
INTRODUCTION
The integrity of the DNA in each cell is continually challenged by hundreds of thousands of insults each day that can alter the sequence or chemical composition of the DNA [1]. These changes include single-strand or double-strand DNA breaks, base damage, bulky adducts, intrastrand and interstrand cross-links and breakdown of the replication fork [1]. To limit severe impacts of DNA damage and replication errors, cells have evolved a plethora of molecular processes that maintain the integrity of the genome [2]. Once DNA is damaged, it can be repaired by at least one of the six major DNA damage repair (DDR) pathways: direct repair, mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), non-homologous end joining (NHEJ) and homologous recombination (HR) [3, 4] (Figure 1). Loss of efficiency of one or more DNA repair pathways can accelerate the rate of accumulation of additional mutations by 100–1,000 times [5]. Unrepaired DNA damage is a major source of potentially mutagenic lesions that drive carcinogenesis [5, 6].
In patients with hereditary non-polyposis colorectal cancer (HNPCC), germ-line defects in mismatch-repair genes (primarily MLH1 and MSH2) confer a lifetime risk of colorectal cancer (CRC) of approximately 80% [7]. Germline mutations in MSH2 and MLH1 account for approximately 60% of HNPCC, while nearly one-third of HNPCC patients do not show MMR gene mutations, which may be attribute to epigenetic silencing [8-11]. Somatic inactivation of the mismatch-repair gene MLH1 occurs in approximately 15% of patients with sporadic colorectal cancers by promoter region methylation in bialleles [12-14]. Heterozygous germline mutations in breast cancer susceptibility gene 1/2 (BRCA1/2) are responsible for a large fraction of hereditary breast cancers. The risk of developing breast cancer by age 70 is 50–70% for BRCA1/2 mutation carriers [6, 15, 16]. While BRCA1 and BRCA2 mutations affect a minority of breast cancer patients (fewer than 5%), BRCA1 and BRCA2 were silenced by promoter region hypermethylation in 9% and 2% of sporadic breast cancer, respectively [17, 18]. O6-methylguanine-DNA methyltransferase (MGMT) is mutated in 17.5% and methylated in 44% of human esophageal squamous cell carcinoma [19, 20]. Thus, aberrant epigenetic changes may play more important roles than gene mutations in DNA damage repair genes to drive carcinogenesis. This review is mainly focused on the role of epigenetics in DNA damage repair genes.
EPIGENETIC CHANGES OF DNA DAMAGE REPAIR GENES IN HUMAN CANCERS
Direct repair genes
The simplest form of DNA repair is the direct reversal of the lesion [21]. MGMT removes alkyl groups from O6-methylguanine that are formed as a result of erroneous methylation by S-adenosylmethionine, and it also removes other alkylations at the O6-position of guanine that are induced by dietary nitrosamines or chemotherapy agents such as temozolomide (TMZ), dacarbazine (DTIC) and carmustine (BCNU) [22, 23]. Unrepaired O6-methylguanine gives rise to O6-methylguanine/thymine mispairing, which is recognized and excised by the DNA MMR system during DNA replication [24, 25]. If O6-methylguanine is not repaired by MGMT and MMR, G:C to A:T transition mutations will lead to carcinogenesis when they occur in cancer-related genes, such as K-ras and p53 [26, 27]. MGMT is frequently methylated in various tumors, including gliomas (40%) [28], diffuse large B-cell lymphoma (36%) [29], colorectal cancer (46%) [30], gastric cancer (36.8%) [31], non-small cell lung cancer (21%) [32], esophageal cancer (44%) [19] and head and neck cancers (60.8%) [33].
Base excision repair pathway
The most versatile of the excision repair systems, BER, is required to repair DNA single-strand breaks and deamination, oxidation, and alkylation-induced DNA base damage that may result from chemotherapy (alkylating agents, topoisomerase I poisons, antimetabolites), radiotherapy, environmental exposure (nitrosamines, ultraviolet light, smoke) or endogenous byproducts of cellular metabolism (reactive oxygen species) [34].
The 8-oxo-G DNA lesion is one of the best-studied types of oxidative DNA damage caused by reactive oxygen species (ROS), and it is weakly cytotoxic but mostly mutagenic [34]. Because 8-oxodG DNA lesions do not block DNA replication, they lead to mispairing with A and result in GC to TA transversions, the most predominant somatic mutations in lung, breast, ovarian, gastric and colorectal cancers [35]. The main components of the BER pathway are glycosylases, endonucleases, DNA polymerases and DNA ligases, with poly(ADP-ribose) polymerase 1 (PARP1) and PARP2 facilitating the process [36]. Mutations and single nucleotide polymorphisms (SNPs) in BER genes are associated with numerous cancers, such as UNG2 (uracil DNA glycosylase) mutations in glioblastoma [37], MED1 (mediator complex subunit 1) mutations in colorectal cancer [38], and OGG1 (8-oxoguanine DNA glycosylase) mutations in lung cancer [39]. The X-ray repair cross-complementing gene 1 (XRCC1) polymorphism, Arg399Gln, increased the occurrence of p53 mutations in human lung cancer [40]. Aberrant methylation of BER genes were found in a variety of cancers. XRCC1 was methylated in 76.4% of human gastric cancers [41]. Aberrant DNA methylation of the TDG (thymine DNA glycosylase) gene is reported in multiple myeloma cells and is related to genomic instability in multiple myeloma [42]. Promoter region hypermethylation of MED1 is reported frequently in colorectal and ovarian cancers [43]. OGG1 is found to be methylated in breast and thyroid cancers [44, 45].
The Werner syndrome gene (WRN), a RecQ family member with both helicase and exonuclease activities, also participates in base excision repair [46, 47]. Germline mutations in WRN are the cause of Werner syndrome, an autosomal recessive disorder characterized by premature aging, genomic instability, and predisposition to cancer [48]. However, somatic mutations of WRN have not been described in sporadic neoplasms [47]. A study in colorectal cancer showed that epigenetic inactivation of WRN leads to loss of WRN-associated exonuclease activity and increased chromosomal instability and apoptosis induced by topoisomerase inhibitors [48]. Promoter region methylation of the WRN gene has been reported frequently in colorectal (37.9%), non-small cell lung (37.5%), gastric (25%), prostate (20%), breast (17.2%), and thyroid (12.5%) cancers, chondro-sarcomas (33.3%), and non-Hodgkin lymphoma (23.7%) [47].
Nucleotide excision repair genes
The NER genes recognize and repair bulky DNA damage that involves more than one nucleotide, including adducts induced by chemical carcinogens, such as tobacco smoke, platinum compounds and photoproducts, or dimers induced by ultraviolent (UV) light [49]. NER abnormalities are present in several rare recessive photosensitive syndromes, including xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD) [50]. XP is the first human disease that was clearly associated with defects in DNA repair. It is a rare autosomal recessive disease caused by biallelic inactivating mutations in genes involved in nucleotide excision repair [51]. Patients with mutations in XP genes are extremely sensitive to UV and have extraordinarily high incidences of non-melanoma skin cancer and melanoma, as well as other solid tumors [49]. In 158 cases of lung cancer samples, promoter region methylation of the XPC (xeroderma pigmentosum group C) gene was found in 53 cases (34%). Interestingly, XPC methylation was more common in nonsmokers (39/94, 41%) than in smokers (14/64, 22%) [52]. The XPC gene was methylated in 32.5% of bladder cancers [53]. ERCC1 (excision repair cross-complementation group 1) was methylated in 37.5% of gliomas [54]. hHR23B (RAD23 homolog B) was methylated in multiple myeloma cells [55]. XPG was methylated in 20% of ovarian cancers [56].
Mismatch repair genes
The MMR system recognizes base–base mismatches and insertion or deletion loops (IDLs) in double-stranded DNA, and it degrades the error-containing region of the newly synthesized strand, allowing the polymerase to correctly re-synthesize the second strand according to the template sequence [48]. The human MMR system includes the MLH1, MLH3, MSH2, MSH6, PMS1 and PMS2 genes [57, 58]. Defective MMR increases mutation rates up to 1,000-fold and leads to microsatellite instability (MSI) resulting in carcinogenesis [22]. The MSI status of a tumor can be detected by five microsatellite markers which are composed of two mononucleotide repeats (BAT25, BAT26) and three dinucleotide repeats (D2S123, D5S346 and D17S250) [59]. If two or more of the five loci show instability, the tumor is MSI-H; if only one locus is unstable, the tumor is MSI-L; and if all five loci are stable, the tumor is MSS [60]. MSI-H is known to occur in more than 90% of HNPCC patients [61]. Inactivation of germline mutations within at least one of four mismatch repair genes (MLH1, MSH2, MSH6, and PMS2) can be found in approximately 70% of cases, and 95% of the mutations occur in hMSH2 or hMLH1 [62]. Loss of expression of the MLH1 or MSH2 genes is approximately 100% associated with the MSI-H phenotype [60]. In population-based studies, the prevalence of MSI among CRCs is approximately 15% [51, 63, 64]. Germline MMR mutations that give rise to HNPCC account for ~3% of all CRCs [63].
