
INTRODUCTION
Malignant pleural effusion (MPE) i.e. the accumulation of fluid in the pleural cavity resulting from tumor cell infiltration of the pleura, is a common manifestation of a wide range of malignancies and is related to short life expectancy and significant morbidity [1, 2]. Current therapies for MPE are merely palliative, associated with side effects or discomfort and benefit only selected patients [3, 4]. Devising novel, safe and effective treatment modalities is therefore urgently needed.
Small GTPases are G-proteins involved in the transduction of signals that promote epithelial cell transformation and cancer cell proliferation, survival, migration and invasion thus being critical to tumor initiation, progression and metastasis [5, 6]. Besides their role in tumor cell biology, they orchestrate several host-related tumor-promoting phenomena such as angiogenesis [7–9], vascular permeability [10, 11], and tumor-associated inflammatory responses [12]. Interestingly, all these aforementioned biological procedures constitute the pathogenetic basis of MPE [13]. We therefore assumed that the inhibition of small GTPases would limit MPE accumulation. Inhibition of the Icmt enzyme prevents post-translational modification of GTPases that is required for their activation and this might be the most effective way to block GTPase activity [14]. In the studies presented here, we aimed at investigating the effects of the Icmt inhibitor cysmethynil [15] in experimental MPE [16, 17] and explore the underlying mechanisms. We hypothesized that cysmethynil would block MPE accumulation by inhibiting angiogenesis and pleural vascular permeability and by modulating tumor-initiated host immune response.
RESULTS
Cysmethynil blocks malignant pleural effusion accumulation in both models and reduces tumor burden in the lung-adenocarcinoma model
In order to examine whether cysmethynil affects MPE accumulation we used two syngeneic murine models: an adenocarcinoma and a mesothelioma-induced MPE. Cysmethynil significantly reduced pleural fluid volume in both models (Figure 1A). Even though cysmethynil reduced pleural fluid accumulation in both models, it impacted pleural tumor burden differently. While it significantly reduced the number of tumor foci in the adenocarcinoma model it did not affect tumor mass in the mesothelioma one (Figure 1B).
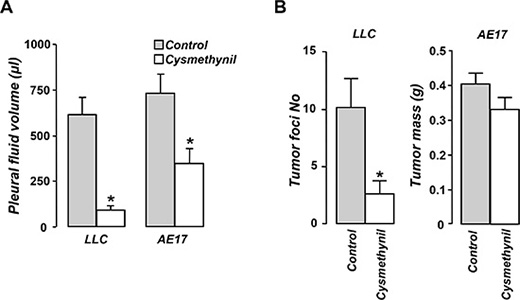
Figure 1: Cysmethynil inhibits pleural fluid accumulation in adenocarcinoma and mesothelioma-induced MPE and reduces adenocarcinoma pleural dissemination. LLC or AE17 cells were intrapleurally injected in syngeneic mice. Mice were intraperitoneally administered with Cysmethynil 0.2 mg/g (body weight) or vehicle thrice per week. 11–14 days later, pleural fluid was retrieved and quantified (A). (B) Mesothelioma tumors were excised and weighed and adenocarcinoma lung implantations were counted under a stereoscope. Data presented as mean ± SEM, n = 6–11, *p < 0.05 compared to vehicle.
Cysmethynil targets pleural vascular permeability and tumor angiogenesis
We subsequently investigated the mechanisms of the effusion-limiting effects of cysmethynil. We thus examined whether cysmethynil affects fundamental components of MPE pathobiology: vascular permeability, angiogenesis and tumor associated inflammatory response [12]. We demonstrated that the inhibitor significantly reduced pleural vascular permeability (Figure 2A) and tumor angiogenesis in both models (Figure 2B and 2C).
