULTS
Akt overexpression downregulates miR301 level in breast cancer cells
In the manuscript, we refer to Akt-1 as Akt, and miR301-3p as miR301. Exiqon miRCURY LNA™ Array was used to conduct microRNA profiling of breast cancer cell lines (MCF7, HEK293, SKBR3) transiently transfected with Akt. The results showed that miR301 expression was downregulated on average 3.5 fold in MCF7 and HEK293. The transient overexpression Akt was confirmed by immunoblotting (Figure 1A). We further confirmed by qPCR, that miR301 expression is significantly decreased in Akt transfected MCF7 and MDAMB468 cells (Figure 1B).
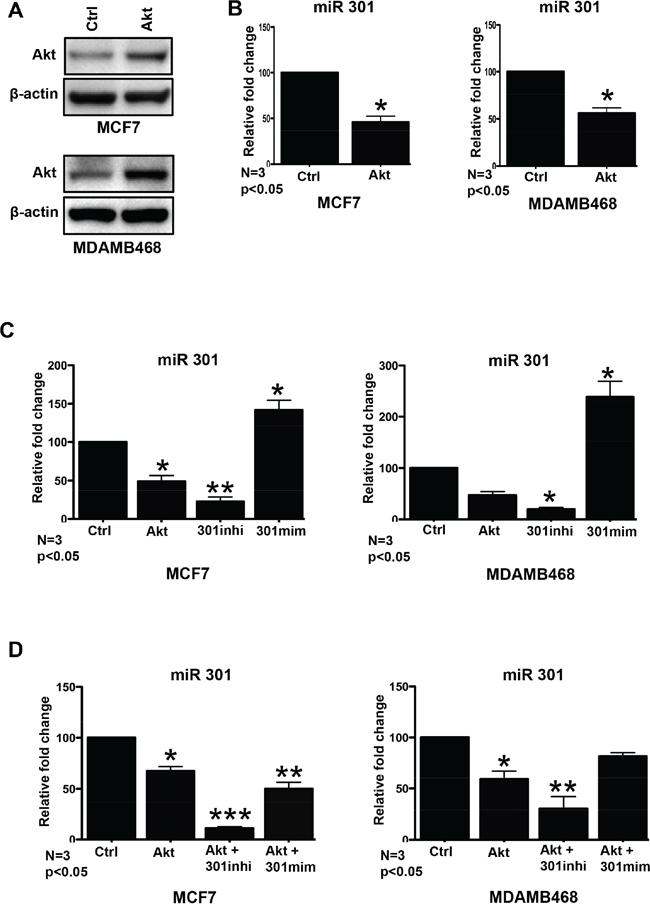
Figure 1: Effect of miR301 on PI3K-Akt pathway. A. In order to study changes in miR levels upon Akt upregulation cells were transiently transfected with Akt. Western blot confirms overexpression of Akt as compared to empty plasmid (control) in MCF7 and in MDAMB468 cells. B. Quantitative RT-PCR shows significant downregulation of miR301 expression mediated by transient transfection with Akt construct, as compared to cells transfected with empty vector, in MCF7 and MDAMB468 cells C. Quantitative RT-PCR shows significant downregulation miR301-expressions by Akt overexpression, even more downregulation by miR301 inhibitor and upregulation by miR301-mimic, as compared to Ctrl (negative control) miR in breast cancer cells. D. Co-transfection of Akt construct with miR301-modulators and Akt alone shows downregulation of miR301 expression in comparison with the control (empty plasmid and Ctrl miR). Since the effect is observed regardless of use of miR301-inhibitor or -mimics, it is likely due to Akt-overexpression, and confirms our results from (A). Akt transfection with miR301-inhibitor showed strong downregulation of miR301 expression compared to control (* p < 0.05).
Once the miR301 downregulation by the transfection of Akt construct was established, we focused on the biological role of miR301. The miR301 specific inhibitor and mimic were utilized to knockdown and boost the miR301 expression in breast cancer cells (Figure 1C). Furthermore, we checked the effect of co-transfection of miR301 inhibitor or mimic with Akt construct and observed that miR301 mimic with Akt showed downregulation of miR301 expression similar to Akt construct alone in MCF7 (Figure 1D). A similar trend was observed in MDAMB468 cells.
Effect of Akt mediated downregulation miR301 on breast cancer cell survival and migration
Given the strong association between miR301 inhibition and Akt, we have investigated whether miR301 inhibition could have any effect on cell survival and migration. Previously, it has been shown that Akt-mediated upregulation of cell survival pathways plays a key role in drug resistance [18, 19, 23]. Here, we evaluated the role of miR301 in metabolic/mitochondrial activity (not necessary proliferation) by MTT assay. MCF7 and MDAMB468 cells were separately co-transfected in 96 well plates, with Akt and miR301-modulators. We observed that downregulation of miR301 expression by Akt and miR301 inhibitor resulted in significant increase in cell viability as compared to control (Figure 2A). Additionally, cell migration was accessed by the scratch assay (known also as ‘wound-healing assay’). Cell migration undeniably increased by transfection with a vector expressing Akt with miR301 inhibitor, as compared to control in MCF7 cells (Figure 2BC).