In contrast to HNPCC, sporadic cancers are rarely found to have mutations in the MLH1 or MSH2 genes. Researchers have confirmed that promoter-region methylation accounts for 80–90% of MLH1 inactivation in sporadic MSI-H colorectal cancer [60]. In a cohort study that included 268 cases of CRC, 46 cases were MSH2-deficient HNPCC, 15 were cases of sporadic MSI CRCs, and 207 were cases of sporadic MSS CRCs. The study found that 80% of MSH2-deficient HNPCC patients harbored germline mutations in MSH2 [65]. However, MSH2 was methylated in 24% (11/46) of MSH2-deficient HNPCC. Moreover, 63% (7/11) of MSH2 methylated CRCs had a simultaneous pathogenic germline MSH2 mutation, but no methylation was found in matched normal tissues, suggesting that methylation may be the required “second hit” in these tumors [65]. MSH2 was not methylated in any of the sporadic CRCs. Promoter region hypermethylation of MLH1 is also found in other sporadic tumors, including adult T-cell leukemia/lymphoma (6%) [66], gastric cancer (21.6%) [67], head and neck squamous cell carcinomas (28.6%) [68], non-small cell lung cancer (55.8%) [69], oral squamous cell carcinoma (76%) [70], esophageal squamous cell carcinoma (23%) [71], pancreatic cancer (23%) [72], ovarian cancer (37.5%) [73], and breast cancer (31.3%) [74].
In addition to somatic methylation, germline epimutation, a condition in which a person has hypermethylation of one allele of MLH1 in somatic cells throughout the body, increased the risk of CRC by 60% for first-degree relatives of MLH1-methylated cases. For second-degree relatives, there was no evidence of an increased CRC risk. Risks were increased for gastric cancer for first and second-degree relatives. The incidence of liver cancer was also increased for first-degree relatives [75].
DNA double-strand breaks (DSB) repair
DSBs do not occur as frequently as the other lesions listed above, and they are difficult to repair and extremely toxic [76]. Damaging agents emanating from endogenous and environmental sources constantly challenge the integrity of DNA [77]. Almost all human cancers are characterized by genomic instability, which is considered to play a key role in the conversion of a normal cell into a premalignant cell [4, 77]. DSBs are considered to be the most hazardous lesions, since a single unrepaired DSB may trigger cell death, whereas a misrepaired DSB may result in chromosomal rearrangements [77, 78]. Interstrand crosslinks (ICLs) represent the most deleterious lesions produced by chemotherapeutic agents such as mitomycin C (MMC), cisplatin and cyclophosphamide [77]. ICL repair is complex and involves the collaboration of several repair pathways, namely Fanconi anemia, NER, translesion synthesis and homologous recombination [79]. Inhibition of repair by compounds that target factors involved in NHEJ or HR will increase the sensitivity of cancer cells to DSB-inducing anticancer agents. There are numerous proteins involved in DSB repair, and inactivation of DSB repair genes is associated with various tumors. XRCC5 is an important component of NHEJ. Promoter region methylation of XRCC5 was reported in 21% of non-small cell lung cancers (NSCLCs) [80]. Genes of the Fanconi Anemia (FA) pathway also play an important role in repair by homologous recombination. The FA pathway is not constitutively active in normal cells, but it is turned on during the S phase of the cell cycle or following DNA damage [81]. FA is a rare chromosomal instability syndrome characterized by aplastic anemia in childhood, susceptibility to leukemia and cancer, and hypersensitivity of FA cells to interstrand DNA crosslinking agents, such as cisplatin and melphalan [81]. There are thirteen FA genes, and one of these genes is identical to the well known breast cancer susceptibility gene, BRCA2 [82]. FA proteins and another breast/ovarian cancer susceptibility protein, BRCA1, cooperate in a DNA repair pathway, which is required for resistance to DNA interstrand crosslinks [81, 83]. FA patients with biallelic mutations in BRCA1 have not been identified, perhaps because biallelic loss of BRCA1 results in embryonic lethality [81]. BRCA1 appears to play a relatively early role in the regulation and promotion of HR. BRCA1 is phosphorylated in response to DNA DSBs by several kinases and likely acts in DNA damage signal transduction [84]. Germline mutations in the BRCA1 and BRCA2 genes are the most important causes of hereditary breast and ovarian cancer. While there is not a direct procedure to estimate the prevalence of BRCA1/2 mutations in the general population, it is estimated that nearly 50% of the mutations would be in BRCA1 and 50% in BRCA2 [85]. Lifetime cancer risks in BRCA mutation carriers are 60-80% for breast cancer and 20-40% for ovarian cancer [86]. Mutations in BRCA genes cannot account for all cases of hereditary breast and ovarian cancer. The remaining cases can be attributed to the involvement of constitutive epimutations or other cancer susceptibility genes [86]. The FANCD1, FANCN and FANCJ genes have been shown to be breast cancer susceptibility genes [81].
In addition to germline mutations in these genes occurring in FA, somatic mutations and epigenetic silencing of these genes occur in a variety of cancers in the general population (non-FA patients) [87, 88]. FANCF promoter methylation has been detected in 21% of ovarian, 17% of breast, 15% of head and neck, 30% of cervical, 14% of non-small cell lung, and 11.4% of gastric cancers [48, 89]. FANCC was methylated in 42% of head and neck squamous cell carcinoma according to PCR-based methylation-sensitive restriction analysis (MSRA) [90]. FANCC was methylated in 1 of 143 acute myeloid leukemia (AML) cases and 3 of 97 acute lymphoblastic leukemia (ALL) cases, while FANCL was found to be methylated in one ALL sample [91]. BRCA1/BRCA2 mutations are often found in hereditary breast and ovarian cancers, while somatic mutations of BRCA1/BRCA2 are rare in sporadic breast and ovarian cancers [92, 93]. BRCA1 methylation is observed in approximately 11–14% of breast cancers and 5%–31% of ovarian cancers [93]. BRCA1 methylation is also found in gastric (1.4%) [89], colorectal (10.75%) [94], non-small cell lung (18.6%) [95], bladder (12.1%) [96] and pancreatic cancers (46%) [97]. BRCA2 methylation is reported in NSCLC (42%) [80].
DNA damage repair pathway and cell cycle checkpoints
The cell cycle may be arrested at either the G1/S or the G2/M transition by cell cycle inhibitors. The resulting cell-cycle arrest allows time for DNA repair, thereby preventing genome duplication or cell division in the presence of damaged DNA. Inducing cell-cycle arrest may reduce the efficacy of DNA-damaging agents used in cancer therapy [77]. The checkpoint with forkhead and ring finger domains (CHFR) gene is an early mitotic checkpoint gene that functions as a key player in controlling chromosomal integrity [98]. CHFR methylation is regarded as a docetaxel sensitive marker [67]. Re-expression of CHFR induced G2/M phase arrest and increased resistance to docetaxel in esophageal cancer cells [99]. CHFR is methylated in CRC (24–53%), gastric cancer (35–52%), esophageal cancer (16.3%) and NSCLC (10–40%) [100]. In docetaxel-treated gastric cancer patients, resistance to docetaxel was found in CHFR unmethylated patients and overall survival was longer in the CHFR methylated group compared to the CHFR unmethylated group [100]. Methylation of p16 has been demonstrated to sensitize gastric cancer to 5-fluorouracil therapy [101]. P16 is a cell cycle regulator that binds specifically to CDK4/6 and inhibits the activity of the CDK4/6-cyclin D1 complex, consequently blocking phosphorylation of retinoblastoma. This allows dissociation of the transcription factor E2F and subsequent transit to the S-phase [101, 102]. Therefore, the cell cycle is disrupted by P16 methylation and the cell number in the S-phase is increased. In this situation, the cancer cell easily absorbs 5-fluorouracil into S-phase cells, where it acts as an antimetabolite agent, leading to the inhibition of cancer cell growth [101]. Thus, selective disruption of the G1/S or G2/M checkpoints sensitizes cancer cells to chemotherapy.