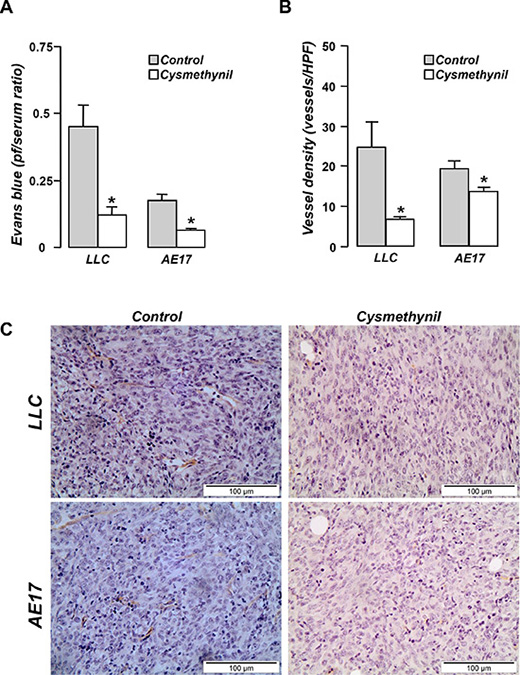
Figure 2: Cysmethynil inhibits pleural vascular hyper-permeability and tumor angiogenesis. (A) MPE-bearing mice received 800 μg of Evans blue i.v. and sacrificed 1 h later. Vascular permeability was evaluated according to the pleural fluid (pf)/serum Evans blue concentration. (B) Tumor tissue sections were stained with endothelial marker CD31 and vascular density was assessed. (C) Representative photographs of CD-31 stained tumor tissue sections at 400x. HPF: High Power Field. Data presented as mean ± SEM, n = 5–7. *p < 0.05 compared to vehicle.
Cysmethynil limits endothelial cell growth, migration and tube formation
In order to elucidate the mechanisms of the in vivo anti-angiogenic effects of the inhibitor we asked whether cysmethynil directly affects critical endothelial cell functions including growth, migration and de novo vessel formation in Matrigel. We found that cysmethynil significantly inhibited endothelial cell viability (Figure 3A) and suppressed their migration by almost 50% (Figure 3B). We then used the matrigel-based system to investigate the ability of endothelial cells to form two-dimensional, capillary-like tubes de novo (Figure 3C). We thus measured the total vessel length and the number of branching points (signaling the size and complexity/maturity of the network) in the tube networks. Cysmethynil-treated endothelial cells formed shorter and less complicated vessel networks than the vehicle-treated ones (Figure 3C).
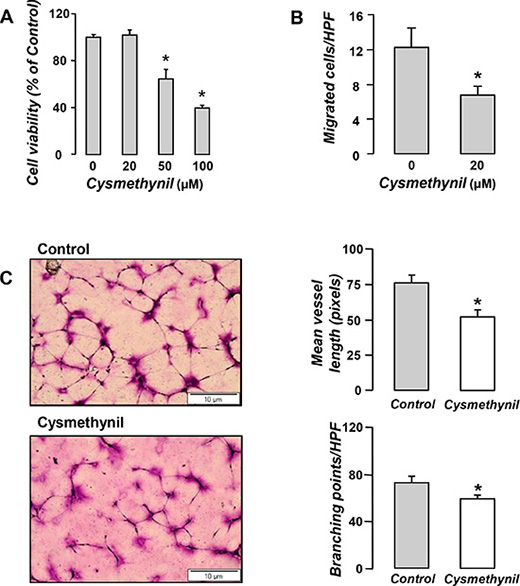
Figure 3: Anti-angiogenic properties of Cysmethynil in vitro. (A) HUVECs were treated with vehicle or cysmethynil (20–100 μM) and 24 h later cell viability was evaluated by MTS. (B) For migratory potential evaluation, HUVECs were loaded onto Transwell inserts in the presence of cysmethynil or vehicle. Lower chambers were filled with full medium. 6 hours later, migrated cells were fixed, stained and counted under a microscope. (C) Serum starved HUVECs were loaded onto Matrigel coated wells and incubated overnight in the presence of cysmethynil (0.5–20 μM) or vehicle. (Left) Representative pictures of the tubular network. (Right) Results of image analysis for network length and branching points using ImageJ (NIH, Bethesda, MD). HPF: High Power Field. Data presented as mean ± SEM, n = 6–9 *p < 0.05 compared to vehicle.
To further assess any effect of cysmethynil in the angiogenic potential of tumor cells we subsequently measured VEGF levels in the tumor lysates, pleural fluids and tumor cell supernatants. No significant difference in VEGF levels was measured in tumors, fluid or cells exposed to the inhibitor compared to the control ones (data not shown) implying that the anti-angiogenic effects of cysmethynil should be more likely ascribed to its direct effects on endothelial cells.