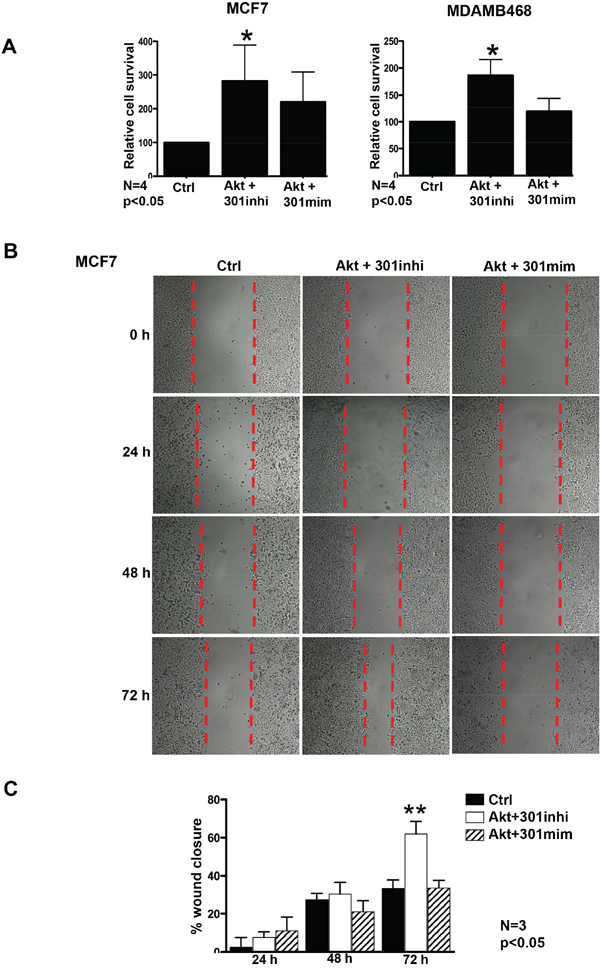
Figure 2: Akt mediated increase in cell survival and migration upon down-regulation of miR301. A. MTT-assay was performed to measure the cell survival upon downregulation of the miR301 with Akt. MCF7 and MDAMB468 breast cancer cells transiently transfected with miR301 inhibitor in the presence of Akt shows a significant increase in cell survival compared to control. B. Scratch assay was preformed to assess cell migration upon inhibition of miR301 in Akt-overexpressing cells. 72h post transfection, the cell migration was highely pronounced in cells co-expressing miR301 inhibitor and Akt, as compared to control, in MCF7 cells (* p < 0.05). C. Quantitative assessment of scratch shows significantly higher wound closure at 72h post transfection, upon miR301 inhibition in the presence of Akt compared to control in MCF7 cells (* p < 0.05).
miR301 inhibition causes increased proliferation of breast cancer cells
We assessed the impact of miR301 inhibition or overexpression, in combination with Akt overexpression, on cell cycle profile in breast cancer cells. As shown in the Figure 3A, miR301 inhibition in cells overexpressing Akt, caused decrease of cells in G1 phase and significant increase in G2 and mitosis as compared to control. Co-transfection of miR301-mimic with Akt presented opposite effect on cell cycle distribution (Figure 3A, Supplementary Figure 1A). We have recently shown [21], that Akt nuclear accumulation causes decrease in G1 phase of the cell cycle by phosphorylation of p21Waf1/Cip1 which leads to decrease p21Waf1/Cip1’s cell cycle inhibitory function. This event ultimately causes the nuclear export of p21Waf1/Cip1 [24]. In the current study, we checked the protein expression by Western blot, and we observed that the co-transfection of miR301 inhibitor with Akt, in breast cancer cells resulted in significant increase in total and phosphorylated p21Waf1/Cip1 and p27kip1 protein levels (Figure 3B and Supplementary Figure 1B).
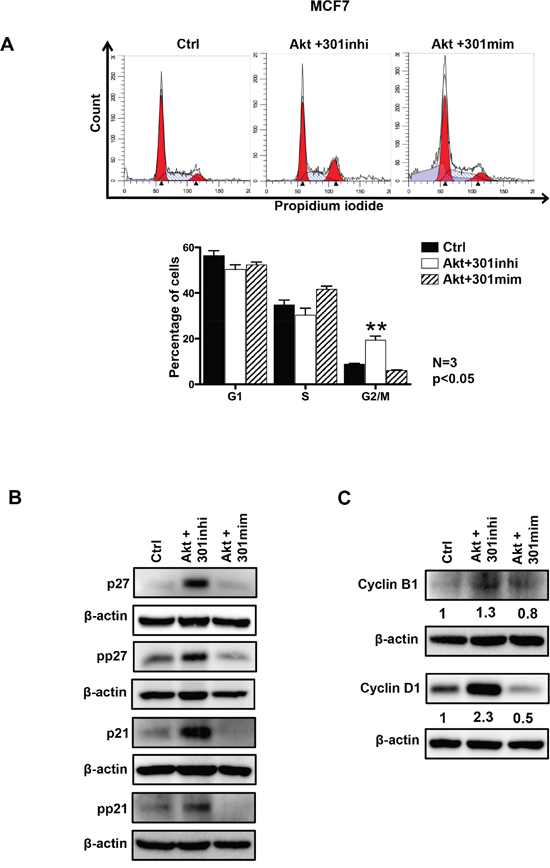
Figure 3: Role of miR301 in the presence of Akt on cell cycle progression. A. Cell cycle analysis shows significant increase in the G2/M phase upon inhibition of miR301 expression in the presence of Akt compared to control and miR301-mimic+Akt in MCF7 breast cancer cells. Lower panel represents quantification of data (A). B. Western blot represents detection of p21Waf1/Cip1 and p27kip1 cell cycle inhibitory proteins. The levels of total and phophorylated p21Waf1/Cip1 and p27kip1 protein were increased upon inhibition of miR301 in the presence of Akt compared to control and miR301-mimic+Akt in MCF7 cells. C. Western blot analysis of Cyclin D1 and Cyclin B1 cell cycle regulatory proteins. Levels of Cyclin D1 and Cyclin B1 were increased upon the expression of miR301 inhibitor in the presence of overexpressed Akt, as compared to control, in MCF7 cells (* p < 0.05).