ABERRANT METHYLATION OF DNA DAMAGE REPAIR GENES MAY SERVE AS PREDICTIVE, DIAGNOSTIC AND PROGNOSTIC MARKERS OF HUMAN CANCER
DNA repair pathways are frequently disrupted through genetic and epigenetic mechanisms. MSI and DNA repair gene mutations have been used as diagnostic and prognostic markers in various tumors [1, 103, 104]. Germ-line mutations in MLH1 and MSH2 suggest a very high risk of colorectal cancer [7]. The prognosis is much better in sporadic colorectal cancers with a mismatch repair deficiency [63]. The overall breast cancer and ovarian cancer risks for BRCA1 mutation carriers by age 70 years are 50-70% and 40-50%, respectively [6]. Thus, detection of the susceptible genes may guide the frequency of cancer surveillance and decision making for prophylactic surgery [7].
DNA methylation represents the epigenetic biomarker with the highest translational potential due to its stable nature and reliable detection technologies [105, 106]. Methylation of DDR genes has been detected in various cancers. MGMT is frequently methylated in various tumors [48]. MGMT methylation is a marker of poor prognosis in human glioma, without treatment with an alkylating agent. While, silencing MGMT by promoter region hypermethylation sensitizes glioma cells to alkylating agents [107]. MGMT methylation is more frequent in the margins of lung and colorectal cancer tissue samples than in the most distant sites from the resected specimen [30, 108]. This suggests that MGMT methylation is a potential early detection marker for lung and colorectal cancers. MLH1 was methylated in human esophageal dysplasia and cancer samples, but it was unmethylated in normal esophageal epithelia [71]. MLH1 methylation may serve as an early detection marker for esophageal cancer. MLH1 methylation was reported to be a good prognostic marker in colorectal and pancreatic cancers [63, 72]. CHFR methylation is associated with an increased risk of disease recurrence and poor survival in NSCLC and CRC [100]. The OGG1 gene is frequently methylated in different cancers, and silencing of its expression by promoter region methylation is associated with poor prognosis in breast cancer [109]. XPC methylation was associated with the malignant behavior of bladder cancer and may predict poor prognosis [53].
ABERRANT EPIGENETIC CHANGES OF DNA DAMAGE REPAIR GENES MAY SERVE AS CHEMO- AND RADIO-THERAPEUTIC SENSITIVE MARKERS
Drug resistance is a serious obstacle to effective cancer therapy [110]. The main mechanism of chemotherapy and radiotherapy is to induce DNA damage directly [5]. Cancer cells can also utilize the DNA repair machinery to process DNA lesions induced by DNA damaging agents in order to maintain cellular survival, which is therefore also an important mechanism of therapeutic resistance [111]. Patients with DDR defects are especially sensitive to these treatments.
Alkylating agents, including BCNU (1,3-bis(2-chloroethyl)-1-nitrosourea), procarbazine, streptozotocin, DTIC and TMZ, are among the most widely used chemotherapeutic drugs in human cancer [39]. Several alkylation sites in DNA have been identified as the targets of action of these compounds, and one of these sites is the O6 position of guanine [26]. Cytotoxic effects of alkylating agents are mediated primarily through methylation of O6-guanine, which leads to DNA double strand breaks and subsequent inhibition of DNA replication and apoptosis [26]. However, the toxicity of alkylating agents is reduced in the presence of MGMT by rapidly reversing the formation of adducts at the O6 position of guanine [26, 112]. For this reason, loss/reduced expression of MGMT can sensitize cancer cells to alkylating agents. In gliomas, enhanced sensitivity to alkylating agents was initially suggested in the subgroup of patients with reduced MGMT activity [41-43]. Since CpG island hypermethylation of MGMT is the major cause of loss of its expression in gliomas, MGMT methylated glioma patients are more sensitive to alkylating agents [19]. Thus, MGMT methylated patients acquired longer overall survival time after BCNU treatment [28, 107]. Similar results were obtained in colorectal cancer by treating MGMT methylated patients with TMZ [23]. Additional studies were reported in different cancer types (Table 1). Similar DNA damage effects can be mediated by radiotherapy through alkylation of O6-guanine. Methylation of MGMT sensitized glioma patients to radiotherapy or combined TMZ and radiotherapy [113, 114].
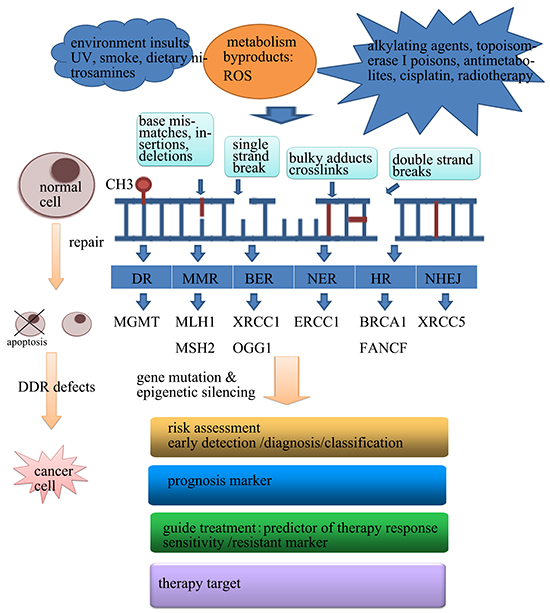
Figure 1: The application of abnormal epigenetic changes of DDR in human cancer.
Table 1: Promoter region methylation of DNA damage repair genes
Repair system |
Gene name |
Tumor type application |
Diagnosis and prognosis Marker |
Sensitive marker |
Ref |
|---|---|---|---|---|---|
Direct reversal |
MGMT |
Gliomas, diffuse large B-cell lymphoma, NSCLC, colorectal, gastric, esophageal and head and neck cancers |
Early detection, poor prognosis |
TMZ, ACNU, BCNU, procarbazine sensitive |
|
BER |
XRCC1 |
Gastric |
[41] |
||
TDG |
Myeloma |
[42] |
|||
MED1 |
Colorectal and ovarian |
[43] |
|||
OGG1 |
Breast and thyroid |
Poor prognosis |
|||
WRN |
NSCLC, colorectal, gastric, prostate, breast and thyroid cancers, chondro-sarcomas, non-Hodgkin lymphoma |
Irinotecan sensitive |
[47] |
||
NER |
XPC |
Bladder cancer |
Poor prognosis |
[53] |
|
ERCC1 |
Glioma |
Poor prognosis |
Cisplatin sensitive |
[54] |
|
hHR23B |
Myeloma cells |
[55] |
|||
XPG |
Ovarian |
Nemorubicin resistance |
[56] |
||
MMR |
MLH1 |
Colorectal, gastric, pancreatic, ovarian and breast cancers, NSCLC, ESCC, adult T-cell leukemia/lymphoma, oral squamous cell carcinoma, head and neck squamous cell carcinomas |
Early detection, good prognosis |
Carboplatin/ cisplatin/ TMZ /epirubicin |
|
MSH2 |
HNPCC |
[65] |
|||
MSH3 |
Esophageal and gastric cancers |
||||
Non-homologous end-joining |
XRCC5 |
NSCLC |
[80] |
||
Homologous recombination |
BRCA1 |
NSCLC, breast, ovarian, gastric, colorectal, bladder and pancreatic cancers |
Diagnosis marker |
Cisplatin, PARPi sensitive |
|
BRCA2 |
NSCLC |
[80] |
|||
SRBC |
Colorectal, lung, pancreatic, gastric and ovarian cancers |
Oxaliplatin resistance |
|||
FA |
FANCF |
NSCLC, ovarian, breast, gastric, cervical and head and neck cancers |
Cisplatin sensitive |
||
FANCC |
Head and neck squamous cell carcinoma, AML, ALL |
||||
FANCL |
ALL |
[91] |
|||
Cell cycle related |
ATM |
Breast cancer, head and neck squamous cell carcinomas |
Radiotherapy sensitive |
||
CHFR |
NSCLC, breast, bladder, colorectal, gastric, nasopharyngeal, esophageal, cervical, hepatocellular and head and neck cancers |
Poor prognosis |
Docetaxel sensitive |
[100] |
|
P16 |
Oral and oropharyngeal squamous cell carcinoma, esophageal, bladder and lung cancers |
Poor prognosis |
5-FU sensitive |
||
RASSF1A |
Breast, ovarian, lung, prostate, colon, bladder and liver cancers |
Early detection, Poor prognosis |
Cisplatin and tamoxifen resistance |
[156] |
|
CHK2 |
Glioma, NSCLC |
||||
Genome “caretaker“ |
FHIT |
Gastric, esophageal, cervical and breast cancers, NSCLC |
Poor prognosis |
||
Antioxidant |
GPX3 |
Non-M3 acute myeloid leukemia, thyroid, liver and head and neck cancers |
Poor prognosis |
Cisplatin resistant |
|
Detoxification of carcinogens |
GST-Pi |
Prostate, breast. kidney, liver and lung cancers |
Poor prognosis |
Doxorubicin sensitive |
Repair of interstrand crosslinks induced by cisplatin leads to drug resistance. The expression of ERCC1 has been related to poor response and survival in cisplatin-treated NSCLC patients [1]. ERCC1 expression is a resistance marker of platinum-based systemic chemotherapy in human gastric and ovarian cancers [115, 116]. Methylation of ERCC1 is associated with cisplatin sensitivity in glioma cell lines [54]. BRCA1/2 deficiency results in cellular sensitivity to cisplatin. BRCA1 methylation has been found to predict significantly higher response rates to cisplatin treatment in patients with triple-negative breast cancer [117]. FANCF methylation is correlated with cisplatin sensitivity in ovarian cancer cells [118]. Demethylation of FANCF leads to acquired cisplatinum resistance [119]. CHFR methylation is a docetaxel sensitive marker in human esophageal, breast, gastric and other cancers [100]. MLH1 methylation is a resistance marker of oxaliplatin in human gastric cancer [67]. WRN methylation is an irinotecan sensitive marker in gastric cancer [120]. Methylation of ATM sensitized glioma and colorectal cancer cells to ionizing radiation therapy [121, 122]. As described above, disruption of DNA repair can promote genetic instability and further lead to tumorigenesis. On the other hand, defects in DNA damage repair sensitize cancer cells to DNA damaging agents. Thus, detection of genetic and epigenetic changes of DDR genes is reasonable before treatment.