Cysmethynil promotes pleural space macrophage homing and their shift towards an M1-phenotype
Tumor associated inflammation plays a major role in malignant pleural effusion accumulation [13]. Although no significant difference was observed in total cellularity of pleural effusions among groups (data not shown), cysmethynil treated animals exhibited increased numbers of pleural space macrophages (Figure 4A). Moreover, analysis of the IL-12/IL-10 expression profile of the pleural macrophages demonstrated a significant shift towards an M1 anti-tumor phenotype in cysmethynil treated animals (Figure 4C). In tumors, the inhibitor-treated mesothelioma-bearing animals had higher numbers of macrophages compared to controls (Figure 4B), while no significant difference was observed in the adenocarcinoma model (Figure 4B). Finally, no significant effect in the intra-tumoral macrophage polarization was observed in both models (Figure 4D).
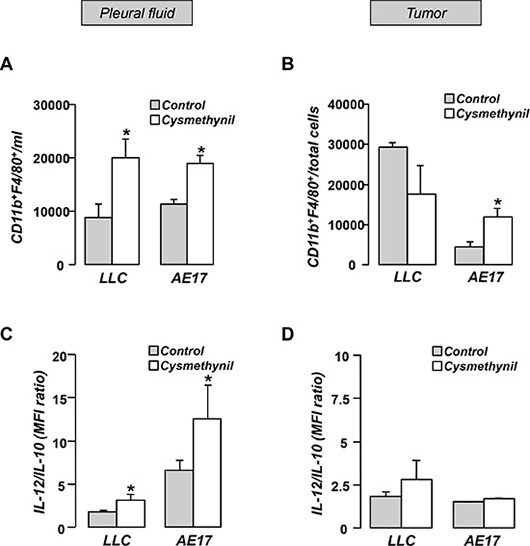
Figure 4: Pleural fluid. (A) and tumors (B) of LLC or AE17 MPE-bearing animals were analyzed for the presence of CD11b+/F4/80+ macrophages. The population was further analyzed for the IL-12/IL-10 expression using flow cytometry (C, D). Data presented as mean ± SEM, n = 6–7 *p < 0.05 compared to vehicle.
Cysmethynil inhibits tumor-induced M2 macrophage polarization and enhances macrophage migration in vitro
We then tested the effect of the inhibitor in macrophage polarization and migration in vitro. When co-cultured with tumor cells, macrophages were polarized towards an M2 phenotype and this alteration was partially reversed by cysmethynil (Figure 5A). In addition, cysmethynil promoted macrophage migration towards tumor cells (Figure 5B).
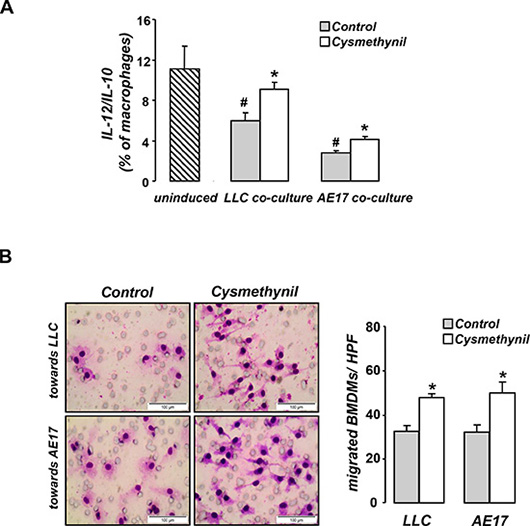
Figure 5: Cysmethynil inhibits tumor-driven M2 polarization (A) and migration (B) of macrophages in vitro. (A) Macrophages were co-cultured with LLC or AE17 cells for 24 h and subsequently treated with vehicle or 20 μm Cysmethynil. 24 h later, macrophages were collected, stained for CD11b/F4/80/IL-12/IL-10 expression and analyzed by flow cytometry. (B) For migration studies, serum starved macrophages treated with vehicle or 20 μm Cysmethynil were loaded on Transwell inserts to migrate towards LLC or AE17 cells. (Left) Representative pictures of migrated cells. (Right) Results of migrated cell counts. HPF: High Power Field. Data presented as mean ± SEM, n = 6–7 *p < 0.05 compared to vehicle, #p < 0.05 compared to uninduced.