To get a broader insight of the effect of miR301 on proliferation, cell cycle regulatory proteins were studied in the miR301 inhibited Akt-overexpressed cells. We observed a strong upregulation of the expression of Cyclin D1, (involved, among others, in the regulation of G1/S transition) upon co-transfection of miR301 inhibitor and Akt (Figure 3C). We further analyzed Cyclin B1 expression, which regulates the S/G2, and mitosis entrance. Cyclin B1 expression was upregulated by Akt and miR301 inhibitor further upregulated its expression in MCF7 cells (Figure 3C).
Effect of miR301 on cell survival of breast cancer cells
We assessed whether the cell survival functions of Akt is enhanced in our model upon inhibition of miR301. Thus, we checked how Akt combined with miR301-modulation affects cell death, using the combination of apoptotic dye Po-Pro and necrotic cell death marker 7-AAD. As shown in Figures 4A, B, cells overexpressing miR301 inhibitor together with Akt showed lower staining for Po-Pro and 7-AAD, implying an increase in cell survival in the presence of miR301 inhibitor with Akt overexpression in MCF7 cells, whereas co-expression of Akt and miR301-mimic had an opposite effect. It is worth to note that although the observed changes were statistically significant (p < 0.05) they were not very strong.
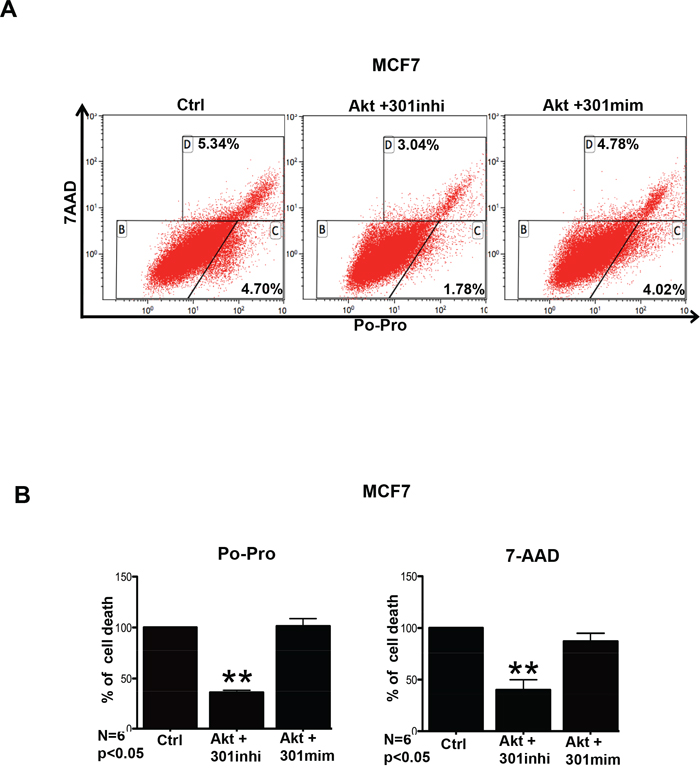
Figure 4: miR301 inhibition in the presence of Akt reduced cell death. A. Cell death was assessed using Po-Pro (apoptotic) and 7-AAD (necrotic) markers. Transient transfection of miR301 inhibitor and Akt resulted in reduced cell death compared to control MCF7 cells. B. Quantitative representation of data from “A” shows significantly decreased in cell death as compared to control (* p < 0.05).
Effect of miR301 on other known miR301-target proteins in breast cancer cells
Our data indicates that miR301 inhibition promotes diverse Akt-mediated effects including proliferation, survival and migration. Thus, we investigated the effect of miR301-modulation on other miR301-targets in our experimental system. Western blot analysis showed the increased protein expression of PTEN, PI3K and FoxF2, as compared to control upon combined miR301 inhibition with Akt overexpression (Figure 5A). Intriguingly, PI3K is the upstream regulator of Akt activity, and plays an important role in Akt activation, whereas PTEN is a negative regulator of Akt pathway. Given the observed similarity in the expression of PI3K and PTEN among miR301 inhibitor with Akt transfected cells, we further checked the protein levels of activated/phosphorylated PI3K and PTEN. We have observed a marked upregulation of phosphorylated PI3K level upon miR301 inhibition in Akt-overexpressed cells. In contrast, the level of phosphorylated PTEN was not affected upon miR301 inhibition or overexpression in the presence of Akt-overexpression (Figure 5A, and Supplementary Figure 2A). We next checked the intracellular localization PTEN upon miR301 inhibition in the presence of Akt-overexpression. miR301 inhibition in Akt-overexpressing cells increases PTEN-nuclear localization. Fraction of PTEN still remains in the cytoplasm (Figure 5B). Co-transfection of miR301-mimic and Akt lead to mostly cytoplasmic localization of PTEN (Figure 5B).