THERAPEUTIC TARGETING OF ABERRANT EPIGENETIC CHANGES IN DNA DAMAGE REPAIR GENES
Demethylation of MMR genes sensitizes cancer cells to DNA damaging agents
MMR recognizes damaged DNA that was induced by DNA damaging agents, and it initiates the cell death machinery to trigger apoptosis [123, 124]. Disruption of MMR by DNA methylation failed to induce apoptosis [125]. In vitro studies have shown that treatment of MLH1-methylated colon cancer cell lines with the demethylating agent 5′-aza-2′-deoxycytidine (5-aza-dC) induced the expression of MLH1 and sensitized cancer cells to 5-fluorouracil (5-FU) [126]. In MLH1 methylated ovarian and colon cancer xenografts, cancer cells were sensitized to carboplatin, cisplatin, TMZ and epirubicin after re-expression of MLH1 by decitabine [127]. Another study demonstrated that demethylation of MLH1 was induced by low-dose 5-aza-dC and the sensitivity of radiotherapy was enhanced in colorectal cancer [128]. In a phase II clinical trial in ovarian cancer patients, decitabine induced MLH1 demethylation, increased the response rate of carboplatin and prolonged progression-free survival [129].
Recently, a novel DNA methylation inhibitor, SGI-110, resensitized a range of platinum-resistant ovarian cancer cells to cisplatin. SGI-110 alone or in combination with cisplatin enhanced antitumor effects in ovarian cancer xenografts [130]. While, SGI-110 is controversial in the treatment of gastric and ovarian cancer in combination with oxaliplatin in patients with MLH1 methylation [67, 131]. It has been reported that MMR proteins do not recognize the adducts formed by oxaliplatin [132]. XPG methylation leads to disruption of the NER system and is related to resistance of nemorubicin, a doxorubicin derivative. Restoration of XPG expression by demethylating agents restored the sensitivity of different cancer cells to nemorubicin [56].
Synthetic lethality therapy in cancer patients with defects in DNA damage repair pathways induced by genetic or epigenetic abnormalities
There are a number of DNA repair pathways that protect cellular DNA from injury. “Cross-talk” between these pathways facilitates a unified guard for genomic integrity. The concept of synthetic lethality originates from studies in drosophila model systems in which a combination of mutations in two or more separate genes leads to cell death [133].
The strategy of synthetic lethality approaches is that defects in two different genes or pathways together result in cell death. While, independently defect does not affect viability [22]. Although only less than 10% of sporadic ovarian cancer and 5% of breast cancer harbored BRCA1 mutations, BRCA1 methylation is observed in approximately 11–14% of breast cancer and 5%–31% of ovarian cancer [93]. HR-defective BRCA1/2-deficient cell lines display dramatically increased sensitivity to inhibition of the single strand break repair enzyme PARP [81]. A phase II clinical trial demonstrated that monotherapy with the PARP inhibitor olaparib (AZD-2281; AstraZeneca) achieved encouraging response rates of 41% and 33% in patients with BRCA1- or BRCA2-mutated advanced breast and ovarian cancers, respectively [77]. Targeting RAD52 can also induce synthetic lethality in BRCA-deficient leukemias [134]. In addition, colorectal cancer cells with defects in MSH2 or MLH1 are sensitized to methotrexate [133, 135]. Many cancer cells acquire defects in a certain DNA repair pathway and become dependent on a compensatory mechanism in order to survive. Thus, pharmacological inhibition of the “backup” pathway in combination with DNA damage will selectively kill cancer cells but spare their normal counterparts [77].
Deficiency of DNA damage repair caused by DNA mutation and methylation opens a new window for immune therapy in human cancers
DNA damage repair deficiency was found in various human cancers by gene mutation or promoter region hypermethylation, including MLH1, MSH2, MGMT, BRCA1, BRCA2 and others. Loss of efficiency of one or more DNA repair pathways can accelerate the rate of accumulation of additional mutations by 100–1,000 times [5]. Somatic mutations have the potential to encode “non-self” immunogenic antigens [136]. The programmed death 1 (PD-1) pathway is a negative feedback system that represses Th1 cytotoxic immune responses. It is up-regulated in many tumors and in their surrounding microenvironment [137, 138]. Blockade of this pathway with antibodies to PD-1 or its ligands has led to remarkable clinical responses in patients with many different types of cancer [136, 139-141]. Epigenetic inactivation of DNA damage repair genes could induce gene mutations and lead to alterations in the expression of tumor-associated self-antigens, therefore altering the antigenicity of the tumor. In a recent study, authors evaluated the clinical activity of pembrolizumab, a PD-1 inhibitor, in colorectal cancer patients with progressive metastatic cancer with or without mismatch-repair deficiency. For mismatch repair-deficient colorectal cancers, the immune-related objective response rate and immune-related progression-free survival rate were 40% and 78%, respectively. While, in mismatch repair-proficient colorectal cancer patients, the immune-related objective response rate and immune-related progression-free survival rate were 0% and 11%, respectively [136]. Whole-exome sequencing revealed that the somatic mutation number in mismatch repair-deficient tumors is much larger than in mismatch repair-proficient tumors, and high somatic mutation loads were associated with prolonged progression-free survival [136]. It was demonstrated in early studies that there is dense immune infiltration and a Th1-associated cytokine-rich environment in mismatch repair-deficient tumors [142]. The mismatch repair–deficient tumor microenvironment strongly expressed several immune checkpoint ligands, including PD-1, PD-L1, CTLA-4, LAG-3 and IDO, to protect the tumor cells from death. Thus, inhibition of these immune checkpoints can enhance T-cell responses and mediate effective antitumor activity in MMR defective cancers [143]. In addition to the MMR system, deficiencies of other DNA damage repair pathways play important roles in tumorigenesis. Thus, both mutational and epigenetic landscapes are necessary to stratify the tumor patients before immune therapy, especially for genes in DNA damage repair pathways.
CONCLUSION & PERSPECTIVE
Precision medicine based on various ‘omics,’ such as genomics, transcriptomics and epigenomics, has already had an impact on the clinical care of cancer patients. Anticancer therapy could be selected on the basis of the molecular phenotype of the cancer. Targeting DNA repair proteins has significantly increased over the past decade. The impact of DNA repair on resistance to cisplatin is well documented in numerous cancers. Immune therapy has reached apparent efficiency in MMR defective cancers. MGMT methylation serves as a sensitive marker for alkylating agents in glioma patients, and CHFR methylation sensitizes esophageal cancer cells to docetaxel. Synthetic lethality is a more exciting approach in patients with DDR defects. PARP inhibitors are the most effective anticancer reagents in BRCA-deficient cancer cells.