Cysmethynil promotes adenocarcinoma cell apoptosis in vivo
Pleural tumor tissues were subsequently analyzed for tumor cell apoptosis (Figure 6A) and proliferation rate (Figure 6B). Cysmethynil promoted tumor cell apoptosis in the adenocarcinoma but not in the mesothelioma model. Proliferation rate did not differ between groups in both models. Cysmethynil was also found to reduce tumor cell viability in vitro (Figure 6C).
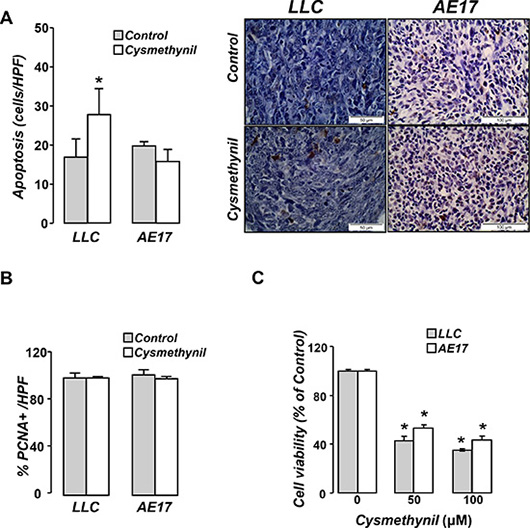
Figure 6: Cysmethynil induces tumor cell apoptosis in vivo and reduces tumor cell viability in vitro. Τumor tissue sections from vehicle or cysmethynil treated animals were analyzed for apoptosis by TUNEL assay (A) while tumor cell proliferation rates were evaluated by PCNA staining (B). Data presented as mean±SEM, n = 7–10 *p < 0.05 from vehicle. HPF: High Power Field. (C) Tumor cells were treated with vehicle or cysmethynil (20–100 μM) and 24 h later cell viability was evaluated by MTS. Data presented as mean ± SEM, n = 5–7. *p < 0.05 compared to vehicle.
DISCUSSION
In the studies presented here we aimed at examining whether cysmethynil, a small GTPase inhibitor, possesses an MPE-limiting capacity, using two experimental MPE models (mesothelioma- and lung adenocarcinoma-induced MPE) established by our group. We demonstrated that cysmethynil: a. Profoundly reduced pleural fluid volume in both models and suppressed pleural adenocarcinoma tumor dissemination while mesothelioma tumor growth was not affected; b. Inhibited pleural vascular permeability and tumor angiogenesis in vivo and reduced endothelial cell proliferation, migration and tube formation in vitro; c. promoted pleural but not tumoral macrophage infiltration, forced pleural macrophages towards an M1 anti-tumor phenotype in vivo, and partially prevented a tumor-cell induced M2 polarization of macrophages in vitro; d. promoted adenocarcinoma but not mesothelioma cell apoptosis in vivo.
This is the first study to show that Icmt inhibition limits MPE. Previous studies have focused on the role of Icmt in tumor initiation and progression [15, 19, 23–26]. Notably, while cysmethynil reduced the size of MPEs in both models, it differentially affected tumor progression: it stimulated apoptosis in vivo and reduced the number of pleural tumor foci in the adenocarcinoma model, but did not affect mesothelioma cell survival in vivo or the size of mesothelioma tumors. While in the in vitro setting, adenocarcinoma cells were more sensitive to the inhibitor than mesothelioma ones, the magnitude of this difference is marginal. It is therefore unlikely that different sensitivity to the compound can explain the divergent in vivo responses. Since adenocarcinoma pleural growth is characterized by disseminated small foci [16] while mesothelioma forms massive tumors [17] it could be speculated that different pharmacokinetics may play a role in the observed different response in vivo. The observation that the MPE size was independent of the effects of the inhibitor in tumor burden, supports the assumption that the mechanisms determining the pleural fluid accumulation and the tumor growth are different. On the other hand, it should be emphasized that our findings should not be interpreted as evidence that mesotheliomas and lung adenocarcinomas in general would be expected to respond to the compound in a fashion similar to that of AE17 and LLC cells, respectively.