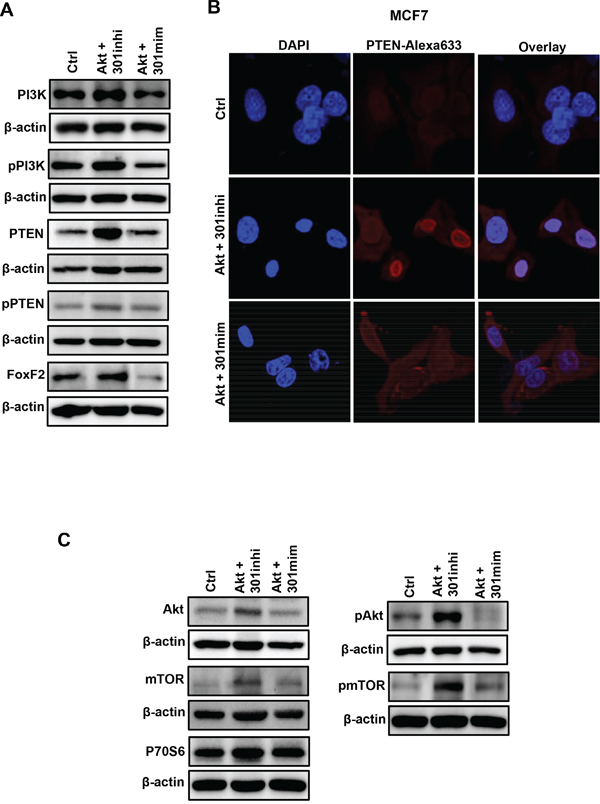
Figure 5: miR301 targeted gene expression in breast cancer cells. A. Western blot analysis of miR301 targets PI3K, PTEN and FoxF2 indicates that total protein levels were significantly increased upon miR301 inhibition in Akt-overexpressing MCF7-cells, as compared to control. Phosphorylated PI3K levels were increased as compared to control and phosphorylated PTEN protein expression was not significantly changed in cells expressing miR301-modulators and Akt. B. Confocal-microscopy images show enhanced nuclear localization of PTEN (red) in MCF7-cells transiently-transfected with miR301 inhibitor and Akt. Transfection with miR301 mimic and Akt caused PTEN’s cytoplasmic localization, in MCF7 cells. For better visualization of nuclei, cells were counterstained with DAPI (blue). C. Western blot analysis shows increased levels of phosphorylated Akt (pAkt), pmTOR and P70S6 in MCF7 cells transiently transfected with miR301 inhibitor and Akt.
The modulation of miR301 had strong effect on PTEN-localization hence we next focused on the functional aspects of PTEN intracellular compartmentalization. Since PTEN localized in the nucleus is unable to dephosphorylate PIP3 to PIP2, it does not counteract the activation of Akt-activation [15]. This observation was further confirmed by increased protein levels and phosphorylation of Akt and its downstream target, mTOR, and to a lesser extend P70S6 (Figure 5C, and Supplementary Figure 2B).
DISCUSSION
In the last decade, deregulatory role of miRs have been widely accepted and reported in majority of cancers [25]. Evidence suggests that miR expression pattern is strongly associated with the specific characteristics of malignant cells, and could be used for distinguishing normal cells and malignant ones [26–28]. Previous studies have revealed that miR has potential to serve as either tumor promoter or suppressor and modulate diverse cellular pathways by downregulation or upregulation of various genes. In the present study, we have shown that miR301 expression is significantly decreased in breast cancer cells upon Akt transient transfection. Previous studies have shown that deregulation of the PI3K-Akt-mTOR signaling pathway contributes to numerous hallmarks of tumor progression, like tumor-angiogenesis, cell proliferation, survival and migration in various cancers [29]. These effects are usually mediated via inhibition of pro-apoptotic signaling molecules and enhancement of the cancer stem-like cells survival [21, 30]. As miR and Akt are known to have diversified cellular functions in oncogenesis, we aimed to explore the miRs role on functional aspects of Akt in human breast cancer cells. Our data shows that overexpression of Akt in breast cancer cells caused downregulation of miR301 expression. Since miR301 mostly counteracts Akt effects, miR301-downregulation upregulates PI3K-Akt pathway. The inhibition of miR301-expression is associated with an increased Akt mediated cell survival, proliferation and migration (Figure 6). These pleiotropic effects of miR301 on PI3K-Akt pathway are directly correlated with the modulation of oncogenic targets including PI3K, PTEN and FoxF2 (Figure 5A). In the current study, PI3K emerges to be the crucial target of miR301 in breast cancer cells. PI3K synthesizes the PIP3 from PIP2, which attracts to the plasma membrane and promotes activation of Akt [16]. Phosphorylated Akt affects the activity of over 800 proteins. It is also the key modulator of mTOR/P70S6 kinase cascade; key autophagy regulators [18, 19, 31, 32]. Our result shows significant increase of Akt activity, mTOR, and P70S6-phosphorylation in cells transfected with miR301-inhibitor that overexpressed Akt. These findings indicate that miR301 plays an important role in the negative modulation of PI3K-Akt pathway.