ACKNOWLEDGMENT
This work was supported by the following grants: National Basic Research Program of China (973 Program No. 2012CB934002, 2015CB553904); National High-tech R&D Program of China (863 Program No. SS2012AA020314, SS2012AA020821, SS2012AA020303); National Key Scientific instrument Special Programme of China (Grant No. 2011YQ03013405); National Science Foundation of China (Grant No. 81490753).
CONFLICTS OF INTEREST
JGH is a consultant to MDxHealth. The other authors declare no conflicts of interest.
REFERENCES
1. Postel-Vinay S, Vanhecke E, Olaussen KA, Lord CJ, Ashworth A and Soria JC. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nature reviews Clinical oncology. 2012; 9:144-155.
2. Martin SA, Lord CJ and Ashworth A. Therapeutic targeting of the DNA mismatch repair pathway. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16:5107-5113.
3. Plummer R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clinical cancer research. 2010; 16:4527-4531.
4. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001; 411:366-374.
5. Abbotts R, Thompson N and Madhusudan S. DNA repair in cancer: emerging targets for personalized therapy. Cancer management and research. 2014; 6:77-92.
6. Dhillon KK, Swisher EM and Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Science. 2011; 102:663-669.
7. Markowitz SD and Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. The New England journal of medicine. 2009; 361:2449-2460.
8. Toyota M and Suzuki H. Epigenetic drivers of genetic alterations. Advances in genetics. 2010; 70:309-323.
9. Lynch HT and de la Chapelle A. Hereditary colorectal cancer. The New England journal of medicine. 2003; 348:919-932.
10. Suter CM, Martin DI and Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nature genetics. 2004; 36:497-501.
11. Chan TL, Yuen ST, Kong CK, Chan YW, Chan AS, Ng WF, Tsui WY, Lo MW, Tam WY, Li VS and Leung SY. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nature genetics. 2006; 38:1178-1183.
12. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM and Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer research. 1997; 57:808-811.
13. Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA and Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:6870-6875.
14. Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD and Markowitz SD. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:8698-8702.
15. King MC, Marks JH and Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science (New York, NY). 2003; 302:643-646.
16. Chen S and Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. Journal of clinical oncology. 2007; 25:1329-1333.
17. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG and Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast cancer research. 2006; 8:R38.
18. Suijkerbuijk KP, Fackler MJ, Sukumar S, van Gils CH, van Laar T, van der Wall E, Vooijs M and van Diest PJ. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Annals of oncology. 2008; 19:1870-1874.
19. Wang L, Zhu D, Zhang C, Mao X, Wang G, Mitra S, Li BF, Wang X and Wu M. Mutations of O6-methylguanine-DNA methyltransferase gene in esophageal cancer tissues from Northern China. International journal of cancer. 1997; 71:719-723.
20. Zhang L, Lu W, Miao X, Xing D, Tan W and Lin D. Inactivation of DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation and its relation to p53 mutations in esophageal squamous cell carcinoma. Carcinogenesis. 2003; 24:1039-1044.
21. Broustas CG and Lieberman HB. DNA Damage Response Genes and the Development of Cancer Metastasis. Radiation research. 2014; 181:111-130.
22. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nature reviews Cancer. 2012; 12:801-817.
23. Inno A, Fanetti G, Di Bartolomeo M, Gori S, Maggi C, Cirillo M, Iacovelli R, Nichetti F, Martinetti A, de Braud F, Bossi I and Pietrantonio F. Role of MGMT as biomarker in colorectal cancer. World journal of clinical cases. 2014; 2:835-839.
24. Su Y, Yin L, Liu R, Sheng J, Yang M, Wang Y, Pan E, Guo W, Pu Y, Zhang J and Liang G. Promoter methylation status of MGMT, hMSH2, and hMLH1 and its relationship to corresponding protein expression and TP53 mutations in human esophageal squamous cell carcinoma. Medical oncology (Northwood, London, England). 2014; 31:784.
25. Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM and Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:6443-6447.
26. Esteller M and Herman JG. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 2004; 23:1-8.
27. Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB and Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer research. 2000; 60:2368-2371.
28. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB and Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. The New England journal of medicine. 2000; 343:1350-1354.
29. Esteller M, Gaidano G, Goodman SN, Zagonel V, Capello D, Botto B, Rossi D, Gloghini A, Vitolo U, Carbone A, Baylin SB and Herman JG. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. Journal of the National Cancer Institute. 2002; 94:26-32.
30. Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR and Issa JP. MGMT promoter methylation and field defect in sporadic colorectal cancer. Journal of the National Cancer Institute. 2005; 97:1330-1338.
31. Ksiaa F, Ziadi S, Amara K, Korbi S and Trimeche M. Biological significance of promoter hypermethylation of tumor-related genes in patients with gastric carcinoma. Clinica chimica acta. 2009; 404:128-133.
32. Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF and Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer research. 2001; 61:249-255.
33. Pierini S, Jordanov SH, Mitkova AV, Chalakov IJ, Melnicharov MB, Kunev KV, Mitev VI, Kaneva RP and Goranova TE. Promoter hypermethylation of CDKN2A, MGMT, MLH1, and DAPK genes in laryngeal squamous cell carcinoma and their associations with clinical profiles of the patients. Head & neck. 2014; 36:1103-1108.
34. Li J, Braganza A and Sobol RW. Base excision repair facilitates a functional relationship between Guanine oxidation and histone demethylation. Antioxidants & redox signaling. 2013; 18:2429-2443.
35. Fortini P, Pascucci B, Parlanti E, D’Errico M, Simonelli V and Dogliotti E. 8-Oxoguanine DNA damage: at the crossroad of alternative repair pathways. Mutation research. 2003; 531:127-139.
36. Sweasy JB, Lang T and DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell cycle. 2006; 5:250-259.
37. Moon YW, Park WS, Vortmeyer AO, Weil RJ, Lee YS, Winters TA, Zhuang Z and Fuller BG. Mutation of the uracil DNA glycosylase gene detected in glioblastoma. Mutation research. 1998; 421:191-196.
38. Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, Masciullo V, Genuardi M, Paravatou-Petsotas M, Bassi DE, Ruggeri BA, Klein-Szanto AJ, Testa JR, Neri G and Bellacosa A. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nature genetics. 1999; 23:266-268.
39. Chevillard S, Radicella JP, Levalois C, Lebeau J, Poupon MF, Oudard S, Dutrillaux B and Boiteux S. Mutations in OGG1, a gene involved in the repair of oxidative DNA damage, are found in human lung and kidney tumours. Oncogene. 1998; 16:3083-3086.
40. Casse C, Hu YC and Ahrendt SA. The XRCC1 codon 399 Gln allele is associated with adenine to guanine p53 mutations in non-small cell lung cancer. Mutation research. 2003; 528:19-27.
41. Wang P, Tang JT, Peng YS, Chen XY, Zhang YJ and Fang JY. XRCC1 downregulated through promoter hypermethylation is involved in human gastric carcinogenesis. Journal of digestive diseases. 2010; 11:343-351.
42. Peng B, Hurt EM, Hodge DR, Thomas SB and Farrar WL. DNA hypermethylation and partial gene silencing of human thymine- DNA glycosylase in multiple myeloma cell lines. Epigenetics. 2006; 1:138-145.
43. Howard JH, Frolov A, Tzeng CW, Stewart A, Midzak A, Majmundar A, Godwin A, Heslin M, Bellacosa A and Arnoletti JP. Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer. Cancer biology & therapy. 2009; 8:94-100.
44. Guan H, Ji M, Hou P, Liu Z, Wang C, Shan Z, Teng W and Xing M. Hypermethylation of the DNA mismatch repair gene hMLH1 and its association with lymph node metastasis and T1799A BRAF mutation in patients with papillary thyroid cancer. Cancer. 2008; 113:247-255.
45. Ronneberg JA, Fleischer T, Solvang HK, Nordgard SH, Edvardsen H, Potapenko I, Nebdal D, Daviaud C, Gut I, Bukholm I, Naume B, Borresen-Dale AL, Tost J and Kristensen V. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Molecular oncology. 2011; 5:61-76.
46. Harrigan JA, Wilson DM, 3rd, Prasad R, Opresko PL, Beck G, May A, Wilson SH and Bohr VA. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic acids research. 2006; 34:745-754.
47. Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, Guerrero D, Castells A, von Kobbe C, Bohr VA and Esteller M. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:8822-8827.
48. Hegi ME, Sciuscio D, Murat A, Levivier M and Stupp R. Epigenetic deregulation of DNA repair and its potential for therapy. Clinical cancer research. 2009; 15:5026-5031.
49. Neumann AS, Sturgis EM and Wei Q. Nucleotide excision repair as a marker for susceptibility to tobacco-related cancers: a review of molecular epidemiological studies. Molecular carcinogenesis. 2005; 42:65-92.
50. de Boer J and Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000; 21:453-460.
51. Boland CR and Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010; 138:2073-2087 e2073.
52. Wu YH, Tsai Chang JH, Cheng YW, Wu TC, Chen CY and Lee H. Xeroderma pigmentosum group C gene expression is predominantly regulated by promoter hypermethylation and contributes to p53 mutation in lung cancers. Oncogene. 2007; 26:4761-4773.
53. Yang J, Xu Z, Li J, Zhang R, Zhang G, Ji H, Song B and Chen Z. XPC epigenetic silence coupled with p53 alteration has a significant impact on bladder cancer outcome. The Journal of urology. 2010; 184:336-343.
54. Chen HY, Shao CJ, Chen FR, Kwan AL and Chen ZP. Role of ERCC1 promoter hypermethylation in drug resistance to cisplatin in human gliomas. International journal of cancer. 2010; 126:1944-1954.
55. Peng B, Hodge DR, Thomas SB, Cherry JM, Munroe DJ, Pompeia C, Xiao W and Farrar WL. Epigenetic silencing of the human nucleotide excision repair gene, hHR23B, in interleukin-6-responsive multiple myeloma KAS-6/1 cells. The Journal of biological chemistry. 2005; 280:4182-4187.
56. Sabatino MA, Marabese M, Ganzinelli M, Caiola E, Geroni C and Broggini M. Down-regulation of the nucleotide excision repair gene XPG as a new mechanism of drug resistance in human and murine cancer cells. Molecular cancer. 2010; 9:259.
57. Silva FC, Valentin MD, Ferreira Fde O, Carraro DM and Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo medical journal. 2009; 127:46-51.
58. Li GM. Mechanisms and functions of DNA mismatch repair. Cell research. 2008; 18:85-98.
59. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. Journal of the National Cancer Institute. 2004; 96:261-268.
60. Haydon AM and Jass JR. Emerging pathways in colorectal-cancer development. The Lancet Oncology. 2002; 3:83-88.
61. Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, Onda A, Okumura Y, Kishi N, Iwama T, Mori T, Koike M, Ushio K, Chiba M, Nomizu S, Konishi F, Utsunomiya J and Miyaki M. Molecular nature of colon tumors in hereditary nonpolyposis colon cancer, familial polyposis, and sporadic colon cancer. Gastroenterology. 1996; 111:307-317.
62. Wheeler JM, Loukola A, Aaltonen LA, Mortensen NJ and Bodmer WF. The role of hypermethylation of the hMLH1 promoter region in HNPCC versus MSI+ sporadic colorectal cancers. Journal of medical genetics. 2000; 37:588-592.
63. Sinicrope FA and Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clinical cancer research. 2012; 18:1506-1512.
64. Poynter JN, Siegmund KD, Weisenberger DJ, Long TI, Thibodeau SN, Lindor N, Young J, Jenkins MA, Hopper JL, Baron JA, Buchanan D, Casey G, Levine AJ, Le Marchand L, Gallinger S, Bapat B, et al. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer epidemiology, biomarkers & prevention. 2008; 17:3208-3215.
65. Nagasaka T, Rhees J, Kloor M, Gebert J, Naomoto Y, Boland CR and Goel A. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer research. 2010; 70:3098-3108.
66. Matsushita M, Takeuchi S, Yang Y, Yoshino N, Tsukasaki K, Taguchi H, Koeffler HP and Seo H. Methylation of the MLH1 gene in hematological malignancies. Oncology reports. 2005; 14:191-194.
67. Li Y, Yang Y, Lu Y, Herman JG, Brock MV, Zhao P and Guo M. Predictive value of CHFR and MLH1 methylation in human gastric cancer. Gastric cancer. 2015; 18:280-287.
68. Tawfik HM, El-Maqsoud NM, Hak BH and El-Sherbiny YM. Head and neck squamous cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 gene. American journal of otolaryngology. 2011; 32:528-536.
69. Wang YC, Lu YP, Tseng RC, Lin RK, Chang JW, Chen JT, Shih CM and Chen CY. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. The Journal of clinical investigation. 2003; 111:887-895.
70. Gonzalez-Ramirez I, Ramirez-Amador V, Irigoyen-Camacho ME, Sanchez-Perez Y, Anaya-Saavedra G, Granados-Garcia M, Garcia-Vazquez F and Garcia-Cuellar CM. hMLH1 promoter methylation is an early event in oral cancer. Oral oncology. 2011; 47:22-26.
71. Guo M, Ren J, House MG, Qi Y, Brock MV and Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clinical cancer research. 2006; 12:4515-4522.
72. House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Cameron JL, Hruban RH, Maitra A and Yeo CJ. Prognostic value of hMLH1 methylation and microsatellite instability in pancreatic endocrine neoplasms. Surgery. 2003; 134:902-908; discussion 909.
73. V S, Bhagat R, C SP, V RP and Krishnamoorthy L. Microsatellite instability, promoter methylation and protein expression of the DNA mismatch repair genes in epithelial ovarian cancer. Genomics. 2014; 104:257-263.
74. Murata H, Khattar NH, Kang Y, Gu L and Li GM. Genetic and epigenetic modification of mismatch repair genes hMSH2 and hMLH1 in sporadic breast cancer with microsatellite instability. Oncogene. 2002; 21:5696-5703.
75. Levine AJ, Win AK, Buchanan DD, Jenkins MA, Baron JA, Young JP, Long TI, Weisenberger DJ, Laird PW, McCall RL, Duggan DJ and Haile RW. Cancer risks for the relatives of colorectal cancer cases with a methylated MLH1 promoter region: data from the Colorectal Cancer Family Registry. Cancer prevention research (Philadelphia, Pa). 2012; 5:328-335.
76. Jackson SP and Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071-1078.
77. Huhn D, Bolck HA and Sartori AA. Targeting DNA double-strand break signalling and repair: recent advances in cancer therapy. Swiss medical weekly. 2013; 143:w13837.
78. Sawan C, Vaissiere T, Murr R and Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutation research. 2008; 642:1-13.
79. Scharer OD. DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. Chembiochem. 2005; 6:27-32.
80. Lee MN, Tseng RC, Hsu HS, Chen JY, Tzao C, Ho WL and Wang YC. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clinical cancer research. 2007; 13:832-838.
81. D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. The New England journal of medicine. 2010; 362:1909-1919.
82. Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature reviews Genetics. 2007; 8:735-748.
83. Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M and D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell. 2001; 7:249-262.
84. Tutt AN, Lord CJ, McCabe N, Farmer H, Turner N, Martin NM, Jackson SP, Smith GC and Ashworth A. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harbor symposia on quantitative biology. 2005; 70:139-148.
85. Neamatzadeh H, Shiryazdi SM and Kalantar SM. BRCA1 and BRCA2 mutations in Iranian breast cancer patients: A systematic review. Journal of research in medical sciences. 2015; 20:284-293.
86. Kobayashi H, Ohno S, Sasaki Y and Matsuura M. Hereditary breast and ovarian cancer susceptibility genes (review). Oncology reports. 2013; 30:1019-1029.
87. van der Heijden MS, Yeo CJ, Hruban RH and Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer research. 2003; 63:2585-2588.
88. Ahmad SI, Hanaoka F and Kirk SH. Molecular biology of Fanconi anaemia--an old problem, a new insight. BioEssays. 2002; 24:439-448.
89. Guo H, Yan W, Yang Y and Guo M. [Promoter region methylation of DNA damage repair genes in human gastric cancer]. Zhonghua yi xue za zhi. 2014; 94:2193-2196.
90. Ghosh A, Ghosh S, Maiti GP, Mukherjee S, Mukherjee N, Chakraborty J, Roy A, Roychoudhury S and Panda CK. Association of FANCC and PTCH1 with the development of early dysplastic lesions of the head and neck. Annals of surgical oncology. 2012; 19:S528-538.