We here demonstrate for the first time, that Icmt, besides inhibiting tumor cell growth, may impact certain host-related hallmarks of cancer, i.e. vascular changes and tumor-associated immune response. The anti-angiogenic and the anti-hyperpermeability properties of the drug are probably central to its anti-MPE activity, since vascular changes represent sine qua non components of MPE pathogenesis [13]. Cysmethynil blocked in vitro endothelial cell proliferation, migration and tube formation, suggesting a direct effect of the inhibitor on endothelial cells. These vascular effects of the inhibitor should be possibly ascribed to its small GTPase-inhibitory properties. In fact, activation of endothelial cell RhoA, RhoG, Rac1 and cdc42 has been shown to stimulate several steps of the angiogenic process [8, 9] and endothelial cell RhoA mediates induction of vascular hyper-permeability [10, 11].
Another essential aspect of MPE pathobiology is the host immune/inflammatory response to pleural tumors [13]. We observed increased numbers of pleural space macrophages in the inhibitor-treated animals, a finding that comes in agreement with the previously observed accumulation of pan-Rho-deficient macrophages in the peritoneal cavity in a peritonitis model [27]. In addition, MPE macrophages were shifted towards an M1 phenotype in cysmethynil-treated animals compared to the control ones and, in vitro, the inhibitor suppressed tumor cell-driven M2 macrophage polarization. Even though the role of M2 macrophages in tumor progression has been long established [28], there is no direct experimental proof that they promote MPE. However, this is likely to be the case since they secrete pro-angiogenic and vascular-permeability-enhancing mediators which are implicated in MPE pathobiology [13]. Moreover, pleural fluid M2 abundance has been associated with worst prognosis in patients with MPE [29]. Surprisingly, while cysmethynil uniformly increased MPE macrophage numbers in both models, only mesothelioma tumors were found to be affected in this fashion. In addition, while the inhibitor induced M1-polarization of fluid macrophages, the phenotype of tumor macrophages was not affected. The reason of this discrepancy remains elusive. However, this anatomic compartment-dependent effect denotes that the effects of the inhibitor on macrophages might be substantially influenced by other factors released in specific in vivo micro-environments.
This proof-of-principle study supporting the notion that Icmt inhibitor may block MPE formation holds certain clinical implications. First, our findings may pave the road for clinical development of Icmt inhibitors [26] and clinical testing in patients with MPE. Second, the fact that the inhibitor targets host-related aspects of MPE pathobiology, may suggest that its activity is irrespective of specific genetic profile of the tumors that cause the effusion. Third, drugs targeting host-related cancer hallmarks might be less prone to the emergence of resistance, since host cells are not characterized by genomic instability as tumor cells are [30–31]. Fourth, inducing tumor-associated macrophages to acquire an M1-phenotype might be a novel immunotherapy strategy [32] and cysmethynil could be used for this purpose though capable to target only pleural and not tumoral macrophages. Fifth, accumulating data outline the role of Rho and Ras in chemosensitivity of tumor cells [33–35]. Therefore, cysmethynil may display synergy with standard cytotoxic agents, a possibility that should be tested in future studies.
In conclusion, we here demonstrated that cysmethynil curtails experiential MPE formation, irrespective of its in vivo activity on the growth of the pleural tumors. Its effects are mostly related to inhibition of new vessel formation and vascular hyper-permeability and to an increase of M1-macrophage homing. The clinical development and testing of cysmethynil as a palliative pharmacological intervention for patients with MPE might be therefore worthy of pursuit.
MATERIALS AND METHODS
Tumor cell lines
Murine lung adenocarcinoma cells (Lewis Lung Cells, LLC) were purchased from American Type Collection Cultures (Manassas, VA). AE17 murine mesothelioma cells [18] were generated by Dr B. Robinson and kindly provided by Dr YCG Lee, Perth, Western Australia. Cells were maintained in Dulbecco’s Modified Eagle Medium (containing 10% heat-inactivated FCS, L-glutamine and 100 mg/l penicillin and streptomycin (Gibco, Grand Island, NY). Both cell lines used in the present study were periodically monitored for mycoplasma presence. Their morphology was examined on a regular basis. The effects of the inhibitor in tumor cell viability were assessed using the MTS (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulphophenyl]-2H-tetrazolium, inner salt) (Promega, Madison, WI) assay.