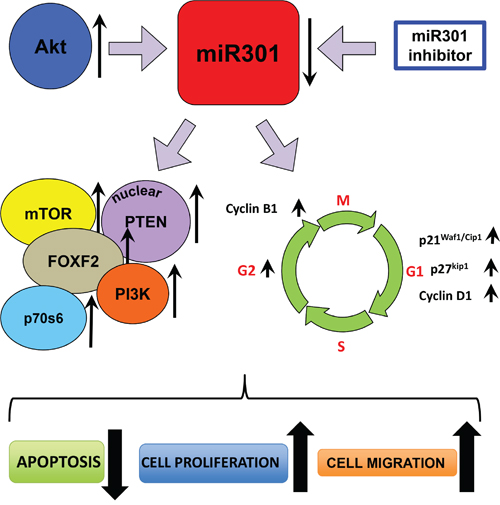
Figure 6: Schematic outline of the miR-301’s functions in breast cancer cells. In the proposed model overexpression of Akt causes down-regulation of miR301. This effect can be also reached and strengthened by applying miR301 specific inhibitor. Down-regulation of miR301 leads to the increase of expression of PI3K, FOXF2, mTOR, p70s6 and nuclear localized PTEN. At the same time several cell cycle related proteins (p21Waf1/Cip1, p27kip1, Cyclin D1 and Cyclin B1) are also overexpressed, which results in higher proliferation rate and accumulation of cells in G2 phase. This all cause increase of cell migration and prevents the induction of apoptosis.
One of the prominent targets of miR301 is PTEN, a tumor suppressor gene and negative regulator of PI3K-Akt pathway. Shi W., et al has recently shown that there is a strong correlation between miR301 and PTEN expression in human breast cancer patients [11]. Phosphorylated PTEN counteracts PI3K-signalling by converting PIP3 back to PIP2, on plasma membrane [33]. In numerous cancer cell types large proportion of PTEN localizes into the nucleus [34]. Nuclear PTEN is unable to counteract PI3K-signalling. Intriguingly, nuclear PTEN function is not yet clearly understood, however, a recent study reported that nuclear-localized PTEN does not dephosphorylate PIP3 [15]. We show that miR301 inhibition both enhances PTEN expression and its nuclear localization. miR301 inhibition has however no effect on PTEN-phosphorylation in breast cancer cells.
Our study also sheds new light on the potential functions of FoxF2 an another target of miR301. FoxF2 is a transcription factor involved in the regulation of different cellular functions [35]. Its role in cancer is not completely understood. Previous studies have reported that there is a correlation between FoxF2 and Wnt5a expression [36]. Wnt5a’s role in human cancer is controversial; it can function both as cancer negative regulator [37] and oncogenic factor [38] in a context-dependent manner. Our work shows significant increase of FoxF2 expression upon miR301 inhibition when Akt expression is upregulated. Thus, our data suggest FoxF2’s role as a tumor promoter, however further studies are required to clarify this aspect.
One of the foremost functions of PI3K-Akt is the induction of cell proliferation through the phosphorylation of cell cycle inhibitory proteins p21Waf1/Cip1 and p27kip1 [39, 40]. Akt also leads to an increase in the levels of cell cycle promoters: cyclin D1 and cyclin B1 [41–43]. Since Akt may affect the status of cell cycle-regulating proteins, we have investigated whether miR301 inhibition in cells overexpressing Akt affects cell cycle progression in breast cancer cells. Indeed, the miR301 inhibition in conjunction with Akt-overexpression, shortens the G0/G1 phase and relatively increases the percentage of cells in G2. In agreement with the above, we have observed increased phosphorylation of p21Waf1/Cip1 and p27kip1 upon miR301 inhibition, leading to p21Waf1/Cip1 and p27kip1 cytoplasmic translocation and removal of its inhibitory effect on cell cycle progression. Based on these evidences, we further looked for the role of miR301 in the cell cycle. miR301 inhibition in Akt-overexpressing cells lead to an increase in Cyclin D1 and -B1 protein expression. Thus, miR301 inhibition enhances Akt-mediated promotion of proliferation.
In conclusion, our study has shown a novel Akt-PI3K pathway inhibitory role of miR301 in breast cancer cells through regulation of PI3K, PTEN and FoxF2. The resulting phenotype triggered by miR301 inhibition includes increased cell survival, migration and proliferation. The data also suggest that the miR301-analogues could serve as leads for the development of PI3K/Akt pathway modulators.
MATERIALS AND METHODS
Cell culture and reagents
Breast cancer cell lines: MCF7, MDAMB468, SKBR3 and HEK293 were cultured in DMEM media (PAA, Pasching, Austria) containing 10% fetal bovine serum (PAA, Pasching, Austria) and 1% penicillin-streptomycin (Gibco, USA) and incubated at 37°C with 5% CO2 in a humidified atmosphere.
Antibodies
The primary antibodies used in the study: pPI3K110 obtained from Bioss Antibodies (USA), PTEN, pPTEN, P70S6, Cyclin B1, pAkt, Akt1, pmTOR and mTOR from Cell Signaling (Beverly, USA), FoxF2, PI3K110, Cyclin D1, ß-actin, p27 and p-p27 from Abcam (Cambridge, UK), and p21 and p-p21 from Santa-Cruz (USA). The secondary antibodies: Alexafluor 633 obtained from Life Technologies, anti-rabbit HRP-conjugate from Biorad (USA) and anti-mouse HRP-conjugate from GE Heath Care (Buckinghamshire, UK).