91. Hess CJ, Ameziane N, Schuurhuis GJ, Errami A, Denkers F, Kaspers GJ, Cloos J, Joenje H, Reinhardt D, Ossenkoppele GJ, Zwaan CM and Waisfisz Q. Hypermethylation of the FANCC and FANCL promoter regions in sporadic acute leukaemia. Cellular oncology. 2008; 30:299-306.
92. Stefansson OA, Villanueva A, Vidal A, Marti L and Esteller M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics. 2012; 7:1225-1229.
93. Turner N, Tutt A and Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nature reviews Cancer. 2004; 4:814-819.
94. Laskar RS, Talukdar FR, Choudhury JH, Singh SA, Kundu S, Dhar B, Mondal R and Ghosh SK. Association of HPV with genetic and epigenetic alterations in colorectal adenocarcinoma from Indian population. Tumour biology. 2015; 36:4661-4670.
95. Harada H, Miyamoto K, Yamashita Y, Nakano K, Taniyama K, Miyata Y, Ohdan H and Okada M. Methylation of breast cancer susceptibility gene 1 (BRCA1) predicts recurrence in patients with curatively resected stage I non-small cell lung cancer. Cancer. 2013; 119:792-798.
96. Yu J, Zhu T, Wang Z, Zhang H, Qian Z, Xu H, Gao B, Wang W, Gu L, Meng J, Wang J, Feng X, Li Y, Yao X and Zhu J. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clinical cancer research. 2007; 13:7296-7304.
97. Guo M, Jia Y, Yu Z, House MG, Esteller M, Brock MV and Herman JG. Epigenetic changes associated with neoplasms of the exocrine and endocrine pancreas. Discovery medicine. 2014; 17:67-73.
98. Scolnick DM and Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000; 406:430-435.
99. Yun T, Liu Y, Gao D, Linghu E, Brock MV, Yin D, Zhan Q, Herman JG and Guo M. Methylation of CHFR sensitizes esophageal squamous cell cancer to docetaxel and paclitaxel. Genes & cancer. 2015; 6:38-48. doi:10.18632/genesandcancer.46.
100. Derks S, Cleven AH, Melotte V, Smits KM, Brandes JC, Azad N, van Criekinge W, de Bruine AP, Herman JG and van Engeland M. Emerging evidence for CHFR as a cancer biomarker: from tumor biology to precision medicine. Cancer metastasis reviews. 2014; 33:161-171.
101. Mitsuno M, Kitajima Y, Ide T, Ohtaka K, Tanaka M, Satoh S and Miyazaki K. Aberrant methylation of p16 predicts candidates for 5-fluorouracil-based adjuvant therapy in gastric cancer patients. Journal of gastroenterology. 2007; 42:866-873.
102. Serrano M, Hannon GJ and Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993; 366:704-707.
103. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, Nathanson KL, Laitman Y, Kushnir A, Paluch-Shimon S, Berger R, Zidan J, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. Jama. 2015; 313:1347-1361.
104. Yamamoto H and Imai K. Microsatellite instability: an update. Archives of toxicology. 2015; 89:899-921.
105. Mulero-Navarro S and Esteller M. Epigenetic biomarkers for human cancer: the time is now. Critical reviews in oncology/hematology. 2008; 68:1-11.
106. Heyn H, Mendez-Gonzalez J and Esteller M. Epigenetic profiling joins personalized cancer medicine. Expert review of molecular diagnostics. 2013; 13:473-479.
107. Jacinto FV and Esteller M. MGMT hypermethylation: a prognostic foe, a predictive friend. DNA repair. 2007; 6:1155-1160.
108. Guo M, House MG, Hooker C, Han Y, Heath E, Gabrielson E, Yang SC, Baylin SB, Herman JG and Brock MV. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clinical cancer research. 2004; 10:5131-5136.
109. Fleischer T, Edvardsen H, Solvang HK, Daviaud C, Naume B, Borresen-Dale AL, Kristensen VN and Tost J. Integrated analysis of high-resolution DNA methylation profiles, gene expression, germline genotypes and clinical end points in breast cancer patients. International journal of cancer. 2014; 134:2615-2625.
110. Dhillon KK, Swisher EM and Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011; 102:663-669.
111. Longley DB and Johnston PG. Molecular mechanisms of drug resistance. The Journal of pathology. 2005; 205:275-292.
112. Rabik CA, Njoku MC and Dolan ME. Inactivation of O6-alkylguanine DNA alkyltransferase as a means to enhance chemotherapy. Cancer treatment reviews. 2006; 32:261-276.
113. Minniti G, Scaringi C, Arcella A, Lanzetta G, Di Stefano D, Scarpino S, Bozzao A, Pace A, Villani V, Salvati M, Esposito V, Giangaspero F and Enrici RM. IDH1 mutation and MGMT methylation status predict survival in patients with anaplastic astrocytoma treated with temozolomide-based chemoradiotherapy. Journal of neuro-oncology. 2014; 118:377-383.
114. Rivera AL, Pelloski CE, Gilbert MR, Colman H, De La Cruz C, Sulman EP, Bekele BN and Aldape KD. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro-oncology. 2010; 12:116-121.
115. Liu YP, Ling Y, Qi QF, Zhang YP, Zhang CS, Zhu CT, Wang MH and Pan YD. The effects of ERCC1 expression levels on the chemosensitivity of gastric cancer cells to platinum agents and survival in gastric cancer patients treated with oxaliplatin-based adjuvant chemotherapy. Oncology letters. 2013; 5:935-942.
116. Bogush TA, Popova AS, Dudko EA, Bogush EA, Tyulyandina AS, Tyulyandin SA and Davydov MI. [ERCC1 as a Marker of Ovarian Cancer Resistance to Platinum Drugs]. Antibiotiki i khimioterapiia = Antibiotics and chemoterapy. 2015; 60:42-50.
117. Stefansson OA and Esteller M. Epigenetic modifications in breast cancer and their role in personalized medicine. The American journal of pathology. 2013; 183:1052-1063.
118. Olopade OI and Wei M. FANCF methylation contributes to chemoselectivity in ovarian cancer. Cancer cell. 2003; 3:417-420.
119. D’Andrea AD. The Fanconi Anemia/BRCA signaling pathway: disruption in cisplatin-sensitive ovarian cancers. Cell cycle. 2003; 2:290-292.
120. Wang L, Xie L, Wang J, Shen J and Liu B. Correlation between the methylation of SULF2 and WRN promoter and the irinotecan chemosensitivity in gastric cancer. BMC gastroenterology. 2013; 13:173.
121. Roy K, Wang L, Makrigiorgos GM and Price BD. Methylation of the ATM promoter in glioma cells alters ionizing radiation sensitivity. Biochemical and biophysical research communications. 2006; 344:821-826.
122. Kim WJ, Vo QN, Shrivastav M, Lataxes TA and Brown KD. Aberrant methylation of the ATM promoter correlates with increased radiosensitivity in a human colorectal tumor cell line. Oncogene. 2002; 21:3864-3871.
123. Wyatt MD and Wilson DM, 3rd. Participation of DNA repair in the response to 5-fluorouracil. Cellular and molecular life sciences. 2009; 66:788-799.
124. Li GM. The role of mismatch repair in DNA damage-induced apoptosis. Oncology research. 1999; 11:393-400.
125. Karran P and Bignami M. DNA damage tolerance, mismatch repair and genome instability. BioEssays. 1994; 16:833-839.
126. Fujita H, Kato J, Horii J, Harada K, Hiraoka S, Shiraha H, Sakaguchi K and Shiratori Y. Decreased expression of hMLH1 correlates with reduced 5-fluorouracil-mediated apoptosis in colon cancer cells. Oncology reports. 2007; 18:1129-1137.
127. Plumb JA, Strathdee G, Sludden J, Kaye SB and Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer research. 2000; 60:6039-6044.
128. Hofstetter B, Niemierko A, Forrer C, Benhattar J, Albertini V, Pruschy M, Bosman FT, Catapano CV and Ciernik IF. Impact of genomic methylation on radiation sensitivity of colorectal carcinoma. International journal of radiation oncology, biology, physics. 2010; 76:1512-1519.
129. Glasspool RM, Brown R, Gore ME, Rustin GJ, McNeish IA, Wilson RH, Pledge S, Paul J, Mackean M, Hall GD, Gabra H, Halford SE, Walker J, Appleton K, Ullah R and Kaye S. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. British journal of cancer. 2014; 110:1923-1929.
130. Fang F, Munck J, Tang J, Taverna P, Wang Y, Miller DF, Pilrose J, Choy G, Azab M, Pawelczak KS, VanderVere-Carozza P, Wagner M, Lyons J, Matei D, Turchi JJ and Nephew KP. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clinical cancer research. 2014; 20:6504-6516.