Animal studies
Mice were purchased form BSRC Al. Fleming (Vari, Greece) or Hellenic Pasteur Institute (Athens, Greece) and were housed at the Animal Model Research Unit of Evangelismos Hospital, (Athens, Greece). Experiments were approved by the Veterinary Administration Bureau, Prefecture of Athens, Greece under compliance to the national law and the EU Directives. Adenocarcinoma LLC (1.5 × 105) or mesothelioma AE17 (5 × 105) cells were intrapleurally injected to 8–10 week-old male, C57BL/6 syngeneic mice and cysmethynil (purchased from Duke University, Durham, NC) was intraperitoneally administered at 0.2 mg/g three times weekly starting at the 4th day of tumor cell injection since pleural tumors are already evident at this time point. This regimen was previously found to be effective in murine models of PC3 prostate cancer cells [19] and HepG2 hepatocellular carcinoma cells [15] and in our hands did not affect mouse welfare status when tested in preliminary experiments. Animals were euthanized 13 days post injection at this point usually exhibiting distress and impaired motility. Pleural fluid was retrieved and volume was measured. To determine pleural tumor burden, the thoracic cavity was opened and all of the mesothelioma masses were carefully excised (until achieving “clean” pleural surfaces) and weighed or lungs were harvested in order for the adenocarcinoma visceral pleural foci to be counted under a stereoscope (Stemi DV4, Zeiss, Germany).
Histology and flow cytometry
Formalin-fixed, paraffin-embedded tumor tissue sections were stained with anti-Proliferating Cell Nuclear Antigen (PCNA) (Santa Cruz Biotechnology, CA), or anti-CD31 (Abcam, Cambridge, MA) as previously described [20]. Tumor cell apoptosis was evaluated by TUNEL assay (Roche, Penzberg, Germany) [20]. For flow cytometry analysis, pleural fluid and tumor cells were fixed, permeabilised and stained with anti-CD45-APC-Cy-7 (pan-leucocyte marker), anti-CD11b-FITC (mature myeloid cell marker), anti-F4/80-PerCP (tumor-associated macrophages), anti-IL10-PE (prototype cytokine of pro-tumor M2 phenotype) and anti-IL12-APC (prototype cytokine of anti-tumor M1 phenotype). The M1/M2 ratio was used as a marker of macrophage polarization. Samples were analyzed using the multicolor flow cytometer BD FacsCanto II. Flow cytometric data were analyzed using the BD FACSDiva Version 6.1.3 Software (BD Bioscience, Athens, Greece). Inflammatory cells were selected based on their forward and side scatter profiles and their CD45 positive staining.
In vivo vascular permeability assay
In order to assess pleural vascular permeability, MPE-bearing animals were intravenously injected with 200 μl, of the albumin-binding Evans blue dye (4 mg/ml) upon anesthesia and sacrificed 1 hour later. Pleural fluid and blood were collected, centrifuged and supernatants’ Evans blue levels were determined by measurement at 630 nm. Concentration of the dye was interpolated using a standard curve and permeability was evaluated by the ratio of pleural fluid/serum Evans blue concentration. This ratio expresses the degree of albumin leakage from the circulation to the pleural space through a leaky pleural vasculature.
In vitro macrophage polarization and migration assays
Bone marrow-derived macrophages from C57BL/6 mice were generated using standard techniques [21] and were seeded at 1 × 105 cells/1,5 ml in the lower compartments of (0.4 μm) Transwell system with an equal population of LLC or AE17 cells loaded on the upper chambers. Macrophages were treated with 20 μM (non-toxic dose) cysmethynil or vehicle 24 h later and left for another 24 h. At the end of the incubation time, macrophages were collected, fixed, permeabilised and stained for CD11b, F4/80 and IL-10, IL-12 presence for flow cytometric analysis.
For migration studies macrophages were serum starved overnight and treated with vehicle or 20 μM cysmethynil. Two hours later, cells were collected and loaded onto the upper compartments of Transwells (8-μm pore size), while the day before lower compartments were seeded with 3 × 104 LLC or AE17 cells. All compartments were serum deprived. Migrated cells were fixed, stained and counted under a microscope.
In vitro endothelial cell viability, migration and tube formation
Human Umbilical Vein Endothelial Cells (HUVEC) were isolated as previously described [23] and maintained in M199 full medium (15% FCS, 200 μg/ml ECGS and 5U/ml heparin). HUVECs were used until passage 2. HUVEC viability was assessed using the MTS assay. For migration studies HUVECs were serum starved overnight, treated with vehicle or 5 μM (non-toxic dose) cysmethynil and loaded on the upper compartments of Transwells at 1 × 105 cells/chamber. Lower chambers were filled with full medium and 6 hours later, migrated cells were fixed, stained and counted under a microscope.