Plasmids and transient transfection
The cells were co-transected using X-treme GENE HP DNA Transfection Reagent (Roche, Mannheim, Germany) according to manufacture’s instructions. Akt1 cDNA was cloned into pLVX-Tight-Puro (Clontech) plasmid as previously reported [21] and empty plasmid pLVX-Tight-Puro was used as control. mirVana 301miR inhibitor, mirVana 301miR mimic as a positive control and mirVana miR negative control (Ambion) were all transfected at a final concentration 30 nmol/L.
Terminology explanation: miR301 mimic- miR301 mimic is small, chemically modified doubled-stranded RNA molecule that mimics the actions of endogenous miR301; miR301 inhibitor – miR301 inhibitor is small, chemically modified single-stranded RNA molecule designed to specifically bind and inhibit endogenous miR301; Negative control – The negative control is a random sequence of miR molecule which has been extensively tested in human cell lines and validated not to produce identifiable effect of known miR function. This negative control transfected sample has been used as a baseline for evaluating the effect of control and experimental miR301 mimic or inhibitor expression.
Target identification by miR profiling
MCF7, SKBR3 and HEK293 cells were transfected with Akt or empty plasmid (control). In a pilot experiment, transient transfection efficiency was assessed by transfections with pEGFP (Invitrogen) vector and subsequent measurement of green fluorescence by flow cytometry. The following efficiency was achieved: MCF7 40%, HEK293 55%, and SKBR3 25%. RNA was isolated and sent to EXIQON for microRNA profiling by miRCURY LNA™ Array. microRNA with greater than 2-fold expression change relative to control were considered as potential targets and selected ahead for further investigation. The investigated here miR301 was down-regulated about 3.5 fold in two (MCF7, HEK293) tested cell lines. It was the most down-regulated miR, and thus it was chosen for further investigation.
Quantitative real time-PCR analysis of miRs
For the quantitative real time-PCR, total RNA was extracted using High Pure miRNA isolation Kit (Roche, Germany) according to the manufacturer’s instructions from co-transfected breast cancer cells. Total RNA concentration was measured by NanoDrop™ spectrophotometer (Thermo Scientific, USA). Total RNA was reverse transcribed into cDNA using TaqMan® MicroRNA Reverse Transcription kit (Life Technologies Ltd, USA) using CFX96™ real-time PCR detection system (Biorad, USA). Next, cDNA was added to the reaction mix containing TaqMan® Fast Universal PCR Master Mix and TaqMan MicroRNA Assay for miR301. All experimental procedures were performed according to the manufacturer’s protocols. Each reaction was performed in triplicates using a CFX96™ real-time PCR detection system (Biorad, USA). RNU6b was used as internal control to normalize the amount of cDNA between different samples. The expression of miR-301 was calculated by applying the 2−ΔΔCt method, where ΔCT = (CTmiR- CTmiR control) and CT (i.e. threshold cycle) indicates the fractional cycle number at which the amount of amplified target reaches a fixed threshold [44].
Cell migration assay
For scratch migration assay, miR301 inhibitor and mimic with Akt co-transfected cells were scratched (wound) using a sterile standard 200 ml tip in 6-well plate. Serial images were taken at different time points using JuLi Smart Fluorescence cell imager in the bright field. The degree of ingrowth of the cells into the “wound” (scratch) indicated the proliferative capacity of cells [23].
Cell survival assay
Cell survival and proliferation was assessed by 3-(4,5-dimethyl-2-thiazolyl) 2,5-diphenyl-2H tetrazolium bromide (MTT) assay. Cells (5000/well) were plated 24h prior to the experiments in a 96-well plate. 48 hours after co-transfection with miR301 inhibitor, mimic, and with Akt, cells were incubated with 10 μl 5 mg/ml MTT solution (Sigma Aldrich) for 3h. Next, plates were centrifuged and supernatant was discarded, formazan crystals were dissolved in DMSO:ethanol (1:1 ratio). The readings were performed at 570 and 630 nm using a spectrophotometer as described previously [23]. Data was always collected in triplicates.
Cell cycle analysis
The experiments were preformed as described previously [45], with minor modifications. Briefly: cells were trypsinized and fixed with ice cold 70% ethanol overnight at -20°C and washed with PBS and resuspended in 50 μg/ml RNAse for 30min at 37°C. Then samples were incubated with 100 μg/ml propidium iodide (Sigma) for 10min and measured by flow cytometry (Gallios, Beckman Coulter Inc.). Kaluza software (Beckman Coulter Inc.) was used to analyze the results.
Po-Pro and 7AAD cell death assay
Co-transfected cells were trypsinized and washed with PBS. Cells were then resuspended in PBS and treated with Po-Pro and 7AAD dyes (Life Technologies Ltd, USA) for 30 min according to the manufacturer’s instruction. The fluorescence intensities of the samples were quantified using Gallios flow cytometer (Beckman Coulter Inc.) and the data was analyzed using Kaluza software (Beckman Coulter Inc.) [31].
Western blot
The procedure was performed similarly as described previously [46]. Co-transfected cells were washed with PBS and lysed with RIPA buffer with protease inhibitors (cOm-plete, Roche Diagnostics, Mannheim, Germany). Cell debris was removed by centrifuging at 10,000 g for 10 min. Protein concentration was determined by performing Bradford assay. Then, equal amount of proteins were loaded into 10% polyacrylamide gel and ran at 100 V for 3-4 h. Further, proteins were transferred to PVDF membrane (Millipore, Darmstadt, Germany) at 80 V for 2-3 h. Blocking was done by using 5% non-fat milk. Next, membranes were incubated with primary antibody overnight at 4 °C. The blots were washed 3 times with 1× TBST and incubated in their respective secondary antibodies for 1 h at RT. Blots were washed again 3 times with 1 × TBST and developed with Amersham ECL plus (GE Technologies, Buckinghamshire, UK).