131. Strathdee G, MacKean MJ, Illand M and Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene. 1999; 18:2335-2341.
132. Martin LP, Hamilton TC and Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clinical cancer research. 2008; 14:1291-1295.
133. McLornan DP, List A and Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. The New England journal of medicine. 2014; 371:1725-1735.
134. Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng H, Roy D, Slupianek A, Rassool FV, Wasik MA, Childers W, Copland M, Muschen M, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood. 2013; 122:1293-1304.
135. Martin SA, McCarthy A, Barber LJ, Burgess DJ, Parry S, Lord CJ and Ashworth A. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO molecular medicine. 2009; 1:323-337.
136. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015; 372:2509-2520.
137. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012; 366:2443-2454.
138. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124-128.
139. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. The New England journal of medicine. 2015; 372:311-319.
140. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013; 369:134-144.
141. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, Chen DS and Vogelzang NJ. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014; 515:558-562.
142. Young J, Simms LA, Biden KG, Wynter C, Whitehall V, Karamatic R, George J, Goldblatt J, Walpole I, Robin SA, Borten MM, Stitz R, Searle J, McKeone D, Fraser L, Purdie DR, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. The American journal of pathology. 2001; 159:2107-2116.
143. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, Vogelstein B, Sears CL, Anders RA, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer discovery. 2015; 5:43-51.
144. Vogelsang M, Paccez JD, Schafer G, Dzobo K, Zerbini LF and Parker MI. Aberrant methylation of the MSH3 promoter and distal enhancer in esophageal cancer patients exposed to first-hand tobacco smoke. Journal of cancer research and clinical oncology. 2014; 140:1825-1833.
145. Kim HG, Lee S, Kim DY, Ryu SY, Joo JK, Kim JC, Lee KH and Lee JH. Aberrant methylation of DNA mismatch repair genes in elderly patients with sporadic gastric carcinoma: A comparison with younger patients. Journal of surgical oncology. 2010; 101:28-35.
146. Moutinho C, Martinez-Cardus A, Santos C, Navarro-Perez V, Martinez-Balibrea E, Musulen E, Carmona FJ, Sartore-Bianchi A, Cassingena A, Siena S, Elez E, Tabernero J, Salazar R, Abad A and Esteller M. Epigenetic inactivation of the BRCA1 interactor SRBC and resistance to oxaliplatin in colorectal cancer. Journal of the National Cancer Institute. 2014; 106:djt322.
147. Zochbauer-Muller S, Fong KM, Geradts J, Xu X, Seidl S, End-Pfutzenreuter A, Lang G, Heller G, Zielinski CC, Gazdar AF and Minna JD. Expression of the candidate tumor suppressor gene hSRBC is frequently lost in primary lung cancers with and without DNA methylation. Oncogene. 2005; 24:6249-6255.
148. Lee JH, Byun DS, Lee MG, Ryu BK, Kang MJ, Chae KS, Lee KY, Kim HJ, Park H and Chi SG. Frequent epigenetic inactivation of hSRBC in gastric cancer and its implication in attenuated p53 response to stresses. International journal of cancer. 2008; 122:1573-1584.
149. Tong SY, Ki KD, Lee JM, Kang MJ, Ha TK, Chung SI, Chi SG and Lee SK. Frequent inactivation of hSRBC in ovarian cancers by promoter CpG island hypermethylation. Acta obstetricia et gynecologica Scandinavica. 2010; 89:629-635.
150. Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, Fletcher O, Dos Santos Silva I, Peto J, Boshoff C, Narod S and Petronis A. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Human molecular genetics. 2009; 18:1332-1342.
151. Brennan K, Garcia-Closas M, Orr N, Fletcher O, Jones M, Ashworth A, Swerdlow A, Thorne H, Riboli E, Vineis P, Dorronsoro M, Clavel-Chapelon F, Panico S, Onland-Moret NC, Trichopoulos D, Kaaks R, et al. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer research. 2012; 72:2304-2313.
152. Das M, Saikia BJ, Sharma SK, Sekhon GS, Mahanta J and Phukan RK. p16 hypermethylation: a biomarker for increased esophageal cancer susceptibility in high incidence region of North East India. Tumour biology. 2015; 36:1627-1642.
153. Toyooka S, Matsuo K and Gazdar AF. DNA methylation in lung cancer. The New England journal of medicine. 2008; 358:2513; author reply 2514.
154. Auerkari EI. Methylation of tumor suppressor genes p16(INK4a), p27(Kip1) and E-cadherin in carcinogenesis. Oral oncology. 2006; 42:5-13.
155. Al-Kaabi A, van Bockel LW, Pothen AJ and Willems SM. p16INK4A and p14ARF gene promoter hypermethylation as prognostic biomarker in oral and oropharyngeal squamous cell carcinoma: a review. Disease markers. 2014; 2014:260549.
156. Hesson LB, Cooper WN and Latif F. The role of RASSF1A methylation in cancer. Disease markers. 2007; 23:73-87.
157. Wang H, Wang S, Shen L, Chen Y, Zhang X, Zhou J, Wang Z, Hu C and Yue W. Chk2 down-regulation by promoter hypermethylation in human bulk gliomas. Life sciences. 2010; 86:185-191.
158. Kim DS, Kim MJ, Lee JY, Lee SM, Choi JE, Lee SY and Park JY. Epigenetic inactivation of checkpoint kinase 2 gene in non-small cell lung cancer and its relationship with clinicopathological features. Lung cancer. 2009; 65:247-250.
159. Li W, Deng J and Tang JX. Combined effects methylation of FHIT, RASSF1A and RARbeta genes on non-small cell lung cancer in the Chinese population. Asian Pacific journal of cancer prevention. 2014; 15:5233-5237.
160. Liu L, Sun L, Li C, Li X, Zhang Y, Yu Y and Xia W. Quantitative detection of methylation of FHIT and BRCA1 promoters in the serum of ductal breast cancer patients. Bio-medical materials and engineering. 2015; 26 :S2217-2222.
161. Leal M, Lima E, Silva P, Assumpcao P, Calcagno D, Payao S, Burbano RR and Smith M. Promoter hypermethylation of CDH1, FHIT, MTAP and PLAGL1 in gastric adenocarcinoma in individuals from Northern Brazil. World journal of gastroenterology. 2007; 13:2568-2574.
162. Banzai C, Nishino K, Quan J, Yoshihara K, Sekine M, Yahata T and Tanaka K. Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. International journal of clinical oncology. 2014; 19:127-132.
163. Lee EJ, Lee BB, Kim JW, Shim YM, Hoseok I, Han J, Cho EY, Park J and Kim DH. Aberrant methylation of Fragile Histidine Triad gene is associated with poor prognosis in early stage esophageal squamous cell carcinoma. European journal of cancer. 2006; 42:972-980.
164. Zhao H, Li J, Li X, Han C, Zhang Y, Zheng L and Guo M. Silencing GPX3 Expression Promotes Tumor Metastasis in Human Thyroid Cancer. Current protein & peptide science. 2015; 16:316-321.
165. Zhou JD, Yao DM, Zhang YY, Ma JC, Wen XM, Yang J, Guo H, Chen Q, Lin J and Qian J. GPX3 hypermethylation serves as an independent prognostic biomarker in non-M3 acute myeloid leukemia. American journal of cancer research. 2015; 5:1786-1794.
166. Cao S, Yan B, Lu Y, Zhang G, Li J, Zhai W, Guo W and Zhang S. Methylation of promoter and expression silencing of GPX3 gene in hepatocellular carcinoma tissue. Clinics and research in hepatology and gastroenterology. 2015; 39:198-204.
167. Chen B, Rao X, House MG, Nephew KP, Cullen KJ and Guo Z. GPx3 promoter hypermethylation is a frequent event in human cancer and is associated with tumorigenesis and chemotherapy response. Cancer letters. 2011; 309:37-45.
168. Maldonado L, Brait M, Loyo M, Sullenberger L, Wang K, Peskoe SB, Rosenbaum E, Howard R, Toubaji A, Albadine R, Netto GJ, Hoque MO, Platz EA and Sidransky D. GSTP1 promoter methylation is associated with recurrence in early stage prostate cancer. The Journal of urology. 2014; 192:1542-1548.
169. Heyn H and Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nature reviews Genetics. 2012; 13:679-692.
170. Yan W, Herman JG and Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. 2016;8:119-33.