For endothelial cell tube formation, 24-well plates were coated with 40 μl Matrigel (growth factor reduced, BD Bioscience, Athens, Greece) and incubated for 1 h at 37°C. Serum deprived HUVEC were loaded onto pre- coated wells at a density of 4 × 104 cells/well and incubated for 18 h in the presence of cysmethynil concentrations that do not affect cell viability (0.5–20 μM) or vehicle. Following incubation tubes were fixed, stained and tube network was imaged at 200X magnification under a phase contrast microscope. Images were subsequently analyzed for network length and branching points using ImageJ (NIH, Bethesda, MD).
Statistics
All values are presented as mean ± standard error of mean (SEM). Differences between groups were evaluated using the Student’s t-test or the one-way ANOVA with least square difference post-hoc tests, as appropriate. P values < 0.05 were considered significant. Statistical analysis was performed using the Statistical Package for the Social Sciences v.13.0.0 (IMB, Armonk, NY).
ACKNOWLEDGMENTS
We thank Dr E. Aravidou and Z. Kollia (Research Unit for animal standards, Evangelismos Hospital, Athens, Greece) for professional veterinarian and animal care assistance, respectively.
GRANT SUPPORT
This work was supported by the ‘Thorax’ Foundation (Athens, Greece).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Sahn SA. Malignant Pleural Effusions. In: Fishman AP, Elias JA, Fishman JA, Grippi MA, Kaiser LR, Senior RM, eds. Fishman’s Pulmonary Diseases and Disorders. 3rd ed. New York, NY: McGraw Hill; 1998:1429–1438.
2. Light RW. Pleural Diseases. Philadelphia, PA: Lippincott, Williams and Wilkins; 2001:108–134.
3. Burgers JA, Kunst PW, Koolen MG, Willems LN, Burgers JS, van den Heuvel M. Pleural drainage and pleurodesis: implementation of guidelines in four hospitals. Eur Respir J. 2008; 32:1321–1327.
4. Roberts ME, Neville E, Berrisford RG, Antunes G, Ali NJ; BTS Pleural Disease Guideline Group. BTS Pleural Disease Guideline Group. Management of a malignant pleural effusion: British Thoracic Society Pleural Disease Guideline 2010. Thorax. 2010; 65:32–40.
5. Etienne-Manneville S and Hall A. Rho GTPases in cell biology. Nature. 2002; 12:629–635.
6. Buongiorno P, Bapat B. Rho GTPases and cancer. Prog Mol Subcell Biol. 2005; 40:29–53.
7. Coleman ML1, Marshall CJ, Olson MF. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol. 2004; 5:355–366.
8. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004; 265:23–32.
9. Bryan BA, D’Amore PA. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cellular and Molecular Life Sciences. 2007; 64:2053–2065
10. Adamson RH, Curry FE, Adamson G, Liu B, Jiang Y, Aktories K, Barth H, Daigeler A, Golenhofen N, Ness W, Drenckhahn D. Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol. 2002; 539:295–308.
11. Baumer Y, Burger S, Curry FE, Golenhofen N, Drenckhahn D, Waschke J. Differential role of Rho GTPases in endothelial barrier regulation dependent on endothelial cell origin. Histochem Cell Biol. 2008; 129:179–191.
12. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011; 11:761–774.
13. Stathopoulos GT, Kalomenidis I. Malignant pleural effusion: tumor-host interactions unleashed. Am J Respir Crit Care Med. 2012; 186:487–92.
14. Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Gomes AQ, Seabra MC, Young SG. Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J Biol Chem. 2001; 276:5841–5845.
15. Wang M, Hossain MS, Tan W, Coolman B, Zhou J, Liu S, Casey PJ. Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene. 2010; 29:4959–4970.
16. Stathopoulos GT, Zhu Z, Everhart MB, Kalomenidis I, Lawson WE, Bilaceroglu S, Peterson TE, Mitchell D, Yull FE, Light RW, Blackwell TS. Nuclear factor-kappaB affects tumor progression in a mouse model of malignant pleural effusion. Am J Respir Cell Mol Biol. 2006; 34:142–150
17. Vazakidou ME, Magkouta S, Moschos C, Psallidas I, Pappas A, Psarra K, Kalomenidis I. Temsirolimus targets multiple hallmarks of cancer to impede mesothelioma growth in vivo. Respirology. 2015; 20:1263–71
18. Jackaman C, Bundell CS, Kinnear BF, Smith AM, Filion P, van Hagen D, Robinson BW, Nelson DJ. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol. 2003; 171:5051–63.