Immunocytochemistry
The immunocytochemistry was performed similarly as described previously [47]. Cells were grown in 12-well plates on coverslips and they were co-transfected with plasmids as described in the result section. Unless stated otherwise, 48 h post-transfection cells were washed with PBS, fixed with 4% paraformadehyde for 30 min at room temperature (RT), and washed 3 times with PBS. Next, cells were permeabilized with 0.1% Triton X for 10 min at RT, blocked with 1% BSA and washed 3 times with PBS. Then, cells were incubated with primary antibody overnight at 4°C and followed by washing with PBS. Next, cells were incubated with respective secondary antibody for 1h at RT and washed 3 times with PBS. Cells were counterstained with DAPI and mounted on a slide. Images were captured using confocal microscope (Zeiss).
Bioinformatics and statistics
Target genes for miR301 were predicted by the following two computer-aided algorithms: TargetScan (http://www.targetscan.org) and PicTar (http://pictar.mdc-berlin.de). All the statistics (one way and two way ANOVA) were performed using Prism (version 5.0d) and p-value < 0.05 was considered as statistically significant, unless mentioned otherwise. Unless mentioned otherwise, all the presented data represents an average of a minimum of 3 independent experiments.
Abbreviations
miR, microRNA; PH, pleckstrin-homology; PI3K, phoshatidylinositol-3-kinase; PTEN, phosphatase and tensin homolog; mTOR, mammalian target of rapamycin; DMEM, dulbecco’s modified Eagles’s medium; RT, room temperature; RIPA, radioimmunoprecipitation assay; PVDF, polyvinylidene fluoride; FBS, fetal bovine serum; 7-AAD, 7-aminoactinomycin D.
ACKNOWLEDGMENTS
MJL kindly acknowledges the support from Cancerfonden (2013/391). ACP thankfully acknowledges the support from GeCONiI (POIG.02.03.01-24-099/13).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends in molecular medicine. 2014; 20:460-469.
2. Mattiske S, Suetani RJ, Neilsen PM, Callen DF. The oncogenic role of miR-155 in breast cancer. Cancer epidemiology, biomarkers & prevention. 2012; 21:1236-1243.
3. Song B, Wang C, Liu J, Wang X, Lv L, Wei L, Xie L, Zheng Y, Song X. MicroRNA-21 regulates breast cancer invasion partly by targeting tissue inhibitor of metalloproteinase 3 expression. Journal of experimental & clinical cancer research. 2010; 29:29.
4. Xu XH, Li DW, Feng H, Chen HM, Song YQ. MiR-300 regulate the malignancy of breast cancer by targeting p53. International journal of clinical and experimental medicine. 2015; 8:6957-6966.
5. Kong X, Li G, Yuan Y, He Y, Wu X, Zhang W, Wu Z, Chen T, Wu W, Lobie PE, Zhu T. MicroRNA-7 inhibits epithelial-to-mesenchymal transition and metastasis of breast cancer cells via targeting FAK expression. PloS one. 2012; 7:e41523.
6. Rivas MA, Venturutti L, Huang YW, Schillaci R, Huang TH, Elizalde PV. Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development. Breast cancer research. 2012; 14:R77.
7. Cheng CW, Wang HW, Chang CW, Chu HW, Chen CY, Yu JC, Chao JI, Liu HF, Ding SL, Shen CY. MicroRNA-30a inhibits cell migration and invasion by downregulating vimentin expression and is a potential prognostic marker in breast cancer. Breast cancer research and treatment. 2012; 134:1081-1093.
8. Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ, Schmittgen TD. Expression profiling identifies microRNA signature in pancreatic cancer. International journal of cancer. 2007; 120:1046-1054.
9. Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, Roberts LR, Schmittgen TD. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clinical cancer research. 2008; 14:419-427.
10. Ma X, Yan F, Deng Q, Li F, Lu Z, Liu M, Wang L, Conklin DJ, McCracken J, Srivastava S, Bhatnagar A, Li Y. Modulation of tumorigenesis by the pro-inflammatory microRNA miR-301a in mouse models of lung cancer and colorectal cancer. Cell Discovery. 2015; 1:15005.
11. Shi W, Gerster K, Alajez NM, Tsang J, Waldron L, Pintilie M, Hui AB, Sykes J, P’ng C, Miller N, McCready D, Fyles A, Liu FF. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer research. 2011; 71:2926-2937.
12. Gonsalves CS, Li C, Malik P, Tahara SM, Kalra VK. Peroxisome Proliferator alpha mediated transcription of miR-301a and miR-454 and their host gene SKA2 regulate Endothelin-1 and PAI-1 expression in sickle cell disease. Biosci Rep. 2015; 35. pii: e00275.
13. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nature reviews Molecular cell biology. 2010; 11:252-263.
14. Baker SJ. PTEN enters the nuclear age. Cell. 2007; 128:25-28.
15. Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, Lucocq J, Downes CP. Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. Journal of cell science. 2006; 119:5160-5168.
16. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature reviews Cancer. 2002; 2:489-501.
17. Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes & development. 2000; 14:1027-1047.
18. Altomare DA, Khaled AR. Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling. Current medicinal chemistry. 2012; 19:3748-3762.
19. Davis NM, Sokolosky M, Stadelman K, Abrams SL, Libra M, Candido S, Nicoletti F, Polesel J, Maestro R, D’Assoro A, Drobot L, Rakus D, Gizak A, Laidler P, Dulinska-Litewka J, Basecke J, et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: possibilities for therapeutic intervention. Oncotarget. 2014; 5:4603-4650. doi: 10.18632/oncotarget.2209.
20. Martelli AM, Faenza I, Billi AM, Manzoli L, Evangelisti C, Fala F, Cocco L. Intranuclear 3’-phosphoinositide metabolism and Akt signaling: new mechanisms for tumorigenesis and protection against apoptosis? Cellular signalling. 2006; 18:1101-1107.
21. Jain MV, Jangamreddy JR, Grabarek J, Schweizer F, Klonisch T, Cieslar-Pobuda A, Los MJ. Nuclear localized Akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1). Cell cycle. 2015; 14:2109-2120.
22. Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004; 15:177-182.
23. Jangamreddy JR, Jain MV, Hallbeck AL, Roberg K, Lotfi K, Los MJ. Glucose starvation-mediated inhibition of salinomycin induced autophagy amplifies cancer cell specific cell death. Oncotarget. 2015; 6:10134-10145. doi: 10.18632/oncotarget.3548.
24. Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nature medicine. 2002; 8:1145-1152.
25. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nature reviews Genetics. 2009; 10:704-714.
26. Cummins JM, Velculescu VE. Implications of micro-RNA profiling for cancer diagnosis. Oncogene. 2006; 25:6220-6227.
27. Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006; 25:6170-6175.
28. Tricoli JV, Jacobson JW. MicroRNA: Potential for Cancer Detection, Diagnosis, and Prognosis. Cancer research. 2007; 67:4553-4555.
29. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005; 24:7455-7464.
30. West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug resistance updates. 2002; 5:234-248.
31. Chaabane W, Cieslar-Pobuda A, El-Gazzah M, Jain MV, Rzeszowska-Wolny J, Rafat M, Stetefeld J, Ghavami S, Los MJ. Human-gyrovirus-Apoptin triggers mitochondrial death pathway--Nur77 is required for apoptosis triggering. Neoplasia. 2014; 16:679-693.
32. Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K, Schweizer F, Lengerke C, Davoodpour P, Palicharla VR, Maddika S, Los M. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. Journal of cellular and molecular medicine. 2013; 17:12-29.
33. Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007; 128:129-139.
34. Lian Z, Di Cristofano A. Class reunion: PTEN joins the nuclear crew. Oncogene. 2005; 24:7394-7400.
35. Gerin I, Bommer GT, Lidell ME, Cederberg A, Enerback S, Macdougald OA. On the role of FOX transcription factors in adipocyte differentiation and insulin-stimulated glucose uptake. The Journal of biological chemistry. 2009; 284:10755-10763.
36. Ormestad M, Astorga J, Landgren H, Wang T, Johansson BR, Miura N, Carlsson P. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development. 2006; 133:833-843.
37. Jonsson M, Dejmek J, Bendahl PO, Andersson T. Loss of Wnt-5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer research. 2002; 62:409-416.
38. Fernandez-Cobo M, Zammarchi F, Mandeli J, Holland JF, Pogo BG. Expression of Wnt5A and Wnt10B in non-immortalized breast cancer cells. Oncology reports. 2007; 17:903-907.
39. Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nature medicine. 2002; 8:1153-1160.
40. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature cell biology. 2001; 3:245-252.
41. Badve S, Collins NR, Bhat-Nakshatri P, Turbin D, Leung S, Thorat M, Dunn SE, Geistlinger TR, Carroll JS, Brown M, Bose S, Teitell MA, Nakshatri H. Subcellular localization of activated AKT in estrogen receptor- and progesterone receptor-expressing breast cancers: potential clinical implications. The American journal of pathology. 2010; 176:2139-2149.
42. Los M, Maddika S, Erb B, Schulze-Osthoff K. Switching Akt: from survival signaling to deadly response. BioEssays. 2009; 31:492-495.
43. Lee SR, Park JH, Park EK, Chung CH, Kang SS, Bang OS. Akt-induced promotion of cell-cycle progression at G2/M phase involves upregulation of NF-Y binding activity in PC12 cells. Journal of cellular physiology. 2005; 205:270-277.
44. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402-408.
45. Ghavami S, Sharma P, Yeganeh B, Ojo OO, Jha A, Mutawe MM, Kashani HH, Los MJ, Klonisch T, Unruh H, Halayko AJ. Airway mesenchymal cell death by mevalonate cascade inhibition: integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochimica et biophysica acta. 2014; 1843:1259-1271.
46. Shakeri R, Hosseinkhani S, Los MJ, Davoodi J, Jain MV, Cieslar-Pobuda A, Rafat M, Ardestani SK. Role of the salt bridge between glutamate 546 and arginine 907 in preservation of autoinhibited form of Apaf-1. International journal of biological macromolecules. 2015; 81:370-374.
47. Cieslar-Pobuda A, Back M, Magnusson K, Jain MV, Rafat M, Ghavami S, Nilsson KP, Los MJ. Cell type related differences in staining with pentameric thiophene derivatives. Cytometry Part A. 2014; 85:628-635.