19. Wang M, Tan W, Zhou J, Leow J, Go M, Lee HS, Casey PJ. A small molecule inhibitor of isoprenylcysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J Biol Chem. 2008; 283:18678–18684
20. Psallidas I, Stathopoulos GT, Maniatis NA, Magkouta S, Moschos C, Karabela SP, Kollintza A, Simoes DC, Kardara M, Vassiliou S, Papiris SA, Roussos C, Kalomenidis I. Secreted phosphoprotein-1 directly provokes vascular leakage to foster malignant pleural effusion. Oncogene. 2013; 32:528–535.
21. Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008; Chapter 14:Unit 14.1.
22. Hatziapostolou M, Katsoris P, Papadimitriou E. Different inhibitors of plasmin differentially affect angiostatin production and angiogenesis. Eur J Pharmacol. 2003; 460:1–8.
23. Bergo MO, Gavino BJ, Hong C, Beigneux AP, McMahon M, Casey PJ, Young SG. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J Clin Invest. 2004; 113:539–550
24. Wahlstrom AM, Cutts BA, Liu M, Lindskog A, Karlsson C, Sjogren AK, Andersson KM, Young SG, Bergo MO. Inactivating Icmt ameliorates K-RAS-induced myeloproliferative disease. Blood. 2008; 112:1357–1365.
25. Teh JT, Zhu WL, Ilkayeva OR, Li Y, Gooding J, Casey PJ, Summers SA, Newgard CB, Wang M. Isoprenylcysteine carboxylmethyltransferase regulates mitochondrial respiration and cancer cell metabolism. Oncogene. 2015; 34:3296–3304.
26. Ramanujulu PM, Yang T, Yap SQ, Wong FC, Casey PJ, Wang M, Go ML. Functionalized indoleamines as potent, drug-like inhibitors of isoprenylcysteine carboxyl methyltransferase (Icmt). Eur J Med Chem. 2013; 63:378–86.
27. Königs V, Jennings R, Vogl T, Horsthemke M, Bachg AC, Xu Y, Grobe K, Brakebusch C, Schwab A, Bähler M, Knaus UG, Hanley PJ. Mouse macrophages completely lacking Rho subfamily GTPases (RhoA, RhoB, and RhoC) have severe lamellipodial retraction defects, but robust chemotactic navigation and altered motility. J Biol Chem. 2014; 289:30772–30784.
28. Quatromoni J, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012; 4:376–389
29. Yang L, Wang F, Wang L, Huang L, Wang J, Zhang B, Zhang Y. CD163+ tumor-associated macrophage is a prognostic biomarker and is associated with therapeutic effect on malignant pleural effusion of lung cancer patients. Oncotarget. 2015; 6:10592–10603. doi: 10.18632/oncotarget.3547.
30. Wang M, Khoo YM, Zhou J, Casey P, Lee HS. A high-performance liquid chromatography method for the quantification of cysmethynil, an inhibitor of isoprenylcysteine carboxylmethyl transferase, in mouse plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 15; 877:553–7.
31. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martín-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 28:358–365.
32. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013; 19:1264–1272.
33. Sharma S, Santiskulvong C, Bentolila LA, Rao J, Dorigo O, Gimzewski JK. Correlative nanomechanical profiling with super-resolution F-actin imaging reveals novel insights into mechanisms of cisplatin resistance in ovarian cancer cells. Nanomed. Nanotechnol. Biol. Med. 2012; 8:757–766.
34. Sharma S, Santiskulvong C, Rao, J, Gimzewski JK, Dorigo O. The role of Rho GTPase in cell stiffness and cisplatin resistance in ovarian cancer cells. Integr. Biol. Quant. Biosci. Nano Macro. 2014; 6:611–617
35. Salaroglio IC, Campia I, Kopecka J, Gazzano E, Sara O, Ghigo D, Riganti C. Zolendronic acid overcomes chemoresistance and immunosuppression of malignant mesothelioma. Oncotarget. 2014; 6:1028–1142. doi: 10.18632/oncotarget.2731.